
Abstract
The incidence and mortality rates of cutaneous melanoma continue to increase worldwide, despite the deployment of targeted therapies. Recently, there has been rapid growth and development in our understanding of epigenetic mechanisms and their role in cancer pathobiology. Epigenetics—defined as the processes resulting in heritable changes in gene expression beyond those caused by alterations in the DNA sequence—likely contain the information that encodes for such phenotypic variation between individuals with identical genotypes. By altering the structure of chromatin through covalent modification of DNA bases or histone proteins, or by regulating mRNA translation through non-coding RNAs, the epigenome ultimately determines which genes are expressed and which are kept silent. While our understanding of epigenetic mechanisms is growing at a rapid pace, the field of melanoma epigenomics still remains in its infancy. In this Pathology in Focus, we will briefly review the basics of epigenetics to contextualize and critically examine the existing literature using melanoma as a cancer paradigm. Our understanding of how dysregulated DNA methylation and DNA demethylation/hydroxymethylation, histone modification, and non-coding RNAs affect cancer pathogenesis and melanoma virulence, in particular, provides us with an ever-expanding repertoire of potential diagnostic biomarkers, therapeutic targets, and novel pathogenic mechanisms. The evidence reviewed herein indicates the critical role of epigenetic mechanisms in melanoma pathobiology and provides evidence for future targets in the development of next-generation biomarkers and therapeutics.
Similar content being viewed by others

EPIGENETICS: A RENAISSANCE IN CANCER PATHOBIOLOGY
Billions of dollars have been invested into our understanding of classic human genetics and its influence on phenotype and disease. Yet, variations in the DNA sequence alone do not explain the subtle phenotypic differences observed between monozygotic twins nor can they completely explain the pathobiologic differences that separate benign from malignant cellular proliferations. Epigenetics—defined as the molecular mechanisms that regulate heritable changes in gene expression without causing any changes to the DNA sequence—provides key insights into the underpinnings of such phenotypic, morphologic, and pathobiologic differences. By altering the structure of chromatin through covalent modification of DNA bases or histone proteins or by regulating mRNA translation through non-coding RNAs (ncRNA), the epigenome reserves ultimate determination over which genes are expressed and which are kept silent. This ‘higher level' of gene regulation may even provide a mechanistic link between how factors such as the environment, gender, and aging influence our individual phenotype as well as our own unique susceptibilities to cancers such as melanoma, a prototype of an aggressive human malignancy.
One key difference between the genome and the epigenome is that the latter may potentially be more therapeutically reversible than mutations affecting the genetic code itself. Given that distinct subsets of malignant melanoma are driven by heterogeneous genetic mutations, this virulent form of human cancer is a prime example for examining the interplay between genetic and epigenetic events. Despite the deployment of therapies directed at specific genomic mutations in melanoma, the incidence and mortality rates from this deadly disease continue to increase worldwide—faster than that of any other potentially preventable cancer. Our understanding of how dysregulated DNA methylation and DNA demethylation/hydroxymethylation, and histone modification, as well as ncRNAs, affect cancer pathogenesis and melanoma virulence, in particular, is growing at a rapid pace and provides us with an ever-expanding repertoire of potential diagnostic biomarkers, therapeutic targets, and novel pathogenic mechanisms. We believe that this flourishing body of evidence points strongly towards prioritization of the cancer epigenome over a solely genome-centric viewpoint when considering the best translational approaches to virulent cancers like melanoma. In this Pathobiology in Focus, we provide a brief overview of the current understanding of epigenetic mechanisms with special attention to the cancer epigenome in melanoma, and explore the direct diagnostic and therapeutic implications and applications of these novel insights. It is critical to unravel and harness the immense power of the epigenome and direct its further clinical application in the setting of personalized medicine, particularly for cancers like melanoma, where existing diagnostic and therapeutic strategies all too often fall short.
EPIGENETICS: FOUNDATION AND PRINCIPLES
First introduced by English biologist Conrad Waddington in 1939, the term ‘epigenetics’ is derived from the Greek ‘epigenesis,’ connoting ‘changes in gene activity during development.’1 During a time when genetics and developmental biology were studied independently, Waddington and others stressed the critical relationship between these two emerging fields.2 Soon it became clear that fundamental features of embryology and development demanded explanation beyond that provided by the genetic ‘code.’ One, for instance, was how pluripotent cells could differentiate into specialized cells, such as fibroblasts and lymphocytes, and despite sharing identical genotypes, stably maintain their distinct biologic phenotypes through generations of cell division.1, 3
Historically, observations that were not easily explained through genetic terms but had a heritable component were considered to be ‘epigenetic’ phenomena. As we understand it today, however, ‘epigenetics’ refers more precisely to the molecular mechanisms whereby gene expression is reversibly modified in a heritable manner without changes in the DNA sequence. Such mechanisms enable the differentiation of embryonic and adult stem cells, as well as the dedifferentiation and acquisition of pluripotency by somatic cells, potentially as a consequence of environmental stimuli and cues. Moreover, epigenetic mechanisms are also likely to contribute to the development and function of self-renewing ‘cancer stem cells’ (CSCs). Epigenetic regulation of gene expression occurs by altering the structure and conformation of chromatin, thereby affecting the ability of transcriptional machinery to access genes and their promoters, as well as by affecting the stability of mRNA transcripts. The principal epigenetic mechanisms include DNA methylation, histone modifications, and ncRNA regulation, and we will briefly review their principles here (Figure 1). The discussion focusing on epigenetic alterations in melanoma will begin under the header ‘Epigenetics and Melanoma.’

Summary of the three primary epigenetic mechanisms. (1) DNA methylation. (2) Histone posttranslational modifications. (3) RNA-based mechanisms, including miRNAs and large non-coding RNAs (lncRNAs). Note: This diagram does not illustrate its mechanisms of binding and silencing mRNAs. Reprinted with permission from Lippincott Williams & Wilkins.
DNA Methylation and Hydroxymethylation
In 1975, the first suggestion that DNA methylation could exert strong effects on gene expression came from two groups working independently to uncover the ‘molecular switch’ that turned genes on or off during development.4, 5 That ‘switch’ was once thought to be DNA methylation, which occurs at the carbon-5 position of cytosine to form 5-methylcytosine (5-mC), otherwise known as the ‘fifth base.’6 Today, it is understood that this methylation does not constitute a simple ‘switch’ and that multiple additional tightly orchestrated epigenetic mechanisms cooperate to silence or activate genes in a context and site-specific manner. However, DNA promoter methylation is known to be critical for stabilizing the silent state of certain genes within terminally differentiated somatic cells. Here, this methylation is thought to act as a target for binding proteins that, together, prevent the reactivation of potentially deleterious germline and pluripotency genes.7
DNA methylation occurs on cytosine residues preceding guanine on the ipsilateral strand (thus forming a ‘CpG’ dinucleotide pair, wherein ‘p’ signifies the phosphodiester bond linking the two). CpG dinucleotide pairs are known to exist in regions enriched in CpG repeats (0.5–4 kb in length) called ‘CpG islands.’8 CpG islands are present in or near approximately 40% of mammalian gene promoters,9 making them prominent targets for methylation, although recent studies may suggest otherwise.7 Current understanding, however, indicates that while promoter methylation is associated with gene silencing,10 methylation within gene bodies correlates positively with transcription.11 Despite significant advances in our knowledge of how CpG dinucleotide and CpG island methylation influence gene expression, the precise mechanisms through which this occurs remains incomplete. However, it is known that the enzymatic methylation of cytosine is performed by DNA methyltransferases (DNMTs),12 of which our genomes have encoded several that perform similar functions in different biological contexts.13, 14 Their function is critical during development and cellular differentiation, as well as for the faithful intergenerational propagation of specific methylation patterns, genomic imprinting, transcriptional repression of retrotransposons in both germ and somatic cells, and X-chromosome inactivation, among others.
In contrast to DNA methylation, the mechanisms underlying DNA demethylation are even less well understood.15 DNA demethylation has been known to occur passively, specifically by the programmed failure to transmit a certain methylation pattern during a round of cell division.15 However, active DNA demethylation in mammalian systems has only very recently been recognized, with accumulating evidence indicating that this occurs through the sequential, iterative oxidation of the methyl group of 5-mC and removal of the final modified group by thymine DNA glycosylase and the base excision repair pathway to yield cytosine from 5-mC (Figure 2).15 The first and most critical step of this reaction involves oxidation of 5-mC to 5-hydroxymethylcytosine (5-hmC), which is performed by the Ten Eleven Translocase (TET) family dioxygenase enzymes.16 Originally discovered as a human homolog of an enzyme present in Trypanosoma cruzi, the TET family proteins were demonstrated to be 2-oxoglutarate (or α-ketoglutarate, α-KG), iron (II)-dependent oxidases that catalyze this initial oxidation step.17 5-hmC is the most abundant intermediate of the active DNA demethylation pathway18, 19 and its content directly correlates with the level of differentiation in a wide variety of human tissues.20 In addition, both 5-hmC and TET expression/activity are tightly regulated during embryonic stem cell differentiation.21, 22

Active DNA demethylation pathway. The pathway involved in the Ten Eleven Translocase (TET)-dependent generation of 5-hydroxymethylcytosine (5-hmC), an epigenetic mark that is lost in melanoma (pictorially rendered to lower right). DNMT, DNA methyltransferase; TDG, thymine DNA glycosylase; BER, base excision repair; IDH, isocitrate dehydrogenase; SDH, succinate dehydrogenase.
Given its ability to initiate the removal of DNA methyl groups, TET has a putative role in maintaining DNA methylation fidelity by enabling DNA demethylation ‘repair,’23 which has earned it the epithet ‘guardian of CpG islands.’24 This would suggest that loss of TET function may have dire biologic consequences. Indeed, TET has been shown to be the most frequently mutated gene in myelodysplastic syndrome and tightly associate with reduced overall survival25 and that its loss increases the self-renewal capacity of hematopoietic stem cells, leads to their eventual myeloproliferation.26 Furthermore, the loss of 5-hmC has also been very recently documented in a number of solid malignancies, including breast cancer,27 oral squamous cell carcinoma,28 gastrointestinal stromal tumor,29 and hepatocellular carcinoma,30 among others. Given these observations and TET’s close functional relationship to other epigenetic mechanisms,31 one may speculate as to whether TET functions more globally as a ‘guardian of the epigenome.’ Understanding the precise cellular function of the TET family enzymes and the biologic significance of 5-hmC loss and dysregulated DNA demethylation is currently a high priority in cancer biology research.
In addition to TET, evidence also suggests a role for dysfunction of the Krebs cycle enzymes in DNA methylation/hydroxymethylation dysregulation. Isocitrate dehydrogenase (IDH) produces a critical co-factor, α-ketoglutarate (α-KG) for TET enzyme function and is also frequently mutated in a number of cancers.32 Interestingly, IDH mutations not only result in loss of the necessary TET enzyme co-factor but also results in the production of oncometabolite 2-hydroxyglutarate (2-HG).33 2-HG competitively inhibits multiple α-KG-dependent enzymes, including the TET family 5-mC hydroxylases, as well as histone demethylases.34 Mutations in succinate dehydrogenase, particularly in a subset of gastrointestinal stromal tumors, have also been proposed to disrupt TET function through similar mechanisms and, interestingly, are tightly associated with a number of hypermethylated genes.29, 35 Taken together, these data suggest that Krebs cycle and metabolic disarray may be involved in the malignant transformation via loss of TET function and 5-hmC and the hypermethylation of tumor suppressor genes, as will be discussed greater detail below specifically with regard to melanoma.
Chromatin Structure and Histone Modification
While pathologists routinely speak of heterochromatin and hyperchromatic nuclei, it is useful to review the concept of chromatin as a functional DNA scaffold that may respond to external cues and instruct the activity and function of the DNA that it envelops. For over 50 years, we have known that histones may be post-translationally modified. Chromosomal DNA is packaged into nucleosomes with DNA wrapped around highly alkaline histone protein octamers, which consist of subunits (H2A, H2B, H3, and H4) and other variants. Histone modifications may either activate or silence transcription depending on the nature and location of the modification by controlling the accessibility of DNA to the transcriptional machinery and by recruiting or excluding additional protein complexes.36 At least 130 posttranslational modifications of histone proteins have been identified in human cells, the best studied of which include methylation and acetylation.37 Histone methylation was thought to be an irreversible process for many years until the first histone demethylase (KDM1A) was discovered in 2004.38
The physical association of histone modifications with anatomical segments of the genome is, in part, determined by the specific modifier. For example, acetylation events may be found in active promoters and enhancers (as with H3K27ac: acetylated histone H3 on lysine 27), in transcribed gene bodies (as with H3K36me3: trimethylated histone H3 on lysine 36), or in association with heterochromatic or repressed regions (as with H3K9me3 and H3K27me3).39 Interestingly, studies have shown that DNMTs physically associate with histone deacetylases, which reverse histone acetylation, as well as with histone methyltransferases,40 and, together, favor closed chromatin conformations near gene regulatory regions thereby perpetuating ‘silent’ epigenetic states through multiple generations of cell division.41 In addition, certain modifications, such as the methylation of H3K9 (histone 3, lysine 9), tightly associates with aberrant heterochromatin formation and silencing of tumor suppressors, such as cyclin-dependent kinase inhibitor 2a (CDKN2a), in cancer cells.42 Moreover, perturbation of the overall histone profile, or ‘histone code,’ has been shown to have prognostic relevance in various cancers.43
Polycomb group (PcG) proteins are epigenetic repressors that associate with specific posttranslational histone modifications and are essential for the transcriptional regulation of cell differentiation and development.44, 45 PcG-mediated repression is associated with and is thought to involve both biochemical and physical modulation of chromatin structure. Given the recent emergence of complex data that has identified novel components of the PcG proteins and their putative roles in cancer, it is of interest that such histone modifications were actually the last of the three main epigenetic mechanisms to be associated with malignancy.41
Regulation of Gene Expression by ncRNAs
While <2% of the total genomic sequence encodes for proteins, at least 90% of the genome is actively transcribed into ncRNA, otherwise known as the ‘dark matter’ of the genome.46 The first eukaryotic ncRNA to be discovered was a large RNA named H19, originally described as having a putative tumor suppressor function in Wilms’ tumor47 and later found to be involved in the process of genomic imprinting.48 As we understand them today, ncRNAs are a heterogeneous group of RNAs that are generally classified into two groups based on their lengths, ranging anywhere from 18–25 to 10,000 nucleotides in length.49 Small ncRNAs are <200 nucleotides in length and within this category, the microRNAs (miRNAs) are the most well studied.49 miRNAs bind to miRNA response elements contained within their target mRNA transcripts and subsequently recruit the RNA-induced silencing complex, which antagonizes target mRNA stability and/or translation.50 In this manner, ncRNAs regulate a wide variety of complex cellular processes, including gene silencing, gene transcription, DNA imprinting, DNA demethylation, chromatin structure dynamics, and RNA interference.51 A number of oncogenic and tumor suppressive ncRNAs, particularly miRNAs, have also been recently described in melanoma and will be discussed in greater detail below.
The second class of ncRNAs that have recently been described are the long ncRNAs (lncRNAs), which range anywhere from 200 nucleotides to ∼100 kb.46 Unlike the miRNAs, lncRNAs bind other protein complexes and also form a secondary structure, although the primary sequence and molecular factors that influence these dynamics remain unknown.52 However, like the miRNAs, lncRNAs have been implicated in a variety of gene regulatory roles, including chromosome dosage compensation, genomic imprinting, epigenetic regulation, cell cycle control, nuclear and cytoplasmic trafficking, transcription, translation, splicing, and cell differentiation, among others.52 It has become clear that dysregulation of ncRNAs, including the miRNAs and lncRNAs in particular, are a critical factor in the pathobiology of cancer.
This overview, although limited and rudimentary in the context of a rapidly emerging database, bears testimony to the diversity and pleiotropism inherent to epigenetic mechanisms of gene regulation. We will now apply these concepts to a form of human cancer that serves as a paradigm for clinical virulence, mechanistic complexity, and therapeutic challenge: malignant melanoma.
EPIGENETICS AND MELANOMA
Aberrant DNA Methylation
In 1983, Feinberg and Vogelstein53 first reported that ‘substantial hypomethylation’ of CpG dinucleotide was present in human cancer cells. Since this discovery, alterations to DNA methylation throughout the genome have been well documented in cancer. Global genome-wide methylation is now known to be reduced very early in the neoplastic progression of carcinogenesis.54, 55, 56 Teleologically, given the additional cellular and environmental functions required for neoplastic cells to proliferate and eventually metastasize, one may hypothesize that hypomethylation allows previously benign cells to express and experiment with novel gene products to exert a survival advantage. In addition, DNA hypomethylation, specifically in or around centromeric repeats and other repetitive sequences, has been shown to contribute to chromosomal instability.57 In addition to global hypomethylation, cancer cells concurrently and paradoxically display localized hypermethylation of CpG islands of multiple tumor suppressor genes early in carcinogenesis,58, 59, 60 which has been deemed ‘one of the most precocious hits in tumorigenesis.’60 In general, the tendency toward hypermethylation has been described in multiple human cancers, including melanoma, and has also been termed the ‘CpG island methylator phenotype.’61, 62
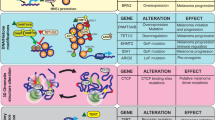
The DNA methylation status of cutaneous melanoma has been extensively studied and has been demonstrated to have prognostic and therapeutic significance. Hypermethylation of specific tumor suppressor genes, as well as those involved in cell-cycle regulation, DNA repair, cell signaling, transcription, and apoptosis, have been reproducibly described in cutaneous melanoma.63 The CDKN2A promoter has been shown to be hypermethylated in a substantial fraction of primary cutaneous melanoma samples and is associated with both increased Ki-67 index and reduced patient survival.64 Of interest, CDKN2A, which encodes negative regulators of cell cycle progression p16 and p14 and is inactivated in the majority of sporadic cutaneous melanomas, is also the most frequently mutated gene inherited in familial cutaneous melanoma.65, 66 In a study of 86 metastatic melanoma specimens, four tumor suppressor genes were found to be frequently hypermethylated.67 Retinoic acid receptor-β2 was the most commonly methylated gene in this series (70% in primary and metastatic melanoma specimens),67 and has also been described to be silenced in multiple other human cancers.68 RAS association domain family protein 1A (RASSF1A), which is critical for mitochondrial apoptosis and cell cycle arrest, was found to be methylated in 57% of melanoma specimens, O6-methylguanine DNA methyltransferase (MGMT, discussed in greater detail below) in 34%, and apoptosis mediator death-associated protein kinase in 19%.67 Indeed, the number of tumor suppressor genes that are hypermethylated in melanoma is accumulating.69 By contrast, specifically hypomethylated genes have been less documented in melanoma.
Besides the regional aberrant methylation status of gene promoters, melanoma also exhibits global hypomethylation within the bulk genome, but the degree is not sufficient to distinguish the benign nevus from melanoma.70 However, we have preliminarily noted that, unlike 5-mC, the loss of 5-hmC by immunohistochemistry can distinguish melanomas from physiologic melanocytes and benign melanocytic proliferations, wherein 5-hmC nuclear immunoreactivity remains high.71 We have also found that a strong correlation exists between the loss of 5-hmC and with the parameters of poor prognosis in melanoma, including Breslow depth, mitotic rate, and ulceration, as well as with lower overall survival, suggesting a potential predictive value of loss of 5-hmC.71 Others have since reproduced these findings.72, 73
In addition, certain subtypes of the nevi, as well as of malignant melanoma, also recapitulate this inverse relationship. The retention of 5-hmC nuclear staining was very recently documented in certain benign nevic subtypes, including the Spitz nevus, whereas the loss of 5-hmC was found to be a feature in multiple melanoma subtypes, including both acral and mucosal melanoma.73 Moreover, the loss of 5-hmC may be common to malignant melanoma of varying etiologies; melanomas arising in chronically sun-damaged skin as well as those arising in sun-protected areas, too, demonstrate this epigenetic phenomenon.73 Further work is clearly indicated, however, to expand on these findings to determine whether retention and loss of 5-hmC are truly universal in benign and malignant melanocytic lesions, irrespective of clinicopathologic variants. Moreover, determination of how other epigenetic alterations involving histone modifications and miRNA expression may relate to tumor subtypes awaits further study. Interestingly, it was recently reported that increasing morphologic atypia in dysplatic melanocytic nevi corresponds to progressive loss of 5-hmC nuclear staining, a finding that also tightly associates with increasing nuclear diameter (Figure 3).74 This provides pathologic evidence that loss of TET function, as evidenced by reduced levels of 5-hmC and resulting epigenomic instability, may be critical to the pathogenesis of melanoma. In addition to melanoma, the loss of 5-hmC has been documented uniformly and universally in a number of human cancers, independent from, but reminiscent of, the global reductions in 5-mC discussed above.20 For example, 5-hmC loss has been reported in cancers of the brain, breast, lung, liver, stomach, pancreas, colon kidney, prostate, ovary, uterus using a variety of methods, including liquid chromatography-mass spectrometry, anti-5hmC antibody-based radio-immune dot blots, and immunohistochemistry.20, 75

Increasingly ‘dysplastic’ melanocytic lesions show progressive loss of immunohistochemical staining for 5-hydroxymethylcytosine (5-hmC). Immunohistochemical staining with 5-hmC demonstrates progressive loss of 5-hmC with progression from benign nevus (no dysplasia) to low- and high-grade dysplastic nevi, and finally to melanoma. Sections at × 200 are shown above with selected areas that are further magnified in the panels directly below. From Larson et al.,74 reprinted with permission from Nature Publishing Group.
Despite these promising observations, the mechanism underlying 5-hmC loss in cancer, in general, and in melanoma, more specifically, remains elusive. Interestingly, whereas IDH1 expression is similar between nevi and melanoma, we find that IDH2 is significantly downregulated in melanoma as are all three TET genes, with the most marked decrease in TET2.71 These data, in part, may shed light on previous findings indicating that mutations in IDH1 or 2 are present in up to 10% of melanomas76 and that 5-hmC levels in IDH1-mutant gliomas compared with wild-type IDH1 do not differ substantially.75 Moreover, overexpression of wild-type IDH2 in a zebrafish melanoma model increases 5-hmC levels and prolongs tumor-free survival compared with mutant IDH2.71 Importantly, overexpression of TET2 reverses the genome-wide 5-hmC distribution from global loss, as is seen in melanoma, toward one resembling a benign nevus-like pattern. In keeping with this, TET2-overexpressing melanoma cells give rise to smaller tumors compared with mutated TET2 melanoma cells,71 and TET2 expression has very recently been shown to be significantly higher in nevi than in superficial spreading melanoma and cutaneous metastatic disease.72 Taken together, TET family enzyme dysfunction and the concomitant loss of 5-hmC and resulting epigenomic instability provide a plausible pathogenic mechanism to explain the inappropriate methylation of tumor suppressor genes, which has been widely observed in various human cancers. Notably, whereas the replicative fidelity of DNA polymerases are well known,77 our understanding of DNMT fidelity is only very recently beginning to emerge.23, 78, 79, 80 Further investigation into this important area of cancer epigenetics promises to shed insights into this critical aspect of the dysregulated cancer epigenome. In light of our preliminary insights into epigenetic fidelity regulation in melanoma, the loss of 5-hmC may be a direct reflection of loss of the TET family ‘guardian’ or fidelity function, which may prove to be central to the epigenetic dysregulation and resultant pathobiology of melanoma and other cancers.
Melanoma Cell Longevity through Histone Modifications
Of the dysregulated epigenetic mechanisms involved in the pathogenesis of melanoma, aberrant histone modifications are among the least documented. While this may be, in part, due to the more challenging laboratory techniques required to delineate histone modifications,81 closer examination of this aspect of the epigenome will likely provide missing links between modifications to DNA bases and their overall influence on chromatin structure and transcriptional regulation. Therapeutic inhibition of histone deacetylase in melanoma cell lines has been shown to improve apoptotic efficiency through upregulated CDK inhibitor p21 expression, suggesting that aberrant histone deacetylation may have pathogenic role in melanoma through the downregulation of apoptotic mechanisms.82 Indeed, histone hypoacetylation has been demonstrated to downregulate other proapoptotic proteins, including the Bcl-2 family proapoptotic proteins (Bim, Bax, and Bak),83 as well as tumor suppressor genes, such as phosphatidylinositol 4,5-bisphosphate 5-phosphatase A, a negatively regulator of the PI3K/Akt signaling pathway.84
In addition to histone hypoacetylation, aberrant histone methylation also appears to have a pathogenic role in melanoma. Increased expression of EZH2 is tightly associated with highly proliferative and aggressive subtypes of melanoma, as well as in cancers of the endometrium, prostate, and breast.85 It is also tightly associated with loss of tumor-suppressive cell cycle inhibitor p16 in melanoma and endometrial carcinoma.85 Interestingly, EZH2 expression in patient melanoma specimens, as demonstrated by immunohistochemistry, has been shown to increase incrementally from benign nevi to melanoma, and is also significantly higher in invasive melanoma than it is in in situ melanoma or in benign melanocytic lesions.86 EZH2 is the subunit of polycomb repressor complex 2 (PRC2) that is responsible for catalyzing the transcriptionally repressive methylation of H3K27, and it appears that EZH2 upregulation in this context represses the expression of tumor suppressor genes.85
Additional histone-modifying enzymes have also demonstrated oncogenic potential in melanoma. The histone methyltransferase SETDB1 (SET Domain, Bifurcated 1) is recurrently amplified in melanoma and accelerates tumor development in zebrafish melanoma models harboring the common BRAF(V600E) mutation.87 SETDB1 catalyzes the trimethylation of histone H3K9 and thereby promotes the repression of target genes.87 SETDB1 overexpression has been shown to cause significant downregulation of a group of genes enriched for the development-regulating homeobox (HOX) genes.87 Dysregulated HOX genes are known to be associated with a number of hematologic malignancies and support the immortalization of leukemic cells.88 Interestingly, whereas the BRAF(V600E) mutation is present both in many melanomas and in benign melanocytic nevi,89 elevated SETDB1 protein is present in human melanomas, but not in nevi or in normal melanocytes.87 Moreover, emerging evidence is further suggestive of a bona fide oncogenic role for SETDB1 in both non-small-cell and small-cell lung cancers.90 Additionally, its overexpression in this context may also correlate with chemosensitivity to clinically approved mithramycin, an antitumoral antibiotic that binds to the minor groove of the DNA double helix, thereby displacing transcriptional activators, and shown to suppress basal SETDB1 promoter activity.90 Taken in aggregate, these data strongly support that dysregulation of the histone modification system contributes to the loss of tumor suppressors or enhanced longevity/proliferative capacity in melanoma and other cancers.
A Role for MiRNAs and Other NcRNAs in Melanoma
The prognostic and pathobiologic importance of ncRNAs in melanoma have been well established and represent an active area of investigation (Table 1).91 Indeed, an array of miRNAs and other ncRNAs have been shown to exhibit either tumor-suppressive capabilities or pro-oncogenic and/or prometastatic potential involving multiple molecular pathways. The tumor-suppressive function of miRNAs, in part, may be mediated through interactions with PcG proteins. miR-200c was shown to be significantly more downregulated in both primary and metastatic melanomas compared with benign melanocytic nevi, and its overexpression in melanoma cell lines appears to result in significantly reduced cell proliferation, migratory capacity, and expression of key transporters involved in melanoma drug resistance.92 Bmi-1 (B lymphoma Mo-MLV insertion region 1 homolog) is a PcG protein component of the PRC1, which, as described earlier, comprises an important class of transcriptional repressors that orchestrate changes in chromatin structure and thereby regulate gene activity.93 Bmi-1, specifically, is an important transcriptional repressor of the Ink4a/Arf locus, which encodes two distinct gene products, including tumor suppressors p16ink4a (p16) and p19Arf (p19).94 p16 inhibits CDK activity and thereby blocks entry into the cell cycle, whereas p19 promotes p53 stability, and in doing so, arrests cell cycle progression and promotes apoptosis.94 Interestingly, overexpression of miR-200c in melanoma cell lines results in significant downregulation of Bmi-1 and shows a similar phenotype to Bmi-1 knockdown melanoma cell lines.92 Moreover, miR-200c overexpression significantly inhibits melanoma xenograft growth and metastasis in vivo, which correlates with diminished expression of Bmi-1 as well as reduced levels of epithelial cadherin (E-cadherin).92 Several other miRNAs, including miR-612, have been demonstrated to suppress the epithelial–mesenchymal transition and metastasis in other human cancers.95 Both miR-200 and E-cadherin are expressed at lower levels at the deep invasive tumor margin and associate clinically with increased melanoma thickness and disease progression.96 Taken collectively, these data suggest that miR-200c exhibits tumor-suppressive function by targeting Bmi-1 and upregulating tumor suppressor and cell adhesion molecules; thus, its downregulation, as observed in primary and metastatic melanoma samples, appears to contribute to the molecular pathogenesis of melanoma.
Several miRNAs have been found to exhibit oncogenic or prometastatic capabilities. Elevated levels of wild-type p53 directly upregulates miR-149 expression, which is also increased in fresh human metastatic melanoma isolates.97 miR-149, in fact, targets and reduces glycogen synthase kinase-3α levels, which increases the expression of antiapoptotic Bcl-2 family protein Mcl-1 known to produce apoptotic resistance in melanoma cell lines.97 Similarly, miRNA-21 is significantly increased in primary melanoma tissues compared with benign nevi and is tightly associated with increased proliferation and decreased apoptosis.98 In addition, a cluster of 14 miRNAs on the X chromosome (miR-506-514 cluster) was found to be consistently and significantly overexpressed in nearly all patient biopsy samples of metastatic melanoma, regardless of mutation status in NRAS or BRAF.99 Notably, inhibition of the expression of this cluster in melanoma cell lines, or one of its subclusters, led to significant abrogation of cell growth, induction of apoptosis, reduced invasiveness, and decreased colony formation in vitro.99 Indeed, a number of miRNAs exhibit oncogenic potential in melanoma through inhibition of apoptosis. In addition, a cooperative network of miRNAs (miRNA-1908, miR-199a-5p, and miR-199a-3p) that endogenously promotes metastatic invasion, angiogenesis, and colonization in melanoma has also been recently identified.100 These miRNAs appear to target apolipoprotein E, which normally suppresses invasion and metastasis.100 Moreover, patients whose primary melanomas express higher levels of miR-199a-3p, miR-199a-5p, or miR-1908 have been shown to have significantly shorter metastasis-free survival times than patients whose primary melanomas express lower levels of each of these miRNAs.100 Interestingly, highly metastatic melanoma cell lines that are pretreated with a cocktail of locked nucleic acids targeting these miRNAs for downregulation show reduced ability to metastasize to multiple distant organs upon their injection into mice.100
Preliminary evidence also implicates several long ncRNAs in the pathobiology of melanoma. HOTAIR, one such lncRNA that has been associated with metastatic behavior, was found to be significantly overexpressed in lymph nodes containing metastatic melanoma compared with matched primary melanoma specimens.101 Moreover, its knock down in cell lines suppressed melanoma cell motility, invasiveness, and extracellular matrix degradation.101 Interestingly, recent evidence suggests that HOTAIR, through direct scaffolding interactions with histone-modifying enzymes, may facilitate changes to chromatin structure.101 Similar roles for other ncRNAs have gained significant attention in the basic science literature, such as Xist, recently shown to silence the X chromosome by exploiting and inducing three-dimensional chromosome structural alterations.102 Additional putative oncogenic lncRNAs have been reported in melanoma, including SPRY4-IT1103 and Llme23,104 and reports of additional lncRNA with either oncogenic or tumor-suppressive roles in melanoma pathobiology will likely follow. Altogether, there is substantial preliminary evidence to suggest that lncRNAs, in addition to miRNAs, are progressively dysregulated and may promote melanomagenesis through the loss of either tumor-suppressive function or promotion of oncogenic or prometastatic molecular pathways. While many facts remain elusive, including the precise mechanisms or drivers underlying their dysfunction, as well as their basic regulatory mechanisms, ncRNAs provide a most bountiful area of further investigation in melanoma and cancer pathogenesis and therapy.
CANCER CELL ‘STEMNESS’ AND THE EPIGENOME
In 2006, the American Association for Cancer Research determined that a CSC is ‘a cell within a tumor that possesses the capacity to self-renew and to cause the heterogeneous lineages of cancer cells that comprise the tumor.’105 First discovered in hematopoietic malignancies in the 1960s and 1970s,106, 107 CSCs have been identified in a variety of solid tumors, including cancers of the breast,108 brain,109 colon,110 and melanoma.111 Although their existence has previously been a matter of debate,112 CSCs, also referred to as cancer-initiating cells,113 are thought to potentially represent oncogenic derivatives of normal-tissue stem or progenitor cells,114, 115 may develop in certain forms of cancer as a consequence of the EMT, and/or evolve spontaneously during tumor progression.116, 117 We have observed that melanoma cells acquire CSC markers with evolution from benign nevi to primary melanoma to metastatic melanoma,74 and, interestingly, a similar progression is observed with respect to loss of 5-hmC, as we have delineated above. This strongly suggests that the acquisition of melanoma ‘stemness’ with tumor progression may be in some way related to the loss of DNA hydroxymethylation (Figure 4). This hypothesis is, in part, grounded in the regulatory role played by DNA methylation in the maintenance and function of embryonic stem cells, which CSCs may, in part, recapitulate.31

Schematic representation of potential interplay between epigenetics (loss of 5-hydroxymethylcytosine (5-hmC)) and melanoma stem cell (MSC) expression during melanoma progression. Melanomagenesis may originate in ‘normal skin,’ or in a pre-existing benign or dysplastic nevus as a result of transformation of melanocytes/nevus cells, a process that appears to be associated with progressive loss of 5-hmC and concomitant heightened activity of self-renewing, stem-like cells. Intravasation of primary melanoma as a consequence of dermal invasion transports cells epigenetically programmed for malignant behavior to lymph nodes and vital organs. Experimental models of metastases reveal cells expressing MSC markers to be relatively devoid of 5-hmC, consistent with epigenetic interplay with the stem cell component of the tumor at the metastatic site. (Lung diagram adapted from courtesy of Patrick J Lynch, medical illustrator; C Carl Jaffe, MD, cardiologist. http://creativecommons.org/licenses/by/2.5/.)
EMT is a complex molecular and cellular process by which epithelial cells lose their differentiated characteristics, including cell–cell adhesion, and acquire mesenchymal features, such as motility, invasiveness, and a heightened resistance to apoptosis.118 This ‘transition’ has been proposed to be instrumental to the acquisition of ‘stemness’ by both non-transformed and tumor cells.119, 120 EMT and acquisition of the ‘stem-like’ phenotype has also been implicated in the development of chemoresistance in many human cancers.118 The loss of E-cadherin, a ‘calcium-dependent transmembrane adhesion’ molecule critical for epithelial cell–cell adhesion, is a hallmark of the EMT.118 Its loss, in part, further stabilizes the mesenchymal state through β-catenin-mediated upregulation of EMT-inducing transcription factors.118 Mechanistically, E-cadherin loss is thought to be either genetic or epigenetic, and epigenetic mechanisms are steadily entering the limelight.121 As melanocytes do not belong to the epithelial lineage, ‘EMT’ cannot be strictly used to describe melanoma pathobiology. Moreover, melanoma stem cells need not coincide with all cells exhibiting an EMT-like phenotype, in that self-renewal resulting in localized tumorigenic rather than purely invasive growth is emblematic of CSC behavior. Indeed, it has been reported that differentiated melanocytes express E-cadherin, which allows them to maintain homophylic adhesion with keratinocytes in the basal layer of the epidermis.116 Of note, the loss of E-cadherin, the quintessential hallmark of the EMT in epithelial tumors, is also present in late-stage, metastatic melanoma to lymph nodes122, 123 and very recently described (2013) to be present in desmoplastic melanoma,117 a tumor that rarely metastasizes. More importantly, the loss of E-cadherin as well as the aberrant expression of neural cadherin (N-cadherin) marks the critical transition from the radial-growth phase to the vertical-growth phase in melanoma,124, 125 an event that is associated with acquisition of potential for metastasis, has also been reported. Nonetheless, the ability of melanoma to show stem cell-driven tumorigenesis at primary and metastatic sites, as well as to toggle between tumorigenesis and EMT-like phenotypes, implicates the likelihood of robust genomic–epigenomic regulatory interactions. In this regard, reprogramming of EMT-inducing transcription factors collaborate with BRAF activation towards the dedifferentiation, loss of E-cadherin, gain of invasive properties, and malignant transformation of melanoma.116
It must be noted that EMT-like behavior, although often reflected by loss of E-cadherin, also requires the upregulation of cell surface molecules necessary for invasion and metastasis, as well as matrix metalloproteases (MMPs), which degrade the extracellular matrix and facilitate invasion of cells with mesenchymal characteristics.126 The expression of αVβ3 integrin, which, in addition to E-cadherin loss and N-cadherin expression, also tightly associates with the transition from the radial- to vertical-growth phase in melanoma.127 Furthermore, this integrin induces the expression of MMP-2, an enzyme that degrades the collagen within the basement membrane.128 While the epigenetic regulation of this integrin and MMP expression has yet to be described in melanoma or other cancers, it is very likely that dysregulated epigenetics are involved in their upregulation in this context. Furthermore, evidence is emerging to support that melanoma stem cells may be oncogenic derivatives of normal-tissue stem or progenitor cells. Latexin, a negative regulator of hematopoietic stem cell populations,129 was recently shown to reduce the risk of old stem cells transforming into CSCs130 and is known to be downregulated in approximately 50% of melanomas.131 Notably, the CpG island promoter of the latexin gene has been shown to be universally hypermethylated in melanoma cell lines and other cancers.131 These findings further demonstrate that epigenetic mechanisms may be critical to the development of melanoma or CSCs. Studies also show that histone modifications have important roles in melanoma stem cells. For instance, a distinct subpopulation of slow-cycling melanoma cells positive for jumonji/ARID1 (JARID1B) was found to be required for continuous tumor growth.132 JARID1B (KDM5B/PLU-1/RBP2-H1) is a member of the highly conserved family of JARID1 H3K4 demethylases, which are known to be involved in development, cancer, and stem cell biology.133 JARID1B is highly expressed in benign nevi, whereas in aggressive primary melanomas and melanoma metastases, single cells comprising 5–10% of the total population have high JARID1B expression.134 JARID1B levels are elevated in highly regenerative tissues as well as in cancer, wherein it regulates the transcription of oncogenes, such as BRCA1 in breast cancer, through direct interaction with promoter sites.135 Indeed, demethylation of H3K4 has been shown to support the transformation of hematopoietic precursors to leukemia stem cells via regulation of the developmental HOX gene family.136 JARID1B has been associated with either positive or negative cell cycle control, depending on the type of cancer (ie, positive cell cycle control in melanoma, negative in breast cancer).137, 138 Interestingly, JARID1B has been shown to have a stabilizing effect on hypophosphorylated pRB,137 which is normally regarded as an immediate-acting antiproliferative mechanism. It has been proposed that JARID1B’s antagonistic effect on proliferation may ultimately permit the maintenance of a slow-cycling tumor sub-population.132 Of interest, recent evidence suggests that this slow-cycling sub-population may be distinct from melanoma subpopulations with ‘stemness’ or EMT-like associated features.132
Finally, evidence points to the involvement of miRNAs both in cancer pathobiology and in the regulation of cancer ‘stemness.’ As discussed above, the miRNAs are a subset of ncRNAs that negatively regulate gene expression by targeting and degrading the mRNA transcript or inhibiting its translation.139 With more than 200 miRNAs described in humans, many have been implicated as putative oncogenes or tumor suppressor genes that are involved in the regulation of ‘stemness’ and metastasis in various human cancers.140 The miR-200 family members (miR-200s, to include miR-200a, miR-200b, miR-200c, miR-141, miR-429), in particular, are key tumor-suppressive regulators of the EMT.141 They control ‘stemness’ by directly targeting transcription factors such as transcriptional repressor of E-cadherin Zeb1/2 (zinc-finger E-box-binding HOX 1/2).142 Interestingly, the miR-200s are downregulated in various cancer types but, more specifically, in cancer cells undergoing the EMT or with other stem-like features, as will be further explored below.143 In addition, a recent study reported that miR-9 is downregulated in metastatic melanoma compared with primary melanomas and that its knockdown in melanoma cell lines enhances cell proliferation and migration capacity.144 Furthermore, overexpression of miR-9 in metastatic melanoma cell lines induces significant downregulation of the NF-κB1-Snail1 pathway and a concomitant increase in E-cadherin expression. Taken together, these data support that epigenetic mechanisms, miRNAs in particular, have a key role in regulating EMT-like changes in melanoma.
More recent evidence has identified miR-22 as a potent proto-oncogenic miRNA that deranges the epigenetic landscape of the cell.145 miR-22 has been shown to enhance the repopulating capacity and stem cell function of hematopoietic stem/progenitor cells.146 In vivo models demonstrate that miR-22 triggers myelodysplastic-like syndromes and hematological malignancies and that its expression correlates directly with poor survival rates.146 Interestingly, miR-22 has also been shown to enhance the EMT by repressing miR-200, leading to the upregulation of Zeb1 and Zeb2, and subsequent repression of E-cadherin expression.31 These results shed light on the possible mechanisms underlying the change from the epithelioid to spindle cell morphology during the first wave of 5-mC loss in mouse cutaneous carcinogenesis observed in a landmark report by Fraga et al.60 miR-22 overexpression has also been shown to instigate higher rates of tumor invasiveness and metastasis, as well as a progressive decrease, in disease-free survival rate in breast cancer mouse models.31 Further analysis has revealed that miR-22 directly targets and reduces the expression of the critical DNA-demethylating enzyme and 5-mC oxidase TET2, resulting in a marked reduction in 5-hmC levels and a concomitant increase in 5-mC levels in the genome of mouse hematopoietic stem/progenitor cells.146 The resulting loss of demethylase function has been shown to lead to genomic hypermethylation and silencing of the miR-200 promoter.146 Indeed, derangement of this miR-22-TET2 pathway has been deemed to be one of the most frequent events in hematologic malignancies.146 Overall, miR-22 appears to have consistent, principal proto-oncogenic potential through the dysregulation of the DNA demethylation apparatus, enhancement of the EMT, and enabling of cancer cell stemness.
EPIGENOMIC BIOMARKER APPLICATIONS IN MELANOMA
Many of the epigenetic markers discussed above have direct diagnostic utility. For example, studies indicate that, in addition to the oncogenic implications of hypermethylated genes, methylation status of certain genes may provide direct prognostic implications in patients with melanoma. Global levels of long-interspersed element-1 (LINE-1) methylation in short-term tumor cell cultures grown from patients with nodal metastatic melanoma have been shown to significantly predict overall survival in patients with stage IIIC cutaneous melanoma.147 Moreover, identification of these epigenetic hallmarks circulating as free DNA in the serum of patients with melanoma using methylation-specific PCR is also an area of active investigation.148 In addition, the loss of 5-hmC, as demonstrated through immunohistochemistry, may aid in distinguishing malignant melanocytic lesions from dysplastic or borderline melanocytic lesions wherein 5-hmC staining is relatively more intense.71, 72, 73 The diagnostic utility and prognostic significance of loss of 5-hmC by immunohistochemistry, as has been demonstrated in melanoma,71 also has been recapitulated in other human tumors, including oral squamous cell carcinoma,28 gastrointestinal stromal tumor,29 and hepatocellular carcinoma.30
miRNAs may also have powerful prognostication potential in melanoma. Patient melanoma specimens expressing lower levels of miRNA-205 by immunohistochemistry have been shown to associate tightly with significantly shorter melanoma-specific survival, independent of melanoma stage, age, gender, or Breslow depth.149 Interestingly, miRNA-205 overexpression in patient melanoma samples has been shown to result in lower levels of Zeb2 expression and increased expression of E-cadherin, suggesting that this particular miRNA may also be involved in suppressing the EMT.150 Indeed, in vitro and in vivo models have demonstrated that miR-205 overexpression impedes melanoma cell migration and invasion.150 Furthermore, miR-205 expression progressively decreases from benign to dysplastic nevi, as well as in melanomas, in both clinical specimens and cell lines.150 Another miRNA, miR-29c, was demonstrated to be significantly downregulated in AJCC stage IV melanoma specimens compared to primary tumors, with elevated expression significantly predicting disease-free and overall survival.151 Several other miRNAs, including miRNA-31152 and miRNA-137,153 also exhibit tumor-suppressive function in melanoma by interfering with a number of oncogenic pathways. Interestingly, both of these miRNAs appear to downregulate EZH2, the histone methyltransferase component of PRC2 discussed above,93 the expression of which progressively increases from benign nevi to dysplastic nevi to localized and metastatic melanoma, where its expression is associated with a poor 5-year prognosis.153 These findings emphasize the relevance of dysregulated epigenetic ‘cross-talk’ mechanisms in the pathobiology of melanoma and demonstrate their tumor-suppressive functions. Moreover, this epigenetic insight offers the potential application of prognostic biomarkers in melanoma and other melanocytic lesions.
In addition, miRNAs may serve as prognostic biomarkers when detected in the circulation. Serum levels of miR-221 has been shown to distinguish between patients with melanoma in situ from those with stage I–IV melanoma.154 Furthermore, several miRNAs detected in the serum of patients at the time of primary melanoma diagnosis have been shown to reflect overall tumor burden and to accurately and significantly predict risk of recurrence.155 Because there exists conflicting data regarding their utility and practical reproducibility of various assays,81 more research and translational development is required before such approaches are brought to the bedside. Nevertheless, miRNAs represent a very appealing epigenomic marker of prognosis and certainly deserve much further exploration.
EPIGENOMIC THERAPEUTIC APPLICATIONS IN MELANOMA
Unlike genomic mutations, epigenetic alterations in cancer are, in principle, therapeutically reversible, and a number of epigenetic therapies have already received FDA approval (Table 2). Sole use of DNMT inhibitors for the treatment of melanoma has yielded mixed results, with early studies suggesting enhanced capacity for experimental metastasis in xenograft models.156 In contrast, very recent preliminary data suggest that HDAC inhibitors in nanomolar concentrations may have some therapeutic benefit.157 While the sole use of these epigenetic therapies in melanoma continues to be an active area of clinical investigation, recent studies have shown great promise for their adjunctive use with various treatment regimens, for example, immuno-, chemo- and radiotherapeutic strategies. For example, DNMT and HDAC inhibitors upregulate the expression of a number of critical melanoma cell surface molecules, including major histocompatibility complex and costimulatory molecules, as well as the melanoma antigen encoding gene (MAGE-1) tumor antigen.158, 159, 160 Animal models demonstrate modest benefits using combined HDAC inhibitors with or without157 adoptively transferred, gp100 melanoma antigen-specific T cells.161 The adjunctive use of these epigenetic therapies to upregulate the expression of such critical cell surface target antigens with existing immunotherapies, including interferon-α, ipilimumab, and melanoma peptide vaccines,162, 163 is a promising area of active investigation.164
In addition to their combinatorial use with immunotherapies, epigenetic agents may also support and enhance the effectiveness of standard chemotherapeutic or radiotherapeutic regimens. Alkylating agents are thought to exert their antitumor activity by inducing either DNA double-strand breaks or interstrand crosslinking.165 However, a DNA repair protein called MGMT can remove alkyl lesions induced by these agents, inhibiting their cytotoxic effects.165 Accordingly, elevated expression of MGMT has been shown to contribute to chemoresistance to alkylating agents in multiple human malignancies, including melanoma,165 and has been attributed to aberrant methylation patterns.166 This has provided the rationale for the combined use of DNMT inhibitors alongside alkylating agents, an approach recently shown to have promising results in phase I/II studies in patients with metastatic melanoma.167 Other epigenetically regulated mediators of chemosensitivity to alkylating agents have also been identified that may be therapeutically upregulated with DNMT inhibitors.168 Furthermore, DNMT and HDAC inhibitors also have the ability to restore apoptotic capacity by upregulating epigenetically silenced effectors such as Apaf-1,169 caspase-8,170 and p16,171 and thereby enhancing chemosensitivity to the DNA-intercalating agent doxorubicin,169 DNA crosslinking agent cisplatin, and topisomerase inhibitor etoposide.171 This combination also has shown promising results in phase I/II clinical trials.172 Given their demonstrated ability to restore the apoptosome in melanoma, HDAC inhibitors also may radiosensitize human melanoma cells.173, 174 Taken together, the potential adjunctive role of DNMT and HDAC inhibitors used in conjunction with conventional chemo-, immuno-, and radiotherapeutic strategies is an active and exciting area of investigation (Table 3).69 Of note, several miRNAs have also demonstrated efficacy in animal models and this class of novel therapeutics is being actively investigated.29, 35 Further characterization of the epigenetic regulation of cell surface molecule expression, apoptotic mediators, and other related pathways is likely to further illuminate this promising area of cancer research.
MELANOMA AND ITS EPIGENOME: LOOKING FORWARD
With the incidence of melanoma increasing worldwide and the consistently poor prognosis associated with advanced cases,175 strategies for earlier detection, risk stratification, and enhanced therapeutic efficacy are desperately needed. Moving beyond a concept focused primarily on accumulated mutations to the DNA sequence as the central driver of carcinogenesis or melanomagenesis, the evidence reviewed herein points to a paradigm shift to consider gene expression also in the context of the epigenome. Such an approach provides an opportunity to explore, identify, and deploy new diagnostic and therapeutic strategies. This, in part, will depend on furthering our understanding of how the discrete categories of epigenetic changes interact with and regulate one another, as well as the mechanisms that disrupt these systems. For example, a recent study demonstrated that BRD4 is significantly upregulated in primary and metastatic melanoma tissues compared with melanocytes and benign nevi.176 BRD4 is a bromodomain and extraterminal domain (BET) family protein that exerts key roles at the interface between chromatin remodeling and transcriptional regulation by binding to acetylated histones and recruiting specific coactivating or corepressing chromatin-modifying enzymes to target promoters.177, 178 Newly developed, cell-permeable small-molecule inhibitors of BET proteins have shown very promising anti-melanoma activity in vivo, regardless of BRAF or NRAS mutational status. This final example illustrates the critical nature of advancing our understanding of epigenetic ‘cross-talk’ mechanisms in melanoma and other cancers. Further investigation into epigenetic fidelity maintenance mechanisms and their dysregulation in melanoma and other cancers will also be critical to our understanding the therapeutic manipulation of the cancer epigenome.
As we have discussed, epigenetic mechanisms such as DNA methylation and hydroxymethylation, histone modifications, and ncRNAs are critical to the regulation of gene expression, phenotypic plasticity, cell cycle regulation, apoptosis, and other critical biologic functions in both normal and cancer cells. Furthermore, distinct epigenetic hallmarks show promise for assisting in distinguishing between benign and malignant lesions under the microscope and in the blood, and may also provide critical prognostic information. We have also illustrated the ways in which the epigenome can be harnessed to unlock the expression of molecules critical to the success of chemo-, immuno-, and radiotherapeutic strategies. In summary, there is justification for great optimism that future advancements in our understanding of the melanoma and cancer epigenome will translate into direct diagnostic and therapeutic benefits for patients who are afflicted by this virulent form of human malignancy.
References
Holliday R . The inheritance of epigenetic defects. Science 1987;238:163–170.
Holliday R . Epigenetics: a historical overview. Epigenetics 2006;1:76–80.
Goldberg AD, Allis CD, Bernstein E . Epigenetics: a landscape takes shape. Cell 2007;128:635–638.
Riggs AD . X inactivation, differentiation, and DNA methylation. Cytogenet Cell Genet 1975;14:9–25.
Holliday R, Pugh JE . DNA modification mechanisms and gene activity during development. Science 1975;187:226–232.
Lister R, Ecker JR . Finding the fifth base: genome-wide sequencing of cytosine methylation. Genome Res 2009;19:959–966.
Guibert S, Weber M . Functions of DNA methylation and hydroxymethylation in mammalian development. Curr Top Dev Biol 2013;104:47–83.
Bird AP . CpG-rich islands and the function of DNA methylation. Nature 1986;321:209–213.
Fatemi M, Pao MM, Jeong S et al. Footprinting of mammalian promoters: use of a CpG DNA methyltransferase revealing nucleosome positions at a single molecule level. Nucleic Acids Res 2005;33:e176.
Maunakea AK, Nagarajan RP, Bilenky M et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010;466:253–257.
Lister R, Pelizzola M, Dowen RH et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009;462:315–322.
Catoni GL . S-adenosylmethionine; a new intermediate formed enzymatically from L-methionine and adenosinetriphosphate. J Biol Chem 1953;204:403–416.
Sen GL, Reuter JA, Webster DE et al. DNMT1 maintains progenitor function in self-renewing somatic tissue. Nature 2010;463:563–567.
Okano M, Bell DW, Haber DA et al. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999;99:247–257.
Kohli RM, Zhang Y . TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013;502:472–479.
Wyatt GR, Cohen SS . A new pyrimidine base from bacteriophage nucleic acids. Nature 1952;170:1072–1073.
Tahiliani M, Koh KP, Shen Y et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009;324:930–935.
Cortellino S, Xu J, Sannai M et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 2011;146:67–79.
He YF, Li BZ, Li Z et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011;333:1303–1307.
Haffner MC, Chaux A, Meeker AK et al. Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers. Oncotarget 2011;2:627–637.
Ito S, D'Alessio AC, Taranova OV et al. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010;466:1129–1133.
Koh KP, Yabuuchi A, Rao S et al. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell 2011;8:200–213.
Williams K, Christensen J, Pedersen MT et al. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature 2011;473:343–348.
Williams K, Christensen J, Helin K . DNA methylation: TET proteins—guardians of CpG islands? EMBO Rep 2012;13:28–35.
Abdel-Wahab O, Mullally A, Hedvat C et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 2009;114:144–147.
Moran-Crusio K, Reavie L, Shih A et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 2011;20:11–24.
Sun M, Song CX, Huang H et al. HMGA2/TET1/HOXA9 signaling pathway regulates breast cancer growth and metastasis. Proc Natl Acad Sci USA 2013;110:9920–9925.
Jawert F, Hasseus B, Kjeller G et al. Loss of 5-hydroxymethylcytosine and TET2 in oral squamous cell carcinoma. Anticancer Res 2013;33:4325–4328.
Mason EF, Hornick JL . Succinate dehydrogenase deficiency is associated with decreased 5-hydroxymethylcytosine production in gastrointestinal stromal tumors: implications for mechanisms of tumorigenesis. Mod Pathol 2013;26:1492–1497.
Liu C, Liu L, Chen X et al. Decrease of 5-hydroxymethylcytosine is associated with progression of hepatocellular carcinoma through downregulation of TET1. PLoS One 2013;8:e62828.
Song SJ, Poliseno L, Song MS et al. MicroRNA-antagonism regulates breast cancer stemness and metastasis via TET-family-dependent chromatin remodeling. Cell 2013;154:311–324.
Yan H, Parsons DW, Jin G et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009;360:765–773.
Yen KE, Bittinger MA, Su SM et al. Mutations: biomarker and therapeutic opportunities. Oncogene 2010;29:6409–6417.
Xu W, Yang H, Liu Y et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011;19:17–30.
Killian JK, Kim SY, Miettinen M et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov 2013;3:648–657.
Kouzarides T . Chromatin modifications and their function. Cell 2007;128:693–705.
Tan M, Luo H, Lee S et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011;146:1016–1028.
Shi Y, Lan F, Matson C et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004;119:941–953.
Li B, Carey M, Workman JL . The role of chromatin during transcription. Cell 2007;128:707–719.
Dobosy JR, Selker EU . Emerging connections between DNA methylation and histone acetylation. Cell Mol Life Sci 2001;58:721–727.
Feinberg AP, Tycko B . The history of cancer epigenetics. Nat Rev Cancer 2004;4:143–153.
Nguyen CT, Weisenberger DJ, Velicescu M et al. Histone H3-lysine 9 methylation is associated with aberrant gene silencing in cancer cells and is rapidly reversed by 5-aza-2'-deoxycytidine. Cancer Res 2002;62:6456–6461.
Seligson DB, Horvath S, Shi T et al. Global histone modification patterns predict risk of prostate cancer recurrence. Nature 2005;435:1262–1266.
Schwartz YB, Pirrotta V . Polycomb silencing mechanisms and the management of genomic programmes. Nat Rev Genet 2007;8:9–22.
Schwartz YB, Pirrotta V . A new world of Polycombs: unexpected partnerships and emerging functions. Nat Rev Genet 2013;14:853–864.
Qi P, Du X . The long non-coding RNAs, a new cancer diagnostic and therapeutic gold mine. Mod Pathol 2013;26:155–165.
Moulton T, Crenshaw T, Hao Y et al. Epigenetic lesions at the H19 locus in Wilms' tumour patients. Nat Genet 1994;7:440–447.
Bartolomei MS, Zemel S, Tilghman SM . Parental imprinting of the mouse H19 gene. Nature 1991;351:153–155.
Costa FF . Non-coding RNAs: lost in translation? Gene 2007;386:1–10.
Bartel DP . MicroRNAs: target recognition and regulatory functions. Cell 2009;136:215–233.
Costa FF . Non-coding RNAs: new players in eukaryotic biology. Gene 2005;357:83–94.
Wapinski O, Chang HY . Long noncoding RNAs and human disease. Trends Cell Biol 2011;21:354–361.
Feinberg AP, Vogelstein B . Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983;301:89–92.
Kim YI, Giuliano A, Hatch KD et al. Global DNA hypomethylation increases progressively in cervical dysplasia and carcinoma. Cancer 1994;74:893–899.
Cravo M, Pinto R, Fidalgo P et al. Global DNA hypomethylation occurs in the early stages of intestinal type gastric carcinoma. Gut 1996;39:434–438.
Soares J, Pinto AE, Cunha CV et al. Global DNA hypomethylation in breast carcinoma: correlation with prognostic factors and tumor progression. Cancer 1999;85:112–118.
Karpf AR, Matsui S . Genetic disruption of cytosine DNA methyltransferase enzymes induces chromosomal instability in human cancer cells. Cancer Res 2005;65:8635–8639.
Herman JG, Latif F, Weng Y et al. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci USA 1994;91:9700–9704.
Yoshiura K, Kanai Y, Ochiai A et al. Silencing of the E-cadherin invasion-suppressor gene by CpG methylation in human carcinomas. Proc Natl Acad Sci USA 1995;92:7416–7419.
Fraga MF, Herranz M, Espada J et al. A mouse skin multistage carcinogenesis model reflects the aberrant DNA methylation patterns of human tumors. Cancer Res 2004;64:5527–5534.
Toyota M, Ahuja N, Ohe-Toyota M et al. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA 1999;96:8681–8686.
Tanemura A, Terando AM, Sim MS et al. CpG island methylator phenotype predicts progression of malignant melanoma. Clin Cancer Res 2009;15:1801–1807.
Patino WD, Susa J . Epigenetics of cutaneous melanoma. Adv Dermatol 2008;24:59–70.
Straume O, Smeds J, Kumar R et al. Significant impact of promoter hypermethylation and the 540 C>T polymorphism of CDKN2A in cutaneous melanoma of the vertical growth phase. Am J Pathol 2002;161:229–237.
Bishop JN, Harland M, Randerson-Moor J et al. Management of familial melanoma. Lancet Oncol 2007;8:46–54.
Walker GJ, Flores JF, Glendening JM et al. Virtually 100% of melanoma cell lines harbor alterations at the DNA level within CDKN2A, CDKN2B, or one of their downstream targets. Genes Chromosomes Cancer 1998;22:157–163.
Hoon DS, Spugnardi M, Kuo C et al. Profiling epigenetic inactivation of tumor suppressor genes in tumors and plasma from cutaneous melanoma patients. Oncogene 2004;23:4014–4022.
Xu XC . Tumor-suppressive activity of retinoic acid receptor-beta in cancer. Cancer Lett 2007;253:14–24.
Sigalotti L, Covre A, Fratta E et al. Epigenetics of human cutaneous melanoma: setting the stage for new therapeutic strategies. J Transl Med 2010;8:56.
Paz MF, Fraga MF, Avila S et al. A systematic profile of DNA methylation in human cancer cell lines. Cancer Res 2003;63:1114–1121.
Lian CG, Xu Y, Ceol C et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell 2012;150:1135–1146.
Gambichler T, Sand M, Skrygan M . Loss of 5-hydroxymethylcytosine and ten-eleven translocation 2 protein expression in malignant melanoma. Melanoma Res 2013;23:218–220.
Uchiyama R, Uhara H, Uchiyama A et al. 5-Hydroxymethylcytosine as a useful marker to differentiate between malignant melanomas and benign melanocytic nevi. J Dermatol Sci 2014;73:161–163.
Larson AR, Dresser KA, Zhan Q et al. Loss of 5-hydroxymethylcytosine correlates with increasing morphologic dysplasia in melanocytic tumors. Mod Pathol, advance online publication, 3 January 2014 doi:10.1038/modpathol.2013.224(e-pub ahead of print).
Jin SG, Jiang Y, Qiu R et al. 5-Hydroxymethylcytosine is strongly depleted in human cancers but its levels do not correlate with IDH1 mutations. Cancer Res 2011;71:7360–7365.
Shibata T, Kokubu A, Miyamoto M et al. Mutant IDH1 confers an in vivo growth in a melanoma cell line with BRAF mutation. Am J Pathol 2011;178:1395–1402.
Preston BD, Albertson TM, Herr AJ . DNA replication fidelity and cancer. Semin Cancer Biol 2010;20:281–293.
Song J, Teplova M, Ishibe-Murakami S et al. Structure-based mechanistic insights into DNMT1-mediated maintenance DNA methylation. Science 2012;335:709–712.
Matje DM, Zhou H, Smith DA et al. Enzyme-promoted base flipping controls DNA methylation fidelity. Biochemistry 2013;52:1677–1685.
Borgaro JG, Benner N, Zhu Z . Fidelity index determination of DNA methyltransferases. PLoS One 2013;8:e63866.
Greenberg ES, Chong KK, Huynh KT et al. Epigenetic biomarkers in skin cancer. Cancer Lett 2014;342:170–177.
Florenes VA, Skrede M, Jorgensen K et al. Deacetylase inhibition in malignant melanomas: impact on cell cycle regulation and survival. Melanoma Res 2004;14:173–181.
Zhang XD, Gillespie SK, Borrow JM et al. The histone deacetylase inhibitor suberic bishydroxamate regulates the expression of multiple apoptotic mediators and induces mitochondria-dependent apoptosis of melanoma cells. Mol Cancer Ther 2004;3:425–435.
Ye Y, Jin L, Wilmott JS et al. PI(4,5)P2 5-phosphatase A regulates PI3K/Akt signalling and has a tumour suppressive role in human melanoma. Nat Commun 2013;4:1508.
Bachmann IM, Halvorsen OJ, Collett K et al. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J Clin Oncol 2006;24:268–273.
McHugh JB, Fullen DR, Ma L et al. Expression of polycomb group protein EZH2 in nevi and melanoma. J Cutan Pathol 2007;34:597–600.
Ceol CJ, Houvras Y, Jane-Valbuena J et al. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature 2011;471:513–517.
Alharbi RA, Pettengell R, Pandha HS et al. The role of HOX genes in normal hematopoiesis and acute leukemia. Leukemia 2013;27:1000–1008.
Karram S, Novy M, Saroufim M et al. Predictors of BRAF mutation in melanocytic nevi: analysis across regions with different UV radiation exposure. Am J Dermatopathol 2013;35:412–418.
Rodriguez-Paredes M, Martinez de Paz A, Simo-Riudalbas L et al. Gene amplification of the histone methyltransferase SETDB1 contributes to human lung tumorigenesis. Oncogene 2013;33:2807–2813.
Bennett PE, Bemis L, Norris DA et al. miR in melanoma development: miRNAs and acquired hallmarks of cancer in melanoma. Physiol Genom 2013;45:1049–1059.
Liu S, Tetzlaff MT, Cui R et al. MiR-200c inhibits melanoma progression and drug resistance through down-regulation of BMI-1. Am J Pathol 2012;181:1823–1835.
Ringrose L, Paro R . Epigenetic regulation of cellular memory by the Polycomb and Trithorax group proteins. Annu Rev Genet 2004;38:413–443.
Jacobs JJ, Scheijen B, Voncken JW et al. Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF. Genes Dev 1999;13:2678–2690.
Tao ZH, Wan JL, Zeng LY et al. MiR-612 suppresses the invasive-metastatic cascade in hepatocellular carcinoma. The J Exp Med 2013;210:789–803.
van Kempen LC, van den Hurk K, Lazar V et al. Loss of microRNA-200a and c, and microRNA-203 expression at the invasive front of primary cutaneous melanoma is associated with increased thickness and disease progression. Virchows Arch 2012;461:441–448.
Jin L, Hu WL, Jiang CC et al. MicroRNA-149*, a p53-responsive microRNA, functions as an oncogenic regulator in human melanoma. Proc Natl Acad Sci USA 2011;108:15840–15845.
Satzger I, Mattern A, Kuettler U et al. MicroRNA-21 is upregulated in malignant melanoma and influences apoptosis of melanocytic cells. Exp Dermatol 2012;21:509–514.
Streicher KL, Zhu W, Lehmann KP et al. A novel oncogenic role for the miRNA-506-514 cluster in initiating melanocyte transformation and promoting melanoma growth. Oncogene 2012;31:1558–1570.
Pencheva N, Tran H, Buss C et al. Convergent multi-miRNA targeting of ApoE drives LRP1/LRP8-dependent melanoma metastasis and angiogenesis. Cell 2012;151:1068–1082.
Tang L, Zhang W, Su B et al. Long noncoding RNA HOTAIR is associated with motility, invasion, and metastatic potential of metastatic melanoma. Biomed Res Int 2013;2013:251098.
Engreitz JM, Pandya-Jones A, McDonel P et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science 2013;341:1237973.
Khaitan D, Dinger ME, Mazar J et al. The melanoma-upregulated long noncoding RNA SPRY4-IT1 modulates apoptosis and invasion. Cancer Res 2011;71:3852–3862.
Wu CF, Tan GH, Ma CC et al. The non-coding RNA llme23 drives the malignant property of human melanoma cells. J Genet Genom 2013;40:179–188.
Clarke MF, Dick JE, Dirks PB et al. Cancer stem cells—perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res 2006;66:9339–9344.
Bruce WR, Van Der Gaag H . A quantitative assay for the number of murine lymphoma cells capable of proliferation in vivo. Nature 1963;199:79–80.
Park CH, Bergsagel DE, McCulloch EA . Mouse myeloma tumor stem cells: a primary cell culture assay. J Natl Cancer Inst 1971;46:411–422.
Al-Hajj M, Wicha MS, Benito-Hernandez A et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 2003;100:3983–3988.
Singh SK, Hawkins C, Clarke ID et al. Identification of human brain tumour initiating cells. Nature 2004;432:396–401.
O'Brien CA, Pollett A, Gallinger S et al. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007;445:106–110.
Schatton T, Murphy GF, Frank NY et al. Identification of cells initiating human melanomas. Nature 2008;451:345–349.
Girouard SD, Murphy GF . Melanoma stem cells: not rare, but well done. Lab Invest 2011;91:647–664.
Ito K, Bernardi R, Pandolfi PP . A novel signaling network as a critical rheostat for the biology and maintenance of the normal stem cell and the cancer-initiating cell. Curr Opin Genet Dev 2009;19:51–59.
Visvader JE, Lindeman GJ . Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer 2008;8:755–768.
White RA, Neiman JM, Reddi A et al. Epithelial stem cell mutations that promote squamous cell carcinoma metastasis. J Clin Invest 2013;123:4390–4404.
Caramel J, Papadogeorgakis E, Hill L et al. A switch in the expression of embryonic EMT-inducers drives the development of malignant melanoma. Cancer Cell 2013;24:466–480.
Concepcion Garrido M, Requena L, Kutzner H et al. Desmoplastic melanoma: expression of epithelial–mesenchymal transition-related proteins. Am J Dermatopathol 2013;36:238–242.
Polyak K, Weinberg RA . Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 2009;9:265–273.
Chaffer CL, Weinberg RA . A perspective on cancer cell metastasis. Science 2011;331:1559–1564.
Gupta GP, Massague J . Cancer metastasis: building a framework. Cell 2006;127:679–695.
Mani SA, Guo W, Liao MJ et al. The epithelial–mesenchymal transition generates cells with properties of stem cells. Cell 2008;133:704–715.
Miller AJ, Mihm MC Jr . Melanoma. N Engl J Med 2006;355:51–65.
Alexaki VI, Javelaud D, Van Kempen LC et al. GLI2-mediated melanoma invasion and metastasis. J Natl Cancer Inst 2010;102:1148–1159.
Danen EH, de Vries TJ, Morandini R et al. E-cadherin expression in human melanoma. Melanoma Res 1996;6:127–131.
Hsu MY, Wheelock MJ, Johnson KR et al. Shifts in cadherin profiles between human normal melanocytes and melanomas. J Invest Dermatol 1996;1:188–194.
Thiery JP . Epithelial–mesenchymal transitions in tumour progression. Nat Rev Cancer 2002;2:442–454.
Danen EH, Ten Berge PJ, Van Muijen GN et al. Emergence of alpha 5 beta 1 fibronectin- and alpha v beta 3 vitronectin-receptor expression in melanocytic tumour progression. Histopathology 1994;24:249–256.
Hofmann UB, Westphal JR, Waas ET et al. Coexpression of integrin alpha(v)beta3 and matrix metalloproteinase-2 (MMP-2) coincides with MMP-2 activation: correlation with melanoma progression. J Invest Dermatol 2000;115:625–632.
Liang Y, Jansen M, Aronow B et al. The quantitative trait gene latexin influences the size of the hematopoietic stem cell population in mice. Nat Genet 2007;39:178–188.
Liang Y, Van Zant G . Aging stem cells, latexin, and longevity. Exp Cell Res 2008;314:1962–1972.
Muthusamy V, Premi S, Soper C et al. The hematopoietic stem cell regulatory gene latexin has tumor-suppressive properties in malignant melanoma. J Invest Dermatol 2013;133:1827–1833.
Roesch A, Fukunaga-Kalabis M, Schmidt EC et al. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010;141:583–594.
Iwase S, Lan F, Bayliss P et al. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell 2007;128:1077–1088.
Roesch A, Becker B, Meyer S et al. Retinoblastoma-binding protein 2-homolog 1: a retinoblastoma-binding protein downregulated in malignant melanomas. Mod Pathol 2005;18:1249–1257.
Scibetta AG, Santangelo S, Coleman J et al. Functional analysis of the transcription repressor PLU-1/JARID1B. Mol Cell Biol 2007;27:7220–7235.
Krivtsov AV, Armstrong SA . MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer 2007;7:823–833.
Roesch A, Becker B, Schneider-Brachert W et al. Re-expression of the retinoblastoma-binding protein 2-homolog 1 reveals tumor-suppressive functions in highly metastatic melanoma cells. J Invest Dermatol 2006;126:1850–1859.
Yamane K, Tateishi K, Klose RJ et al. PLU-1 is an H3K4 demethylase involved in transcriptional repression and breast cancer cell proliferation. Mol Cell 2007;25:801–812.
Bartel DP . MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004;116:281–297.
He L, Thomson JM, Hemann MT et al. A microRNA polycistron as a potential human oncogene. Nature 2005;435:828–833.
Gregory PA, Bracken CP, Bert AG et al. MicroRNAs as regulators of epithelial–mesenchymal transition. Cell Cycle 2008;7:3112–3118.
Park SM, Gaur AB, Lengyel E et al. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev 2008;22:894–907.
Shimono Y, Zabala M, Cho RW et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 2009;138:592–603.
Liu S, Kumar SM, Lu H et al. MicroRNA-9 up-regulates E-cadherin through inhibition of NF-kappaB1-Snail1 pathway in melanoma. J Pathol 2012;226:61–72.
Song SJ, Pandolfi P . MiR-22 in tumorigenesis. Cell Cycle 2014;13:11–12.
Song SJ, Ito K, Ala U et al. The oncogenic microRNA miR-22 targets the TET2 tumor suppressor to promote hematopoietic stem cell self-renewal and transformation. Cell Stem Cell 2013;13:87–101.
Sigalotti L, Fratta E, Bidoli E et al. Methylation levels of the ‘long interspersed nucleotide element-1’ repetitive sequences predict survival of melanoma patients. J Transl Med 2011;9:78.
Marini A, Mirmohammadsadegh A, Nambiar S et al. Epigenetic inactivation of tumor suppressor genes in serum of patients with cutaneous melanoma. J Invest Dermatol 2006;126:422–431.
Hanna JA, Hahn L, Agarwal S et al. In situ measurement of miR-205 in malignant melanoma tissue supports its role as a tumor suppressor microRNA. Lab Invest 2012;92:1390–1397.
Liu S, Tetzlaff MT, Liu A et al. Loss of microRNA-205 expression is associated with melanoma progression. Lab Invest 2012;92:1084–1096.
Nguyen T, Kuo C, Nicholl MB et al. Downregulation of microRNA-29c is associated with hypermethylation of tumor-related genes and disease outcome in cutaneous melanoma. Epigenetics 2011;6:388–394.
Asangani IA, Harms PW, Dodson L et al. Genetic and epigenetic loss of microRNA-31 leads to feed-forward expression of EZH2 in melanoma. Oncotarget 2012;3:1011–1025.
Luo C, Tetteh PW, Merz PR et al. MiR-137 inhibits the invasion of melanoma cells through downregulation of multiple oncogenic target genes. J Invest Dermatol 2013;133:768–775.
Kanemaru H, Fukushima S, Yamashita J et al. The circulating microRNA-221 level in patients with malignant melanoma as a new tumor marker. J Dermatol Sci 2011;61:187–193.
Friedman EB, Shang S, de Miera EV et al. Serum microRNAs as biomarkers for recurrence in melanoma. J Transl Med 2012;10:155.
McMillan TJ, Hart IR . Enhanced experimental metastatic capacity of a murine melanoma following pre-treatment with anticancer drugs. Clin Exp Metast 1986;4:285–292.
Woods DM, Woan K, Cheng F et al. The antimelanoma activity of the histone deacetylase inhibitor panobinostat (LBH589) is mediated by direct tumor cytotoxicity and increased tumor immunogenicity. Melanoma Res 2013;23:341–348.
Weber J, Salgaller M, Samid D et al. Expression of the MAGE-1 tumor antigen is up-regulated by the demethylating agent 5-aza-2'-deoxycytidine. Cancer Res 1994;54:1766–1771.
Serrano A, Garcia A, Abril E et al. Methylated CpG points identified within MAGE-1 promoter are involved in gene repression. Int J Cancer 1996;68:464–470.
De Smet C, De Backer O, Faraoni I et al. The activation of human gene MAGE-1 in tumor cells is correlated with genome-wide demethylation. Proc Natl Acad Sci USA 1996;93:7149–7153.
Vo DD, Prins RM, Begley JL et al. Enhanced antitumor activity induced by adoptive T-cell transfer and adjunctive use of the histone deacetylase inhibitor LAQ824. Cancer Res 2009;69:8693–8699.
Weber JS, Kudchadkar RR, Yu B et al. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J Clin Oncol 2013;31:4311–4318.
Kruit WH, Suciu S, Dreno B et al. Selection of immunostimulant AS15 for active immunization with MAGE-A3 protein: results of a randomized phase II study of the European Organisation for Research and Treatment of Cancer Melanoma Group in Metastatic Melanoma. J Clin Oncol 2013;31:2413–2420.
Jazirehi AR, Arle D . Epigenetic regulation of the TRAIL/Apo2L apoptotic pathway by histone deacetylase inhibitors: an attractive approach to bypass melanoma immunotherapy resistance. Am J Clin Exp Immunol 2013;2:55–74.
Verbeek B, Southgate TD, Gilham DE et al. O6-methylguanine-DNA methyltransferase inactivation and chemotherapy. Br Med Bull 2008;85:17–33.
Christmann M, Pick M, Lage H et al. Acquired resistance of melanoma cells to the antineoplastic agent fotemustine is caused by reactivation of the DNA repair gene MGMT. Int J Cancer 2001;92:123–129.
Tawbi HA, Beumer JH, Tarhini AA et al. Safety and efficacy of decitabine in combination with temozolomide in metastatic melanoma: a phase I/II study and pharmacokinetic analysis. Ann Oncol 2013;24:1112–1119.
Sato Y, Marzese DM, Ohta K et al. Epigenetic regulation of REG1A and chemosensitivity of cutaneous melanoma. Epigenetics 2013;8:1043–1052.
Soengas MS, Capodieci P, Polsky D et al. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature 2001;409:207–211.
Fulda S, Kufer MU, Meyer E et al. Sensitization for death receptor- or drug-induced apoptosis by re-expression of caspase-8 through demethylation or gene transfer. Oncogene 2001;20:5865–5877.
Valentini A, Gravina P, Federici G et al. Valproic acid induces apoptosis, p16INK4A upregulation and sensitization to chemotherapy in human melanoma cells. Cancer Biol Ther 2007;6:185–191.
Daud AI, Dawson J, DeConti RC et al. Potentiation of a topoisomerase I inhibitor, karenitecin, by the histone deacetylase inhibitor valproic acid in melanoma: translational and phase I/II clinical trial. Clin Cancer Res 2009;15:2479–2487.
Munshi A, Kurland JF, Nishikawa T et al. Histone deacetylase inhibitors radiosensitize human melanoma cells by suppressing DNA repair activity. Clin Cancer Res 2005;11:4912–4922.
Munshi A, Tanaka T, Hobbs ML et al. A histone deacetylase inhibitor, enhances the response of human tumor cells to ionizing radiation through prolongation of gamma-H2AX foci. Mol Cancer Ther 2006;5:1967–1974.
Lian CG, Mihm MC, Pierard G et al. Skin cancer. In: Stewart BW, Wild CP, (eds). World Cancer Report 2014. Lyon, France: International Agency for Research on Cancer, WHO. World Health Organization Press, 2014.
Segura MF, Fontanals-Cirera B, Gaziel-Sovran A et al. BRD4 sustains melanoma proliferation and represents a new target for epigenetic therapy. Cancer Res 2013;73:6264–6276.
Matzuk MM, McKeown MR, Filippakopoulos P et al. Small-molecule inhibition of BRDT for male contraception. Cell 2012;150:673–684.
Belkina AC, Denis GV . BET domain co-regulators in obesity, inflammation and cancer. Nat Rev Cancer 2012;12:465–477.
Müller DW, Bosserhoff AK . Integrin beta 3 expression is regulated by let-7a miRNA in malignant melanoma. Oncogene 2008;27:6698–6706.
Bhattacharya A, Schmitz U, Wolkenhauer O et al. Regulation of cell cycle checkpoint kinase WEE1 by miR-195 in malignant melanoma. Oncogene 2013;32:3175–3183.
Felicetti F, Errico MC, Bottero L et al. The promyelocytic leukemia zinc finger-microRNA-221/-222 pathway controls melanoma progression through multiple oncogenic mechanisms. Cancer Res 2008;68:2745–2754.
Igoucheva O, Alexeev V . MicroRNA-dependent regulation of cKit in cutaneous melanoma. Biochem Biophys Res Commun 2009;379:790–794.
Chen J, Zhang X, Lentz C et al. miR-193b Regulates Mcl-1 in Melanoma. Am J Pathol 2011;179:2162–2168.
Penna E, Orso F, Cimino D et al. MicroRNA-214 contributes to melanoma tumour progression through suppression of TFAP2C. EMBO J 2011;30:1990–2007.
Dar AA, Majid S, de Semir D et al. MiRNA-205 suppresses melanoma cell proliferation and induces senescence via regulation of E2F1 protein. J Biol Chem 2011;286:16606–16614.
Reuland SN, Smith SM, Bemis LT et al. MicroRNA-26a is strongly downregulated in melanoma and induces cell death through repression of silencer of death domains (SODD). J Invest Dermatol 2013;133:1286–1293.
Lodygin D, Tarasov V, Epanchintsev A et al. Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle 2008;7:2591–2600.
Kappelmann M, Kuphal S, Meister G et al. MicroRNA miR-125b controls melanoma progression by direct regulation of c-Jun protein expression. Oncogene 2013;32:2984–2991.
Dar AA, Majid S, Rittsteuer C et al. The role of miR-18b in MDM2-p53 pathway signaling and melanoma progression. J Natl Cancer Inst 2013;105:433–442.
Wang HF, Chen H, Ma MW et al. MiR-573 regulates melanoma progression by targeting the melanoma cell adhesion molecule. Oncol Rep 2013;30:520–526.
Acknowledgements
This study is supported by NIH Grant 5P50CA093683 to the SPORE Core at Brigham and Women’s Hospital (GFM). This study is supported, in part, by R01 CA138231, R01 CA158467 (GFM).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
As the incidence and mortality rates of melanoma continue to increase worldwide, there is a pressing need to revisit its fundamental pathobiology. Recent evidence implicates dysregulated epigenetics in the pathogenesis of this virulent malignancy. This review examines the existing literature and discusses its translational potential for developing novel biomarkers and therapeutic targets.
Rights and permissions
About this article

Cite this article
Lee, J., Murphy, G. & Lian, C. Melanoma epigenetics: novel mechanisms, markers, and medicines. Lab Invest 94, 822–838 (2014). https://doi.org/10.1038/labinvest.2014.87
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2014.87
This article is cited by
-
Multi-scale geometric network analysis identifies melanoma immunotherapy response gene modules
Scientific Reports (2024)
-
Altered DNA methylation in kidney disease: useful markers and therapeutic targets
Clinical and Experimental Nephrology (2022)
-
Sulforaphane and iberin are potent epigenetic modulators of histone acetylation and methylation in malignant melanoma
European Journal of Nutrition (2021)
-
Epigenetic reprogramming of primary pancreatic cancer cells counteracts their in vivo tumourigenicity
Oncogene (2019)
-
Emerging Biomarkers in Cutaneous Melanoma
Molecular Diagnosis & Therapy (2018)
