
Abstract
There are two distinct subtypes of dura mater graft-associated Creutzfeldt–Jakob disease (dCJD) with methionine homozygosity at codon 129 of the PRNP gene. The majority of cases is represented by a non-plaque-type (np-dCJD) resembling sporadic CJD (sCJD)-MM1 or -MV1, while the minority by a plaque-type (p-dCJD). p-dCJD shows distinctive phenotypic features, namely numerous kuru plaques and an abnormal isoform of prion protein (PrPSc) intermediate in size between types 1 and 2. Transmission studies have shown that the unusual phenotypic features of p-dCJD are linked to the V2 prion strain that is associated with sCJD subtypes VV2 or -MV2. In this study, we applied protein misfolding cyclic amplification (PMCA) using recombinant human prion protein as a substrate and demonstrated that p-dCJD prions show amplification features that are distinct from those of np-dCJD. Although no amplification of np-dCJD prions was observed with either 129 M or 129 V substrate, p-dCJD prions were drastically amplified with the 129 V substrates, despite the PRNP codon 129 incompatibility between seed and substrate. Moreover, by using a type 2 PrPSc-specific antibody not recognizing PrPSc in p-dCJD, we found that type 2 products are generated de novo from p-dCJD prions during PMCA with the 129 V substrates. These findings suggest that our cell-PMCA is a useful tool for easily and rapidly identifying acquired CJD associated with the transmission of the V2 CJD strain to codon 129 methionine homozygotes, based on the preference for the 129 V substrate and the type of the amplified products.
Similar content being viewed by others

Main
Creutzfeldt–Jakob disease (CJD) is a fatal neurodegenerative disease. The large majority of CJD cases are thought to be caused by a spontaneous conformational change of the normal monomeric isoform of the prion protein (PrPC) into an abnormal isoform (PrPSc), as in sporadic CJD (sCJD) or genetic/familial CJD. On the other hand CJD can also be acquired through PrPSc infection, as in variant CJD or iatrogenic CJD (iCJD). The wide heterogeneity of sCJD clinico-pathological phenotypes depends on both the genotype (methionine (M) or valine (V)) at the polymorphic codon 129 of the PRNP gene and the type (1 or 2) of PrPSc accumulating in the brain.1 Type 1 and type 2 can be distinguished according to the size of the proteinase K-resistant core of the protein (21 and 19 kDa, respectively) on western blots. Based on the polymorphism at codon 129 of the PRNP gene and the type of PrPSc, sCJD patients are classified into six major subtypes: MM1/MV1, MM2 cortical, MM2 thalamic, VV1, VV2, and MV2.1
iCJD is caused by the transmission of prions via cadaveric pituitary hormones, dura mater, and corneal grafts, or contaminated neurosurgical instruments. Many cases of dura mater graft-associated CJD (dCJD) have been reported in Japan.2, 3, 4 There are two distinct phenotypes in dCJD with methionine homozygosity at codon 129. The first is a major group represented by a non-plaque-type dCJD (np-dCJD), the second a minor group represented by a plaque-type dCJD (p-dCJD).5, 6, 7 np-dCJD shares phenotypic characteristics such as diffuse synaptic-PrP deposition and type 1 PrPSc with sCJD-MM1 or -MV1, the most common sCJD phenotype (denoted as M1 strain in transmission studies).4, 8 In contrast, p-dCJD is characterized by unusual phenotypic features such as the presence of numerous kuru plaques and a unique PrPSc type with an electrophoretic mobility of about 20 kDa, which is intermediate in size between types 1 and 2 (type i PrPSc).9
We previously demonstrated that the transmission properties of p-dCJD prions resemble those of sCJD-VV2 or -MV2 prions and, in particular, that the transmission of sCJD-VV2 prions to humanized knock-in mice carrying the 129 MM genotype produce type i PrPSc and kuru plaques, indicating that this subtype of dCJD is caused by cross-sequential transmission of sCJD-VV2 or -MV2 (denoted as V2 strain),8 the second most common CJD strain, to individuals with codon 129 methionine homozygosity.9, 10, 11 Interestingly, type i PrPSc and kuru plaques in patients homozygous for methionine at codon 129 have only been observed in acquired prion diseases, especially in p-dCJD.9, 12, 13, 14, 15
A correct diagnosis and classification of cases is of critical importance for CJD surveillance centers worldwide aiming to identify potentially new disease forms or risk factors. Although PrPSc typing may be considered a tool for identification of the CJD etiology (eg, sporadic vs acquired) when no reliable evidence of CJD infection is available from clinical records, it is rather difficult to differentiate type i from type 1 PrPSc in routine western blot analysis due to the subtle differences in their electrophoretic mobility. In a transmission study with knock-in humanized mice, we have recently demonstrated, for example, that two cases with atypical CJD-MM with plaques, reported as sCJD in the literature, were actually acquired CJD cases caused by the V2 strain.16 Therefore, the most reliable current approach to identify acquired CJD is to carry out expensive and time-consuming experimental transmission studies. Under these circumstances, this study was undertaken to establish a new, rapid, and reliable method for the differentiation of acquired CJD from sCJD with PMCA using cell lysates containing exogenously expressed human PrPC as a substrate (denoted as cell-PMCA).17 We investigated the relative amplification efficiency of PrPSc and characterized PrPSc amplified products in patients with p-dCJD, np-dCJD, and sCJD. Based on the results obtained, we propose that p-dCJD (V2 strain) can be reliably distinguished from np-dCJD (M1 strain) according to distinctive amplification properties.
MATERIALS AND METHODS
Ethic Statement and Brain Tissues
Brain tissues were obtained at autopsy after receiving written informed consent for research use. For each selected case, the results of PrPSc immunohistochemistry, PRNP sequence analysis, and PrPSc typing by western blotting were available in addition to clinical and histopathological data.18, 19, 20 Brain homogenates were prepared from dCJD (np-dCJD or p-dCJD) cases with MM at codon 129, five different sCJD subtypes (MM1, MM2, MV1, MV2, or VV2), two atypical CJD-MM cases with plaques, and nine non CJD controls. Atypical CJD case #1 was a patient who had a medical history of neurosurgery without dura mater grafting, and case #2 was a neurosurgeon. Details about the clinico-pathological and molecular features, including transmission properties of the two atypical sCJD-MM cases with plaques were previously described.16, 21, 22, 23 Brains were homogenized at 10% (w/v) in PMCA buffer (50 mM Phosphate buffer (pH 6.8) containing 4 mM ethylenediaminetetraacetic acid (EDTA), 80 mM NaCl, 1.0% Nonidet P-40, and Complete Protease Inhibitor Cocktail (Roche)) and stored at −80 °C in small aliquots.
Preparation of Substrates
FreeStyle 293F cell (Invitrogen) lysates were used as substrate for cell-PMCA of human prions. Twenty percent (w/v) lysates from a cell line stably expressing human PrPC with methionine at codon 129 were prepared as described previously,17 with some modifications. Cells were homogenized at 20% (w/v) in PMCA buffer and stored at −80 °C in small aliquots. For cells expressing human PrPC with valine at codon 129, a cDNA for chimeric mouse-human PrP with 129 V was constructed as described.24 Since 293F cells endogenously express detectable levels of human 129MPrPC, to minimize the effects of simultaneous expression of both 129MPrPC and 129VPrPC, we prepared a human 129MPrPC cell line by RNA interference-mediated knockdown using short hairpin RNA (shRNA). To target the 5′ untranslated region of human PRNP mRNA (5′-GGACTTAGTGCAACAGGTT-3′), a shRNA expression vector was designed. A double-stranded DNA oligonucleotide encoding the chosen shRNA with the sense (5′-GATCCGGACTTAGTGCAACACCTTTAGTGCTCCTGGTTGAACCTGTTGCACTAAGTCCTTTTTTA-3′) and antisense sequences (5′-AGCTTAAAAAAGGACTTAGTGCAACAGGTTCAACCAGGAGCACTAAACCTGTTGCACTAAGTCCG -3′) was cloned into the expression vector pBAsi-hUp pur (Takara bio Inc, Japan) after digestion with BamHI and XhoI. The generated plasmids were linearized with ScaI (Takara) and transfected into 293F cells using Lipofectamine 2000 transfection reagent (Invitrogen), according to the manufacturer’s instruction. Cells expressing the lowest levels of PRNP mRNA were selected by real-time PCR, and maintained in Freestyle293 expression medium (Gibco) supplemented with puromycin. The levels of endogenous expression of either PRNP mRNA or PrPC in the knockdown cells were reduced by 90% or 95%, respectively. Twenty percent (w/v) lysates of 129MPrPC knockdown cell line stably expressing human 129 V- PrPC were prepared as described above. Human 129 M- or 129 V- PrPC concentration in the each of the two 20% (w/v) cell lysates was 5–10 times higher than the concentration of human PrPC in 10% (w/v) brain homogenates from knock-in humanized mice. Cell lysates were stored at −80 °C in small aliquots until use.
Estimation of PrPSc Levels in Brain Homogenates
After partial purification with collagenase digestion and Sarkosyl-NaCl extraction, samples were run in 12% Mini-PROTEAN TGX Precast Gels (Bio-Rad Laboratories), together with an internal positive control with optimal signal intensity, and probed with the primary antibody 3F4 (Signet).25 Undiluted and 10-fold diluted brain samples were loaded together with 10 μl of the control. Western blot signal intensities were measured by densitometry using the Quantity One software and the imaging device VersaDoc 5000 (Bio-Rad Laboratories). The relative concentration of PrPres in each brain homogenate was expressed by the dilution factor needed to equalize the signal intensity of PrPres between the examined samples and the positive control.
Cell-PMCA
Cell-PMCA was performed as previously described,17 with some modifications. For amplification of CJD prions, brain homogenates were combined with the cell lysates, expressing human PrPC carrying either M or V at codon 129, in a 0.1-ml-thin-walled PCR tube. Mixtures were then subjected to rounds of PMCA up to a 10−4 fold dilution (eg, 100, 1000, 10.000 times) using the cell lysate as the diluent. Since the amount of PrPSc accumulated in each brain was extremely variable, brain homogenates were subjected to PMCA reaction after serial dilution in cell lysates without adjusting the amounts of the PrPSc seed. Each PMCA round comprised 48 cycles of sonication (5 pulses of 5 s with 1-s rest) and agitation (1 h at 37 °C) and was carried out using a fully automatic cross-ultrasonic protein activating apparatus (ELESTEIN 070-GOT, Elekon Science, Japan).
Proteinase K Digestion and Western Blotting Analysis
Either before or after PMCA, samples were digested with 50 μg/ml proteinase K at 37 °C for 60 min. The digested samples were subjected to SDS-PAGE and western blot analysis. Anti-PrP monoclonal antibody 3F4 or type 2 PrPSc-specific polyclonal antibody (designated as Tohoku 2 (T2))10 was used as primary antibodies. Anti-rabbit EnVision+ or anti-mouse EnVision+ (Dako) were used as secondary antibodies. Enhanced chemiluminescence (GE Healthcare) was used to visualize the immunoreactivity. Signal intensities at the optimal exposure time were quantified by densitometry using a CCD camera and the imaging device VersaDoc 5000. Amplification factors were obtained by measuring western blot signals of amplified PrPSc/seeded PrPSc.
RESULTS
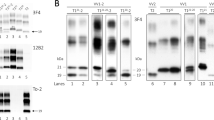
Western blot profiles of PrPres from representative CJD brain homogenates that were used as seeds for cell-PMCA are shown in Figure 1. It was difficult to recognize subtle mobility differences in the non-glycosylated forms between type 1- and type i-PrPres using 15% SDS-PAGE mini gels (Figure 1, lane 3 and 6). Furthermore, as expected, PrPSc type 2-specific antibody (T2) revealed a specific signal in both sCJD-VV2 and -MV2 cases (Figure 1, lane 4 and 8), while it did not recognize PrPSc in both np-dCJD and p-dCJD (Figure 1, lane 2, 3, 6 and 7).

Western blot analysis of PrPres in the brain homogenate seeded for cell-PMCA. Ten percent (w/v) CJD brain homogenates were digested with proteinase K (50 μg/ml at 37 °C for 60 min). Anti-PrP monoclonal antibody 3F4 (upper panel) or type 2 PrPSc-specific polyclonal antibody (T2) (lower panel) was used as the primary antibody to detect PrPres. Numbers show the molecular size standards (kDa). CJD, Creutzfeldt–Jakob disease; np-dCJD, non-plaque-type CJD; p-dCJD, plaque-type CJD; PMCA, protein misfolding cyclic amplification; sCJD, sporadic CJD.
We applied cell-PMCA using recombinant human 129MPrPC or 129VPrPC as a substrate to compare the relative amplification efficiency of PrPSc among 114 CJD patients. The calculated amplification factors for each CJD case are summarized in Table 1 and Supplementary Table 1, and representative western blots are shown in Figure 2. No significant signal of the amplified products was observed using either sCJD-MM1 or -MM2 prions as seed, irrespective of whether the genotype at codon 129 of the substrate was M or V (Figure 2a, upper panel and Supplementary Table 1). In contrast, PrPSc signals from sCJD-VV2 and -MV2 prions were significantly amplified, up to a 10−3-folds dilution, although only with the 129 V substrate (Figure 2e, upper panels). Overall, these results demonstrate that, in the tested PMCA conditions, the V2 strain is amplified more efficiently than the M1 or M2 strains, and that the valine genotype at codon 129 makes the substrate more susceptible to conversion by the V2 strain.

Western blot analysis of cell-PMCA products. PrPSc in the brain homogenates from patients with np-dCJD (a), sCJD-MM1 or -MM2C (b), p-dCJD (c), atypical CJD-MM with plaques (d) and sCJD-VV2 or -MV2K (e) were amplified with substrate lysates prepared from 293F cells stably expressing human 129MPrPC or human 129VPrPC. Ten-fold serial dilutions of CJD brain homogenates were seeded into the substrates, and aliquots were immediately stored at −80 °C (no PMCA) or subjected to 48 cycles of PMCA. Either before or after, the PMCA samples were treated with proteinase K (50 μg/ml at 37 °C for 60 min) and subjected to western blotting using the anti-PrP monoclonal antibody 3F4 (upper panel) or the type 2 PrPSc-specific polyclonal antibody T2 (lower panel). Signals with the T2 antibody in 10−2-fold dilutions of sCJD-VV2 or -MV2 prions in the 129M substrates (e, lower panel) might be the original type 2 PrPSc in the brain homogenates seeded. Each of the results was typical of experiments performed at least two or three times. Numbers show the molecular size standards (kDa). CJD, Creutzfeldt–Jakob disease; np-dCJD, non-plaque-type CJD; p-dCJD, plaque-type CJD; PMCA, protein misfolding cyclic amplification; sCJD, sporadic CJD.
Likewise no efficient amplification of the sCJD-MM1 prions was observed in the nine cases of np-dCJD regardless of whether the codon 129 genotype of substrate was M or V, suggesting that the seeding activity of np-dCJD prions matches that of sCJD-MM1 prions (Figure 2a, upper panel). In contrast, PrPSc from p-dCJD cases was strongly amplified in the presence of 129 V substrate despite the mismatched codon 129 genotype between the seed and substrate (Figure 2c, upper panel), indicating that p-dCJD prions, like sCJD-VV2 or -MV2 prions, have a clear preference for the 129 V genotype.
There was a significant variability in the amplification factor among p-dCJD cases (Table 1), which might reflect an heterogeneity in the PrPSc content among brain homogenates used as seed. Amplification products were detected up to a 10−3-fold dilution in all cases, and up to a 10−4-fold dilution in some cases showing a relatively high PrPSc content in the undiluted homogenate. In the latter group, the calculated amplification factors at a 10−2-fold dilution were likely overestimated due to the excess of PrPSc in the seeded brain homogenate (Table 1, p-dCJD case #3, #6, or #9).
As next step, we characterized the PMCA products using the T2 antibody. While no amplifications were detected with either sCJD-MM1, MM2, or np-dCJD prions (Figures 2a and b, lower panel), amplified products strongly reacting with T2 were detected in all sCJD-VV2 or -MV2 prions seeded with the 129 V substrate (Figure 2e, lower panel). Interestingly, a strong T2-reactive signal was also observed in all amplified products obtained using p-dCJD as seed and the 129 V-PrP as substrate, despite the original PrPSc in p-dCJD being not recognized by T2 (Figure 2c, lower panel). Furthermore, no T2-reactive PMCA product was detected with the 129 M substrate. Taken together, these results clearly show that the amplification characteristics of p-dCJD prions fully differ from those of np-dCJD prions, and rather match those of sCJD-VV2 or -MV2 prions, ie, strong amplification with the 129 V substrate and the generation of type 2 PrPSc products.
We then analyzed the seeding activity and amplified products of two cases affected by atypical CJD-129MM with plaques.21, 22, 23 Both cases showed abundant kuru plaques and type i PrPSc and preferentially transmitted to humanized 129 V/V mice resulting in the accumulation of type 2 PrPSc accumulation in the recipient animals.16 While the PrPSc from both cases did not amplify at all with the 129 M substrates despite the matched codon 129 genotypes, a very efficient amplification was seen with the 129 V substrate (Figure 2d, upper panel). Consistently, a strong PrP signal, also recognized by the T2 antibody (Figure 2d, lower panel), was detected up to a 10−3-fold dilution in both cases (Figure 2d, upper panel).
DISCUSSION
We have demonstrated that p-dCJD prions differ from np-dCJD prions in their PMCA properties, namely the preference for the 129 V substrate and the type of generated amplified products. Indeed, np-dCJD prions were not amplified in our current cell-PMCA system irrespective of codon 129 genotype of the substrates, whereas p-dCJD prions were consistently amplified with the 129 V substrates despite the PRNP codon 129 genotypic incompatibility between seed and substrate. Moreover, the amplified products from the p-dCJD prions with the 129 V substrates were similarly reactive with the PrPSc type 2-specific antibody as those generated by sCJD-VV2 prions, despite the PrPSc in the original p-dCJD homogenate being not reactive.
We previously reported that intermediate type PrPSc with a downward size shift from type 1 and kuru plaques in p-dCJD were phenotypic features associated with the cross-sequential transmission of the V2 sCJD strain to individuals with the codon 129 MM genotype.9, 10, 11, 14 Hence, given the transmission properties of p-dCJD prions, it is conceivable that p-dCJD prions exhibit cell-PMCA amplification properties similar to those of other CJD subtypes associated with the V2 strain. Although the amplification factors varied among the p-dCJD cases because of the variability in PrPSc amount in the seeded brain homogenates, all p-dCJD cases were amplified up to a 10−3-fold dilution with the 129 V substrates.
To test the specificity of cell-PMCA as a new tool to differentiate between the acquired CJD derived from V2 strain and sCJD associated with M1 strain, we also performed cell-PMCA in two atypical CJD-129MM cases with plaques. Although direct evidence of exposure to prion-contaminated brain tissues or surgical instruments was not obtained from the available clinical records of these patients, a transmission study has revealed that the two cases might actually be acquired CJD caused by infection with the V2 sCJD strain.16 In transmission experiments, the two atypical CJD cases exhibited the same properties as p-dCJD prions. Consistent with the results of transmission studies, both atypical CJD cases showed amplification properties identical to those of p-dCJD prions. These findings indicate that acquired CJD derived from the V2 strain can be easily identified by analyzing the characteristic amplification properties, ie, strong amplification with the 129 V substrate and generation of type 2 amplified products in cell-PMCA.
Transmission studies using knock-in humanized mice are a powerful tool to identify the causative origin of acquired prion diseases.11, 16, 26 However, they require an incubation period of over 2 years to yield significant results. Instead, cell-PMCA will help us to rapidly find out acquired CJD associated with the V2 strain within just a week, even without definitive evidence of contact with prion-contaminated tissues.
Except for variant CJD prions,17, 27 human prions have not been efficiently amplified by PMCA,27 whereas some animal prions such as those derived from hamster scrapie,28, 29, 30 bovine spongiform encephalopathy,31 or chronic wasting disease32, 33 can be efficiently amplified. The compatibility of the genotype at codon 129 between the seed and substrate is one of the most important factors for the efficient amplification of human prions.34 However, in the present study the amplification of the M1 or M2 CJD strains failed even with the matching 129 M substrate. This might be due to inadequate PMCA buffer conditions (50 mM phosphate buffer (pH 6.8), 1% (v/v) Nonidet-P-40, 4 mM EDTA, 80 mM NaCl and complete PI), ineffective duration, or strength of sonication (five pulses of 5 s with 1-s rest) in our PMCA apparatus. Thus, although PMCA has not yet been put into practical use as an ultrasensitive assay for the detection of the whole spectrum of human prions, we propose cell-PMCA as a rapid diagnostic method for identifying acquired CJD associated with the V2 strain. To avoid the potential risk of CJD transmission via unidentified routes, further surveillance of acquired CJD cases using cell-PMCA, in addition to careful analyses of the phenotypic features of the patients, is needed.
References
Parchi P, Giese A, Capellari S et al. Classification of sporadic Creutzfeldt–Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 1999;46:224–233.
Brown P, Brandel JP, Sato T et al. Iatrogenic Creutzfeldt-Jakob Disease, final assessment. Emerg Infect Dis 2012;18:901–907.
Brown P, Brandel JP, Preece M et al. Iatrogenic Creutzfeldt-Jakob disease: the waning of an era. Neurology 2006;67:389–393.
Hoshi K, Yoshino H, Urata J et al. Creutzfeldt-Jakob disease associated with cadaveric dura mater grafts in Japan. Neurology 2000;55:718–721.
Kretzschmar HA, Sethi S, Foldvari Z et al. Iatrogenic Creutzfeldt-Jakob disease with florid plaques. Brain Pathol 2003;13:245–249.
Shimizu S, Hoshi K, Muramoto T et al. Creutzfeldt-Jakob disease with florid-type plaques after cadaveric dura mater grafting. Arch Neurol 1999;56:357–362.
Mochizuki Y, Mizutani T, Tajiri N et al. Creutzfeldt-Jakob disease with florid plaques after cadaveric dura mater graft. Neuropathology 2003;23:136–140.
Bishop MT, Will RG, Manson JC . Defining sporadic Creutzfeldt-Jakob disease strains and their transmission properties. Proc Natl Acad Sci USA 2010;107:12005–12010.
Kobayashi A, Asano M, Mohri S et al. Cross-sequence transmission of sporadic Creutzfeldt-Jakob disease creates a new prion strain. J Biol Chem 2007;282:30022–30028.
Kobayashi A, Sakuma N, Matsuura Y et al. Experimental verification of a traceback phenomenon in prion infection. J Virol 2010;84:3230–3238.
Kobayashi A, Asano M, Mohri S et al. A traceback phenomenon can reveal the origin of prion infection. Neuropathology 2009;29:619–624.
Noguchi-Shinohara M, Hamaguchi T, Kitamoto T et al. Clinical features and diagnosis of dura mater graft associated Creutzfeldt Jakob disease. Neurology 2007;69:360–367.
Yamada M, Noguchi-Shinohara M, Hamaguchi T et al. Dura mater graft-associated Creutzfeldt-Jakob disease in Japan: clinicopathological and molecular characterization of the two distinct subtypes. Neuropathology 2009;29:609–618.
Kobayashi A, Matsuura Y, Mohri S et al. Distinct origins of dura mater graft-associated Creutzfeldt-Jakob disease: past and future problems. Acta Neuropathol Commun 2014;2:32.
Nozaki I, Hamaguchi T, Sanjo N et al. Prospective 10-year surveillance of human prion diseases in Japan. Brain 2010;133:3043–3057.
Kobayashi A, Parchi P, Yamada M et al. Transmission properties of atypical Creutzfeldt-Jakob disease: a clue to disease etiology? J Virol 2015;89:3939–3946.
Yokoyama T, Takeuchi A, Yamamoto M et al. Heparin enhances the cell-protein misfolding cyclic amplification efficiency of variant Creutzfeldt-Jakob disease. Neurosci Lett 2011;498:119–123.
Kitamoto T, Shin RW, Doh-ura K et al. Abnormal isoform of prion proteins accumulates in the synaptic structures of the central nervous system in patients with Creutzfeldt-Jakob disease. Am J Pathol 1992;140:1285–1294.
Kitamoto T, Ohta M, Doh-ura K et al. Novel missense variants of prion protein in Creutzfeldt-Jakob disease or Gerstmann-Straussler syndrome. Biochem Biophys Res Commun 1993;191:709–714.
Notari S, Capellari S, Giese A et al. Effects of different experimental conditions on the PrPSc core generated by protease digestion: implications for strain typing and molecular classification of CJD. J Biol Chem 2004;279:16797–16804.
Ishida C, Kakishima A, Okino S et al. Sporadic Creutzfeldt-Jakob disease with MM1-type prion protein and plaques. Neurology 2003;60:514–517.
Schoene WC, Masters CL, Gibbs CJ et al. Transmissible spongiform encephalopathy (Creutzfeldt-Jakob disease). Atypical clinical and pathological findings. Arch Neurol 1981;38:473–477.
Parchi P, Cescatti M, Notari S et al. Agent strain variation in human prion disease: insights from a molecular and pathological review of the National Institutes of Health series of experimentally transmitted disease. Brain 2010;133:3030–3042.
Hizume M, Kobayashi A, Teruya K et al. Human prion protein (PrP) 219K is converted to PrPSc but shows heterozygous inhibition in variant Creutzfeldt-Jakob disease infection. J Biol Chem 2009;284:3603–3609.
Grathwohl KU, Horiuchi M, Ishiguro N et al. Improvement of PrPSc-detection in mouse spleen early at the preclinical stage of scrapie with collagenase-completed tissue homogenization and Sarkosyl-NaCl extraction of PrPSc. Arch Virol 1996;141:1863–1874.
Asano M, Mohri S, Ironside JW et al. vCJD prion acquires altered virulence through trans-species infection. Biochem Biophys Res Commun 2006;342:293–299.
McDowell KL, Nag N, Franco Z et al. Blood reference materials from macaques infected with variant Creutzfeldt-Jakob disease agent. Transfusion 2015;55:405–412.
Soto C, Anderes L, Suardi S et al. Pre-symptomatic detection of prions by cyclic amplification of protein misfolding. FEBS Lett 2005;579:638–642.
Castilla J, Saa P, Hetz C et al. In vitro generation of infectious scrapie prions. Cell 2005;121:195–206.
Saa P, Castilla J, Soto C . Ultra-efficient replication of infectious prions by automated protein misfolding cyclic amplification. J Biol Chem 2006;281:35245–35252.
Murayama Y, Masujin K, Imamura M et al. Ultrasensitive detection of PrP(Sc) in the cerebrospinal fluid and blood of macaques infected with bovine spongiform encephalopathy prion. J Gen Virol 2014;95:2576–2588.
Kurt TD, Perrott MR, Wilusz CJ et al. Efficient in vitro amplification of chronic wasting disease PrPRES. J Virol 2007;81:9605–9608.
Johnson CJ, Aiken JM, McKenzie D et al. Highly efficient amplification of chronic wasting disease agent by protein misfolding cyclic amplification with beads (PMCAb). PLoS One 2012;7:e35383.
Jones M, Peden AH, Wight D et al. Effects of human PrPSc type and PRNP genotype in an in-vitro conversion assay. Neuroreport 2008;19:1783–1786.
Acknowledgements
This study was supported by Grant-in-Aid for Scientific Research from JSPS (AT:15K09307, TK: 25257506), Grants-in-Aid from the Ministry of Health, Labour and Welfare of Japan (AK), a grant from MEXT for the Joint Research Program of the Research Center for Zoonosis Control, Hokkaido University (TK), and a Grant-in-Aid for Scientific Research on Innovative Areas (Brain Protein Aging and Dementia Control) from MEXT (TK). Research for development of decontamination/disinfection procedures against various sCJD subgroups has been supported by a Grant-in-Aid from the Research Committee of Surveillance and Infection Control of Prion Disease, the Ministry of Health, Labor and Welfare of Japan (TK). We thank M Yamamoto for her excellent technical assistance and B Bell for critical review of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
In this paper, the authors introduce a new method for identifying iatrogenic Creutzfeldt-Jakob disease (CJD) using protein misfolding cyclic amplification. Based on the preference for the genotype at codon 129 of substrates and the protease resistance prion protein type of amplified products, CJD cases derived from sporadic CJD-VV2 could be rapidly and easily differentiated from those linked to sporadic CJD-MM1 within just one week
Supplementary information
Rights and permissions
About this article

Cite this article
Takeuchi, A., Kobayashi, A., Parchi, P. et al. Distinctive properties of plaque-type dura mater graft-associated Creutzfeldt–Jakob disease in cell-protein misfolding cyclic amplification. Lab Invest 96, 581–587 (2016). https://doi.org/10.1038/labinvest.2016.27
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2016.27
This article is cited by
-
Sensitive protein misfolding cyclic amplification of sporadic Creutzfeldt–Jakob disease prions is strongly seed and substrate dependent
Scientific Reports (2021)
-
Iatrogenic Creutzfeldt-Jakob disease with Amyloid-β pathology: an international study
Acta Neuropathologica Communications (2018)
-
UK Iatrogenic Creutzfeldt–Jakob disease: investigating human prion transmission across genotypic barriers using human tissue-based and molecular approaches
Acta Neuropathologica (2017)
