
Abstract
There is experimental evidence that some antioxidant flavonoids show therapeutic potential in the treatment of hepatitis C through inhibition of hepatitis C virus (HCV) replication. We examined the effect of treatment with the flavonols quercetin and kaempferol, the flavanone taxifolin and the flavone apigenin on HCV replication efficiency in an in vitro model. While all flavonoids studied were able to reduce viral replication at very low concentrations (ranging from 0.1 to 5 μM), quercetin appeared to be the most effective inhibitor of HCV replication, showing a marked anti-HCV activity in replicon-containing cells when combined with interferon (IFN)α. The contribution of oxidative/nitrosative stress and lipogenesis modulation to inhibition of HCV replication by quercetin was also examined. As expected, quercetin decreased HCV-induced reactive oxygen and nitrogen species (ROS/RNS) generation and lipoperoxidation in replicating cells. Quercetin also inhibited liver X receptor (LXR)α-induced lipid accumulation in LXRα-overexpressing and replicon-containing Huh7 cells. The mechanism underlying the LXRα-dependent lipogenesis modulatory effect of quercetin in HCV-replicating cells seems to involve phosphatidylinositol 3-kinase (PI3K)/AKT pathway inactivation. Thus, inhibition of the PI3K pathway by LY294002 attenuated LXRα upregulation and HCV replication mediated by lipid accumulation, showing an additive effect when combined with quercetin. Inactivation of the PI3K pathway by quercetin may contribute to the repression of LXRα-dependent lipogenesis and to the inhibition of viral replication induced by the flavonol. Combined, our data suggest that oxidative/nitrosative stress blockage and subsequent modulation of PI3K-LXRα-mediated lipogenesis might contribute to the inhibitory effect of quercetin on HCV replication.
Similar content being viewed by others


Main
Hepatitis C virus (HCV) is a positive-sensed, single-stranded RNA virus of the Flaviviridae family.1 The genome of the human HCV encodes a polyprotein post-translationally cleaved by both viral and cellular proteases to produce four structural (core, E1, E2 and p7) and six non-structural (NS2, NS3, NS4A, NS4B, NS5A and NS5B) proteins.2 HCV is a major cause of viral hepatitis, with estimated 150–200 million people infected worldwide. Approximately 70% of patients suffer from persistent infection, which causes chronic hepatitis and induces several complex mechanisms leading to inflammation, insulin resistance, steatosis, fibrosis and hepatocellular carcinoma.3
Although the molecular mechanisms of HCV pathogenesis remain unclear, oxidative/nitrosative stress associated with reactive oxygen and nitrogen species (ROS/RNS) generation is emerging as a key step and a major initiator in the development and the progression of liver damage.4, 5, 6, 7
Hepatic steatosis is often observed in patients with chronic hepatitis C and has been reported to be associated with insulin resistance and progression of fibrosis in the liver.8, 9 The molecular mechanisms of HCV-associated steatosis are poorly understood,10 but they are mediated in large part by changes in lipogenic and oxidative stress-mediated proinflammatory cytokines gene expression associated with HCV proteins.3, 7, 11 Thus, NS5A and core proteins colocalize on lipid droplets and activate various pathways of lipid metabolism, contributing to the development of HCV-associated steatosis.12, 13 Moreover, lipid accumulation is necessary for successful HCV replication.14 In this respect, we recently showed that HCV replication induces liver X receptor (LXR)α-mediated intracellular lipid accumulation, which in turn contributes to the efficient replication of HCV.15
As oxidative stress has a central role in hepatitis C pathogenesis and progression, antioxidants have been proposed as therapeutic agents and drug coadjuvants. Much attention has been given to the potential health-promoting properties of flavonoids due to their reported wide range of activities in the prevention of common diseases, including coronary heart disease, cancer, neurodegenerative disease, gastrointestinal disorders and others.16, 17, 18 Information on the potential benefits of flavonoids in the treatment of hepatitis C is relatively limited and sometimes contradictory,19, 20 although some flavonoids, including quercetin, one of the most abundant flavonol-type flavonoids present in the human diet, seem to inhibit the replication of HCV.21, 22, 23
On the basis of these data, in the current research we explored the effects of different natural antioxidant flavonoids on HCV replication in an in vitro model. We have previously described that important differences exist between flavonoids with different structural features in their capacity to abrogate the generation of different ROS/RNS.24 Therefore, we examined the effect of the flavonols quercetin and kaempferol, the flavanone taxifolin and the flavone apigenin on HCV replication efficiency. Because results obtained indicated that quercetin was the most effective inhibitor of HCV replication, we aimed to investigate the molecular mechanisms involved in the modulation of HCV replication by this flavonol, focusing on its effects on ROS and RNS generation and lipid peroxidation, lipid accumulation and LXRα gene expression. In view of our previous findings showing that phosphatidylinositol 3-kinase (PI3K) pathway inhibition attenuates LXRα upregulation induced by HCV expression,15 we also examined the role of the PI3K/AKT pathway in lipogenesis modulation by quercetin in our in vitro model.
MATERIALS AND METHODS
Cells, Cell Culture and Treatment Protocols
Huh7 cells expressing full-length genotype 1b HCV replicons (HCV-G1) were established as previously described.25 HCV-G1 cells were treated with human interferon (IFN)α-2b (PBL Interferon Source, Piscataway, NJ, USA) to eliminate replicons, and were used as control cured cells (Huh7). Huh7 and their derivatives were grown at 37 °C with a 5% CO2 atmosphere in Dulbecco’s modified Eagle’s medium, supplemented with 10% fetal calf serum, 2 mM L-glutamine and 50 mg/ml gentamycin. HCV-G1 cells were selected by growth in culture medium containing G418. To prevent phenotypic drift, the cultures were used for only 8–10 weeks before reverting to frozen stocks from an early passage.
HCV-G1 cells were treated with the flavonols quercetin and kaempferol, the flavanone taxifolin and the flavone apigenin (Sigma-Aldrich, Madrid, Spain) (Figure 1a) in concentrations ranging from 0.1 to 5 μM for 48 h to evaluate their potential to inhibit HCV replication. Then, to analyze the molecular mechanisms involved in HCV inhibition by quercetin, we treated HCV-G1 cells with 0.1–0.5–1–5 μM quercetin with or without 0.5 U/ml IFNα-2b for 48 h. HCV-replicating cells were also treated with 50 U/ml IFNα-2b as a control of replication inhibition. Finally, we investigated the effect of PI3K chemical inhibition by treating replicon-containing cells with LY294002 (Tocris Bioscience, Bristol, UK).26 Thus, HCV-G1 cells were pretreated for 8 h with 50 μM LY294002 and then with or without quercetin (0.1–5 μM) or IFNα-2b (0.5 and 50 U/ml) for an additional 48 h. The vehicle used for all treatments was DMSO (0.05%). HCV-G1 control cells were incubated with DMSO alone (vehicle-treated cells).

(a) Chemical structure of the flavonols quercetin and kaempferol, the flavanone taxifolin and the flavone apigenin. (b) Effect of the different flavonoids on HCV RNA replication in genomic replicon-containing cells. HCV-G1 cells were incubated with 0.1–5 μM quercetin, kaempferol, taxifolin or apigenin. Samples of 1 μg RNA were analyzed by real-time RT-qPCR using specific primers to determine HCV RNA levels. Histone H3 mRNA levels were used for sample normalization. Data are expressed as HCV copies/μg total RNA. Data are described as the mean values±s.d. of six independent experiments (*P<0.05; **P<0.01; ***P<0.001 vs HCV-G1 vehicle-treated cells).
Cell Viability in Cell Culture
The cell viability was assessed by the mitochondrial function, measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction activity for measuring cell proliferation and cytotoxicity as previously reported.27 Briefly, cells were seeded in a 24-well plate and incubated with the different treatments. After treatment, the cells were incubated with 0.5 mg/ml MTT (Sigma-Aldrich) for 2 h at 37 °C. Subsequently, the media were aspirated and the cells were lysed with DMSO, where after the absorbance was read at 560 nm, with background subtraction at 650 nm, using a microplate reader (Bio-Rad Laboratories, Veenendaal, The Netherlands).
Flow Cytometry and Fluorescence Microscopy
ROS and RNS production, lipid peroxidation and lipid accumulation were assessed by flow cytometry. The ROS and RNS production was analyzed by flow cytometry as the fluorescence of 2′,7′-dichlorofluorescein (DCF) and ethidium (ETH), which are the oxidation products of 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA; Sigma-Aldrich) and dihydroethidium (DHE; Molecular Probes, Leiden, The Netherlands) with a sensitivity for H2O2/NO-based radicals and O2•−, respectively.24 The lipid content and lipid peroxidation in cultured cells were determined using Bodipy 493/503 and Bodipy 581/591 C11, respectively (Invitrogen, Carlsbad, CA, USA). Briefly, cell monolayers were washed twice with PBS and incubated with the corresponding dye solution (5 μM for DCFH-DA and DHE or 1 μg/ml for Bodipy 493/503 and 581/591 C11) for 15–45 min at 37 °C, then washed twice, resuspended in PBS, and analyzed on a FACSCalibur flow cytometer (Becton Dickinson Biosciences, San Jose, CA, USA). Fluorescence of 10,000 cells was analyzed using the Cell Quest software (Becton Dickinson Biosciences). Indeed, intracellular lipid accumulation was corroborated by fluorescence microscopy using a Nikon Eclipse Ti inverted microscope (Nikon, Amstelveen, The Netherlands).15
Triglyceride and Free Fatty Acid Assay
Intracellular triglyceride (TG) and free fatty acids (FFAs) accumulation were evaluated after the lysis of cells. We collected the supernatants of each group to determine the TG and FFA content in the cell lysates. TG and FFA levels were determined with kits from Biovision Research Products (Mountain View, CA, USA) following the guide provided by the company.
Quantitative Real-Time PCR
Total RNA was obtained by using a Trizol reagent (Life Technologies, Carlsbad, CA, USA). First-strand cDNA was synthesized using the High-Capacity cDNA Archive Kit (Applied Biosystems, Weiterstadt, Germany). For gene expression assays, cDNA was amplified using multiplex real-time PCRs on a StepOne Plus (Applied Biosystems).28 TaqMan primers and probes were derived from the commercially available TaqMans® Gene Expression Assays (Applied Biosystems) (Table 1). Relative changes in gene expression levels were determined using the 2−ΔΔCt method. The cycle number at which the transcripts were detectable (CT) was normalized to the cycle number of GAPDH detection, referred to as ΔCT. PCR efficiency was determined by TaqMan analysis on a standard curve for targets and endogenous control amplifications that were highly similar. Replication studies were carried out using a SYBR Green kit (Roche Diagnostics GmbH) and two specific primer sets (5′-CCTGTGAGGAACTACTGTCT-3′ and 5′-CTATCAGGCAGTACCACAAG-3′ for HCV, spanning 255 nucleotides of the 5′ non-translated region; 5′-AAAGCCGCTCGCAAGAGTGCG-3′ and 5′-ACTTGCCTCCTGCAAAGCAC-3′ for histone H3).6 The number of HCV RNA copies was determined by crossing point interpolation into standard curves, which were generated by reverse transcription of serially diluted, in vitro synthesized viral RNA (genomic) followed by quantitative PCR. The total amount of RNA per reaction was kept constant (1 μg) by the addition of Huh7 RNA.
Western Blotting
Protein extraction and western blotting were performed as described,15 using rabbit polyclonal antibodies against LXRα (Abcam, Cambridge, UK), phospho-AKT (Ser 473) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), AKT (Santa Cruz Biotechnology), mouse monoclonal antibody against HCV NS5A protein (ViroStat, Portland, ME, USA) and HCV core protein (Thermo Scientific Pierce, Rockford, IL, USA). Bound primary antibody was detected with HRP (horseradish peroxidase)-conjugated anti-rabbit or anti-mouse antibodies (DAKO, Glostrup, Denmark), and blots were developed using an enhanced chemiluminescence detection system (ECL kit; Amersham Pharmacia, Uppsala, Sweden). The density of the specific bands was quantified with an imaging densitometer (Scion Image, Frederick, MD, USA).
Adenoviral Vectors
The recombinant adenovirus encoding LXRα was constructed as follows: human LXRα cDNA was kindly provided by Dr D Mangelsdorf (UT Southwestern Medical Center, Dallas, Texas). The coding sequence was released from the plasmid pCMX with KpnI-BamHI, and subcloned in the adenoviral shuttle vector pAC/CMVpLpA29 pre-digested with the same restriction enzymes. The resulting shuttle plasmid and the pJM17 were co-transfected into 293 cells to allow homologous recombination and generate the Ad-LXRα adenovirus. Cell monolayers of human hepatoma Huh7 cell line were infected with recombinant adenovirus encoding transcription factor or with an insertless adenoviral vector (Ad-CONT) for 24 h at a multiplicity of infection (MOI) of 15–30 plaque-forming units/cell. LXRα ligand T0901317 (Sigma-Aldrich) (10 μM) was added at 24 h post infection and at 48 h post infection cells were treated with 0.1–5 μM of quercetin or IFNα-2b (0.5 and 50 U/ml). Finally, cells were harvested and analyzed.
Immunofluorescence Analysis
Immunofluorescence analysis was performed as previously described.15 Coverslips were incubated with rabbit anti-LXRα antibody (Abcam) at 4 °C overnight. Thereafter, the secondary antibody donkey anti-rabbit conjugated with FITC (Jackson ImmunoResearch, Baltimore, PA, USA) was applied. Nuclei were stained with DAPI (blue). After washing, the coverslips were mounted on DakoCytomation Fluorescent Mounting Medium (DAKO). The preparations were analyzed with an inverted fluorescence microscope (Nikon Eclipse Ti-U).
Statistical Analysis
Results are expressed as the mean±standard deviation. Significant differences were evaluated by one way analysis of variance (ANOVA) and Newman–Keul’s test. P<0.05 was considered to be significant for a difference.
RESULTS
Quercetin is the Most Effective Inhibitor of HCV Replication
To examine the effect of treatment with different flavonoids on HCV replication efficiency, HCV-G1 cells were cultured with quercetin, kaempferol, taxifolin and apigenin in concentrations ranging from 0.1 to 5 μM or vehicle. At these very low concentrations, flavonoids exhibit no cellular toxicity and the limited antiperoxidative capacity does not interfere with their anti-HCV activity.30 As shown in Figure 1b, all flavonoids were able to significantly reduce the HCV RNA copy number (K0.1: −35%, K1: −43%; T0.1: −32%, T1: −39%; T5: −43%; A0.1: −40%, A1: −30%, vs non-treated cells), but quercetin appeared to be the most effective inhibitor of HCV replication at doses resembling human plasma concentrations upon quercetin supplementation.31
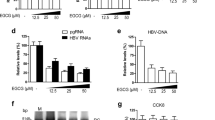
In view of these findings, HCV-G1 cells were incubated with quercetin at concentrations of 0.1, 0.5, 1 and 5 μM alone or combined with IFNα 0.5 (Figure 2). HCV-G1 cells were also treated with IFNα 50 as a control of replication inhibition (−90%, vs non-treated cells). As expected, quercetin significantly decreased HCV replication at all tested concentrations in a dose-dependent manner (Q0.1: −49%, Q0.5: −52%, Q1: −57%, Q5: −61%; Figure 2a) compared with untreated cells. When combined with IFNα 0.5, a higher antiviral effect was observed (Q0.1: −55%, Q0.5: −62%, Q1: −68%, Q5: −72%, vs HCV-G1 vehicle-treated cells), surpassing the HCV inhibitory effect of IFNα 0.5 alone (Q0.5: −24%, Q1: −34%, Q5: −43%, vs HCV-G1 IFN 0.5 cells) (Figure 2a). As shown in Supplementary Figure 1, this antiviral activity is not associated with cytotoxicity and cell proliferation impairment. Similarly, quercetin was able to dose dependently reduce HCV NS5A and core proteins in replicon-containing cells (NS5A, Q0.1: −24%, Q0.5: −27%, Q1: −54%, Q5: −67%; core, Q0.5: −32%, Q1: −50%, Q5: −62%; Figure 2b) compared with untreated cells, showing a marked inhibitory effect when combined with IFNα 0.5 (NS5A, Q0.1: −62%, Q0.5: −67%, Q1: −71%, Q5: −78%; core, Q0.1: −51%, Q0.5: −53%, Q1: −63%, Q5: −70%, vs HCV-G1 vehicle-treated cells). Moreover, combined treatment inhibited HCV replication more than IFN alone (NS5A, Q0.5: −21%, Q1: −32%, Q5: −48%; core, Q0.1: −16%, Q0.5: −19%, Q1: −37%, Q5: −49%, vs HCV-G1 IFN 0.5 cells) (Figure 2b).

Effect of quercetin treatment and its combination with IFNα on viral replication and HCV NS5A and core protein levels. HCV-G1 cells were incubated with quercetin alone or in combination with IFNα-2b (0.5 U/ml) at indicated concentrations for 48 h. (a) Samples of 1 μg RNA were analyzed by real-time RT-qPCR using specific primers to determine HCV RNA levels. Histone H3 mRNA levels were used for sample normalization. HCV-G1 cells were treated with 50 U/ml IFNα-2b as a control of replication inhibition. Results are expressed as HCV RNA copies/μg total RNA. (b) NS5A and core protein levels were analyzed by western blotting. Densitometry analysis of specific bands expressed as the percentage relative to HCV-G1 cells (100%). β-Actin levels were used as a loading control. Molecular weight markers (kDa) are indicated on the right. Photographs are representative of eight independent experiments. Data are described as the mean values±s.d. of eight independent experiments (*P<0.05;**P<0.01; ***P<0.001 vs HCV-G1 vehicle-treated cells, ##P<0.01 vs HCV-G1 IFN 0.5).
Quercetin Inhibits Oxidative and Nitrosative Stress in HCV-G1 Cells
To address the contribution of the antioxidant capacity of quercetin to the inhibition of HCV replication, we investigated the generation of ROS/RNS and lipid peroxidation in replicon-containing cells by flow cytometry. HCV-G1 cells were incubated with quercetin (0.1–5 μM) or just DMSO for 48 h. As shown in Figure 3, viral replication induced oxidative and nitrosative stress, as indicated by the significantly increased generation of different ROS/RNS (DCF: +55%, ETH: +56%) and lipoperoxidation (+350%) compared with Huh7 cells. Analysis of histograms in which the fluorescence was plotted against the relative cells number (Figure 3a and b, top) and quantification of the corresponding fluorescence intensity (Figures 3a–c) indicated that quercetin significantly abrogated the HCV-induced ROS/RNS overproduction (DCF: Q0.5: −13%, Q1: −22%, Q5: −28%; ETH: Q0.1: −29%, Q0.5: −32%, Q1: −34%, Q5: −35%, vs HCV-G1 vehicle-treated cells) and partially reduced the oxidation of lipids increased in HCV-G1 cells (Q0.5: −25%, Q1: −27%, Q5: −30%). Similarly, IFNα 50 significantly decreased ROS/RNS generation (DCF: −30%; ETH: −22%) and lipid peroxidation (−17%) in HCV-replicating cells (Figures 3a–c). These results suggest that the modulation of oxidative and nitrosative stress by quercetin may also have an important role in its anti-HCV activity.

Quercetin inhibits ROS and RNS production and lipid peroxidation induced by viral replication. HCV-G1 cells were incubated with quercetin at indicated concentrations for 48 h, and then ROS and RNS and lipid peroxidation were determined by flow cytometry as described. (a) Representative histograms of DCF fluorescence. The fluorescence is plotted against the number of cells. Results are expressed as DCF relative fluorescence intensity and normalized to HCV-G1 cells (100%). (b) Representative histograms of ETH fluorescence. The fluorescence is plotted against the number of cells. Results are expressed as ETH relative fluorescence intensity and normalized to HCV-G1 cells (100%). (c) Lipid peroxidation expressed as Bodipy 581/591 C11 fluorescence intensity as the percentage relative to HCV-G1 cells (100%). Data are described as the mean values±s.d. of six separate experiments (*P<0.05; **P<0.01; ***P<0.001 vs Huh7, #P<0.05; ##P<0.01 vs HCV-G1 vehicle-treated cells).
Quercetin Decreases HCV-Induced Intracytoplasmatic Lipid Accumulation
To establish the contribution of lipid metabolism modulation to the inhibition of HCV replication by quercetin, we examined the effect of this flavonol on intracytoplasmatic lipid accumulation in HCV-G1 cells. Results of flow-cytometric analysis of Bodipy 493/503-stained HCV-G1 cells are presented in Figure 4a. As shown, lipid accumulation increased significantly in replicon-containing cells (+257%) compared with Huh7 cells. In addition, quercetin treatment significantly decreased HCV-induced intracellular lipid accumulation in a dose-dependent manner (Q0.5: −17.5%, Q1: −22%, Q5: −55%, vs HCV-G1 vehicle-treated cells). Representative fluorescence images of Bodipy 493/503-stained cells, corroborating the results obtained by flow cytometry, are exhibited in Figure 4c. We next analyzed the intracellular TG and FFA accumulation and, as expected, TG and FFA contents were significantly enhanced in replicating cells (+150 and +81%, respectively) compared with Huh7 cells (Figure 4b). Further, HCV-induced TG and FFA concentrations were significantly reduced by quercetin (TG: Q0.5: −31%, Q1: −40%, Q5: −49%; FFA: Q0.5: −23%, Q1: −24%, Q5: −26%, vs HCV-G1 vehicle-treated cells). Finally, IFNα 50 was also able to decrease intracellular HCV-mediated lipid accumulation (−20%) by reducing TG content and FFA concentration (−44 and −36%, respectively) (Figures 4a–c).

HCV-induced intracytoplasmatic lipid accumulation is decreased by quercetin treatment. HCV-G1 cells were incubated with quercetin at indicated concentrations for 48 h, and then intracellular lipid accumulation was determined as described. (a) Representative histograms of Bodipy 493/503 fluorescence. Results are expressed as Bodipy relative fluorescence intensity and normalized to HCV-G1 cells (100%). (b) Triglyceride and free fatty acid assays were performed on cell lysates as indicated in Materials and Methods. Triglyceride and free fatty acid were normalized by protein content. Data are described as the mean values±s.d. of six experiments (*P<0.05; **P<0.01; ***P<0.001 vs Huh7, #P<0.05; ##P<0.01; ###P<0.001 vs HCV-G1 vehicle-treated cells). (c) Representative fluorescent images of Bodipy 493/503-treated cells (green). Nuclei were stained with DAPI (blue). The merged image with green and blue fluorescence is also shown. Photographs shown are typical results of six independent experiments.
Quercetin Downregulates Lipogenic Gene Overexpression Associated with HCV Replication
There is experimental evidence that LXRα induces the expression of lipogenic genes involved in fatty acid synthesis.32 Thus, we investigated the role of the LXRα-dependent pathway in quercetin-mediated inhibition of intracytoplasmatic lipid accumulation. As shown in Figure 5, we observed a significant induction of LXRα gene expression in replicon-containing cells (mRNA, +82%, protein, +55%) compared with Huh7 cells. Regarding gene expression of LXRα-related lipogenic genes, as expected, we also found a significant increase in fatty acid synthase (FAS) mRNA levels in HCV-replicating cells (+94%, vs Huh7) (Figure 5b). Interestingly, quercetin was able to significantly decrease HCV-induced LXRα gene expression at all experimental concentrations (mRNA, Q0.1: −32%, Q0.5: −38%, Q1: −39%, Q5: −40%; protein, Q0.1: −16%, Q0.5: −19%, Q1: −35%, Q5: −34%) (Figures 5a and b). Similarly, mRNA levels of FAS were significantly decreased by quercetin treatment (Q0.1: −16%, Q0.5: −14%, Q1: −17%, Q5: −20%) compared with HCV-G1 vehicle-treated cells (Figure 5b). Noteworthy, IFNα also decreased LXRα induction (IFNα 0.5: mRNA, −25%, protein, −31%; IFNα 50: mRNA, −45%, protein, −58%; Figures 5a and b) and subsequent FAS overexpression (IFNα 0.5: −30%; IFNα 50: −68%) in HCV-G1 cells (Figure 5b).

Quercetin inhibits HCV-induced lipogenic gene overexpression. HCV-G1 cells were incubated with quercetin at indicated concentrations for 48 h, and then LXRα and FAS gene expression was determined as indicated. (a) LXRα protein levels were analyzed by western blotting. Densitometry analysis of specific bands expressed as the percentage relative to HCV-G1 cells (100%). β-Actin levels were used as a loading control. Molecular weight markers (kDa) are indicated on the right. Photographs are representative of six experiments. (b) Bar graphs show LXRα (upper panel) and FAS (bottom panel) mRNA levels determined by real-time RT-qPCR as indicated. Data are described as the mean values±s.d. of six independent experiments (*P<0.05; **P<0.01; ***P<0.001 vs Huh7, #P<0.05; ##P<0.01; ###P<0.001 vs HCV-G1 vehicle-treated cells).
Effect of Quercetin on LXRα Overexpression Induced by Adenoviral Infection
To corroborate the effect of quercetin on LXRα gene expression, human hepatoma Huh7 cells were infected with adenoviral vector encoding LXRα transcription factor. We first examined the effect of adenoviral infection on LXRα gene expression. As exhibited in Supplementary Figure 2, RT-qPCR, immunofluorescence microscopy and western blot assays confirmed the overexpression of the nuclear receptor LXRα in Huh7 cells infected with the adenoviral vector (protein, Huh7 Ad-0.5: +257%, vs Huh7 Ad-CONT). We next analyzed the effect of quercetin treatment on LXRα mRNA levels and intracytoplasmatic lipid accumulation in Huh7 Ad-0.5 cells (Figures 6a and b, respectively). We observed that quercetin was able to dose dependently reduce LXRα overexpression caused by adenoviral infection at all tested concentrations (Q0.1: −27%, Q0.5: −39%, Q1: −46%, Q5: −58%, vs Huh7 Ad-0.5 vehicle-treated cells; Figure 6a), while the overexpression of the nuclear receptor LXRα was not affected by IFNα (Figure 6a). Similarly, quercetin also reduced intracellular lipid accumulation at all concentrations, as depicted in representative fluorescence images of Bodipy 493/503-stained cells (Figure 6b). Together, our results clearly indicate that quercetin exerts an inhibitory effect on LXRα expression and lipid accumulation in our in vitro model of LXRα overexpression as well as in HCV-replicating Huh7 cells. In view of these findings, we suggest that LXRα inhibition by quercetin contributes to the modulation of HCV-induced lipogenesis.

Effect of quercetin treatment on LXRα overexpression induced by adenoviral infection of human hepatoma Huh7 cells. Cells were infected with 0.5 μl of the recombinant adenovirus encoding LXRα (Huh7 Ad-0.5) or with an insertless adenoviral vector (Huh7 Ad-CONT). LXRα ligand was added at 24 h post infection and at 48 h post infection Huh7 Ad-0.5 cells were treated with 0.1–5 μM of quercetin or IFNα-2b (0.5 and 50 U/ml). (a) LXRα mRNA levels were analyzed by RT-qPCR as described. Data are described as the mean values±s.d. of eight separate experiments (***P<0.001 vs Huh7 Ad-CONT, #P<0.05, ##P<0.01 vs Huh7 Ad-0.5 vehicle-treated cells). (b) Intracellular lipids accumulation was determined by fluorescence microscopy using Bodipy 493/503 as described (green). Nuclei were stained with DAPI (blue). Photographs shown are typical results of six independent experiments.
PI3K/AKT Pathway is Involved in the Inhibitory Effect of Quercetin on LXRα-Mediated Lipogenesis in HCV-G1 Cells
To investigate the role of the PI3K/AKT pathway in LXRα-dependent lipogenesis modulation by quercetin, we first studied the effect of quercetin on AKT activation. As shown in Figure 7a, HCV replication induced a significant increase in AKT phosphorylation (+117%) compared with Huh7 cells. Furthermore, quercetin treatment significantly inhibited AKT activation at all experimental concentrations in a dose-dependent manner (Q0.1:−27%, Q0.5: −30%, Q1: −35%, Q5: −43%, vs HCV-G1 vehicle-treated cells). However, IFNα 50 slightly induced AKT phosphorylation (+21%) compared with HCV-G1 cells (Figure 7a). To corroborate that quercetin inhibits HCV-mediated lipogenesis through the PI3K/AKT pathway, we next studied the effects of the PI3K-inhibitor LY294002 alone or combined with quercetin on LXRα gene expression, lipid accumulation and HCV replication efficiency. First, we confirmed that cell viability was not affected by the different treatments (Supplementary Figure 1). As shown in Figures 7b–e, LY294002 treatment caused a significant reduction in HCV replication, LXRα mRNA levels and lipid accumulation (−31, −48, and −66%, respectively, vs HCV-G1 vehicle-treated cells). Moreover, the antiviral effect was higher when LY294002 and quercetin were combined (LY Q0.1: −53%, LY Q0.5: −47%, LY Q1: −52%, LY Q5: −49%, vs HCV-G1 vehicle-treated cells; Figure 7b). Furthermore, additive anti-HCV activity was associated with a greater reduction in LXRα gene expression (LY Q0.1: −55%, LY Q0.5: −59%, LY Q1: −66%, LY Q5: −66%, vs HCV-G1 vehicle-treated cells; Figure 7c) and lipid accumulation in HCV-G1 cells (LY Q0.1: −69%, LY Q0.5: −67%, LY Q1: −82%, LY Q5: −84%; Figures 7d and e). Overall, these results suggest that quercetin-mediated PI3K pathway inactivation may contribute to LXRα-dependent lipogenesis modulation and inhibition of viral replication by the flavonol.

Additive inhibitory effect of quercetin and LY294002 on PI3K/AKT pathway-mediated LXRα-dependent lipogenesis induced by viral replication. (a) HCV-G1 cells were incubated with quercetin at indicated concentrations for 48 h, and then p-AKT (Ser 473) and AKT protein levels were analyzed by western blotting. Densitometry analysis of specific bands expressed as the percentage relative to HCV-G1 vehicle-treated cells (100%). β-Actin levels were used as a loading control. Molecular weight markers (kDa) are indicated on the right. Photographs are representative of six independent experiments. (b–e) HCV-G1 cells were incubated with LY294002 alone or in combination with quercetin or IFNα-2b as previously described in Materials and Methods. Then HCV replication, LXRα gene expression and intracytoplasmatic lipid accumulation were determined as described. (b) Samples of 1 μg RNA were analyzed by real-time RT-qPCR using specific primers to determine HCV RNA levels. Histone H3 mRNA levels were used for sample normalization. Results are expressed as HCV RNA copies/μg total RNA. (c) Bar graph shows LXRα mRNA levels determined by real-time RT-qPCR as described. (d) Intracellular lipid accumulation expressed as Bodipy 493/503 relative fluorescence intensity and normalized to HCV-G1 cells (100%). (e) Representative fluorescent images of Bodipy 493/503-treated cells. Nuclei were stained with DAPI (blue). Photographs shown are typical results of six experiments. Data are described as the mean values±s.d. of six independent experiments (*P<0.05; **P<0.01; ***P<0.001 vs Huh7, ##P<0.01; ###P<0.001 vs HCV-G1 vehicle-treated cells, ††P<0.01; †††P<0.001 vs HCV-G1 LY).
DISCUSSION
The current Food and Drug administration (FDA)-approved standard of care for chronic HCV is pegylated IFNα combined with ribavirin. However, treatment is ineffective in many HCV patients.33 There is experimental evidence that the natural antioxidant flavonoids show therapeutic potential in the treatment of hepatitis C through inhibition of HCV replication.21, 22, 23 The present study suggests for the first time that quercetin might exert an indirect inhibitory effect on HCV replication via oxidative/nitrosative stress blockage and subsequent modulation of PI3K-LXRα-mediated lipogenesis associated with steatosis development and hepatitis C progression.
Our preliminary results indicated that all of the flavonoids studied were able to reduce HCV replication efficiency at very low concentrations. However, quercetin appears to be the most effective modulator of HCV replication capacity in replicon-containing cells. Furthermore, it has been previously indicated that quercetin also shows antiviral activity and decreases HCV particle production in cell culture,21, 22 whereby we focused our study on the molecular mechanisms involved in HCV replication regulation by this flavonol.
In the present research, a dose-dependent inhibitory effect on HCV replication was obtained with quercetin at all tested concentrations. NS5A protein expression, which is essential for HCV replication, was also reduced by quercetin treatment, as previously described.21 Furthermore, the combination of quercetin and IFNα exerted profound inhibitory effects on NS5A protein levels and HCV replication, as shown when curcumin and IFNα were combined.34 Similarly, our results indicate for the first time an inhibitory effect of quercetin on HCV core expression, showing an additive effect when combined with IFNα. The major pathway for the generation of the antiviral response mediated by IFNα involves a combination of different janus kinase (JAK) and signal transducer and activator of transcription (STAT) proteins to lead the transcription of IFN-stimulated genes.35, 36 In this regard, it has been described that quercetin may increase the antiviral gene expression regulated by the IFN-activated JAK-STAT pathway.37 Nevertheless, the molecular mechanisms involved in quercetin-mediated impairment of HCV replication seem to be more complex. Thereby, quercetin may also inhibit the NS5A-driven augmentation of internal ribosomal entry site (IRES)-mediated translation21, 38 and reduce viral production by inhibiting both NS3 and heat-shock proteins essential for HCV replication.22 In addition to these molecular mechanisms, we investigated the contribution of oxidative/nitrosative stress and lipid metabolism modulation to the inhibition of HCV replication by quercetin.
As previously indicated, a strong relationship between oxidative/nitrosative stress and HCV infection has been reported.4, 5, 6, 7 In the present research, quercetin was able to significantly inhibit HCV-induced ROS and RNS formation in HCV-replicating cells through its antioxidant activity. It has been also described that HCV replication induces lipid peroxidation that in turn reduces the amount of HCV RNA.30 In our HCV replicon system, quercetin was able to decrease HCV-induced lipoperoxidation, but only slightly, thus maintaining its ability to inhibit HCV replication. IFNα treatments also reduced HCV-induced ROS/RNS generation and lipid peroxidation as previously indicated in chronic hepatitis C patients.39, 40 The effects of IFNα on redox status seem to be directly mediated by reducing HCV replication.41
Liver steatosis is one of the most important histopathological features in patients with chronic hepatitis C. Both viral and host factors contribute to the development of steatosis, and putative defects caused by ROS/RNS may be involved through abnormalities in lipid metabolism.7, 9, 11 In addition, several reports suggest that lipid biosynthesis affects HCV replication.14, 42, 43 Furthermore, some dietary flavonoids appear to regulate lipogenic genes and fatty acid synthesis in in vivo and in vitro models.44, 45, 46 Thus, it has been previously described that quercetin decreased de novo fatty acid and triacylglycerol synthesis in rat hepatocytes.31 In our study, quercetin was able to dose dependently decrease induced lipid accumulation in replicon-containing cells, mainly by reducing TG content, at least in part by inhibiting core expression, which interferes with TG turnover.47 However, there is increasing evidence on the central role of non-TG lipotoxicity in the pathogenesis of steatosis. In fact, the accumulation of TG in the form of lipid droplets within the liver may be protective whereas toxic metabolites derived from FFAs could lead to steatosis.48 In this regard, in HCV-replicating cells quercetin reduced FFA concentration, responsible for the lipotoxic effects associated with steatosis. IFNα 50 was also able to decrease total intracytoplasmatic lipid and TG accumulation, as described in Huh7 cells expressing HCV genes.49 Similarly, FFA concentration was reduced by IFNα, as previously observed in serum of HCV-infected patients.50
It has been previously described that LXRα induces the expression of lipogenic genes involved in fatty acid synthesis, including its target FAS.32 Recently, we reported that HCV replication induces LXRα-mediated intracellular lipid accumulation which in turn contributes to the efficient replication of HCV.15 In the current study, quercetin was able to significantly decrease HCV-mediated LXRα induction and subsequent FAS overexpression at all experimental concentrations. While it has been previously described that several flavonoids reduced LXRα in non-hepatic cells,51, 52, 53 our results indicate for the first time a role for quercetin in LXRα gene expression and lipid accumulation modulation in LXRα-overexpressing and replicon-containing Huh7 cells. Interestingly, IFNα also reversed LXRα-mediated FAS induction observed in HCV-replicating cells. A similar decrease in FAS expression by IFNα has been previously indicated in in vitro models of HCV.49 However, in the present study IFNα was not able to repress the overexpression of the nuclear receptor LXRα in Huh7 cells infected with the adenoviral vector, suggesting that the effect of IFNα on lipogenesis modulation can be indirectly mediated by its anti-HCV activity.
We have recently shown that LY294002-mediated PI3K pathway inhibition attenuates LXRα upregulation induced by HCV expression,15 which represents a specific mechanism through which HCV infection alters the cellular lipid profile and causes steatosis. In the current research, HCV-induced PI3K/AKT activation was dose dependently inhibited by quercetin, suggesting a role for PI3K signaling pathway activity in quercetin-mediated LXRα modulation. In this respect, LY294002, which is in fact a derivative of quercetin,54 exerted a similar inhibitory effect on LXRα overexpression and lipid accumulation-mediated HCV replication capacity, showing an additive effect when combined with quercetin. Therefore, reduction in PI3K/AKT-LXRα-mediated hepatic fatty acid synthesis may represent an important mechanism underlying the HCV replication modulatory effect of quercetin. Similarly, PI3K/AKT-sterol regulatory element binding protein (SREBP)-1c suppression by curcumin also inhibits HCV replication in an in vitro model.34 In our in vitro model of LXRα overexpression, quercetin could exert its inhibitory effect on LXRα expression and lipid accumulation not only through modulation of PI3K pathway but also the expression of a large number of miRNAs involved in cell signaling and metabolism, which could in turn cause downregulation of CMV-expressed LXRα.55 However, further investigations are required to study this issue.
It has been described that quercetin bound to PI3K results in the inhibition of PI3K activity.56 Otherwise, it has been suggested that HCV gene expression induces activation of PI3K/AKT via oxidative stress and calcium signaling.26 As previously indicated, HCV proteins induce oxidative stress-mediated Ca2+ homeostasis alterations,6 which might underlie the effects of HCV expression on the PI3K/AKT pathway. In fact, it has been shown that quercetin suppressed the endoplasmic reticulum stress caused by Ca2+ dynamics dysregulation by the inhibition of PI3K.57 Thereby, quercetin can also modulate the PI3K/AKT pathway by reducing HCV-related ROS and RNS generation.
The PI3K pathways have emerged as a critical additional component of IFN-induced signaling.58 Thus, the IFN-induced PI3K/AKT pathway is the essential signal for cell survival and thought to be one mechanism for chronic HCV infection.58, 59 In this study, IFNα treatment is able to interfere with the PI3K/AKT pathway as shown by a dose-dependent induction of Ser 473 phosphorylation of AKT. However, the treatment with PI3K inhibitors did not influence IFN-induced anti-HCV replication, indicating that its anti-HCV activity is not PI3K/AKT dependent, as previously reported.60
In conclusion, our results suggest that quercetin might exert its inhibitory effect on HCV replication at least in part through oxidative/nitrosative stress inhibition and subsequent lipid metabolism modulation, by reducing PI3K/AKT-mediated LXRα overexpression and lipid accumulation associated with steatosis development and hepatitis C progression. Thus, the importance of therapeutic approaches focusing on oxidative/nitrosative stress and associated steatosis in HCV patients is expected to increase in the near future.
References
Reed KE, Rice CM . Overview of hepatitis C virus genome structure, polyprotein processing, and protein properties. Curr Top Microbiol Immunol 2000;242:55–84.
Huang Y, Staschke K, De Francesco R et al. Phosphorylation of hepatitis C virus NS5A nonstructural protein: a new paradigm for phosphorylation-dependent viral RNA replication? Virology 2007;20:1–9.
Sheikh M, Choi J, Qadri I et al. Hepatitis C virus infection: molecular pathways to metabolic syndrome. Hepatology 2008;47:2127–2133.
García-Mediavilla MV, Sánchez-Campos S, González-Pérez P et al. Differential contribution of hepatitis C virus NS5A and core proteins to the induction of oxidative and nitrosative stress in human hepatocyte-derived cells. J Hepatol 2005;43:606–613.
Choi J, Ou JH . Mechanisms of liver injury. III. Oxidative stress in the pathogenesis of hepatitis C virus. Am J Gastroenterol Liver Physiol 2006;290:G847–G851.
Dionisio N, García-Mediavilla MV, Sánchez-Campos S et al. Hepatitis C virus NS5A and core proteins induce oxidative stress-mediated calcium signalling alterations in hepatocytes. J Hepatol 2009;50:872–882.
González-Gallego J, García-Mediavilla MV, Sánchez-Campos S . Hepatitis C virus, oxidative stress and steatosis: current status and perspectives. Curr Mol Med 2011;11:373–390.
Lonardo A, Loria P, Adinolfi LE et al. Hepatitis C and steatosis: a reappraisal. J Viral Hepat 2006;13:73–80.
Negro F, Sanyal AJ . Hepatitis C virus, steatosis and lipid abnormalities: clinical and pathogenic data. Liver Int 2009;29:26–37.
Negro F . Hepatitis C virus-induced steatosis: an overview. Dig Dis 2010;28:294–299.
Vidali M, Tripodi MF, Ivaldi A et al. Interplay between oxidative stress and hepatic steatosis in the progression of chronic hepatitis C. J Hepatol 2008;48:399–406.
Yamaguchi A, Tazuma S, Nishioka T et al. Hepatitis C virus core protein modulates fatty acid metabolism and thereby causes lipid accumulation in the liver. Dig Dis Sci 2005;50:1361–1371.
Kim K, Kim KH, Ha E et al. Hepatitis C virus NS5A protein increases hepatic lipid accumulation via induction of activation and expression of PPARgamma. FEBS Lett 2009;583:2720–2726.
Kapadia SB, Chisari FV . Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc Natl Acad Sci USA 2005;102:2516–2566.
García-Mediavilla MV, Pisonero-Vaquero S, Lima-Cabello E et al. Liver X receptor α-mediated regulation of lipogenesis by core and NS5A proteins contributes to HCV-induced liver steatosis and HCV replication. Lab Invest 2012;9:1191–1202.
Jagtap S, Meganathan K, Wagh V et al. Chemoprotective mechanism of the natural compounds, epigallocatechin-3-O-gallate, quercetin and curcumin against cancer and cardiovascular diseases. Curr Med Chem 2009;16:1451–1462.
Tuñón MJ, García-Mediavilla MV, Sánchez-Campos S et al. Potential of flavonoids as anti-inflammatory agents: modulation of inflammatory gene expression and signal transduction pathways. Curr Drug Metab 2009;10:256–271.
González-Gallego J, García-Mediavilla MV, Sánchez-Campos S et al. Fruit polyphenols, immunity and inflammation. Br J Nutr 2012;104:S15–S27.
Polyak SJ, Morishima C, Hawke R . Antiviral effects of silymarin against hepatitis C: the jury is still out. Hepatology 2008;48:345–346.
Rutter K, Scherzer TM, Beinhardt S et al. Intravenous silibinin as “rescue treatment” for on-treatment non-responders to pegylated interferon/ribavirin combination therapy. Antivir Ther 2011;16:1327–1333.
Gonzalez O, Fontanes V, Raychaudhuri S et al. The heat shock protein inhibitor quercetin attenuates hepatitis C virus production. Hepatology 2009;50:1756–1764.
Bachmetov L, Gal-Tanamy M, Shapira A et al. Suppression of hepatitis C virus by the flavonoid quercetin is mediated by inhibition of NS3 protease activity. J Viral Hepat 2012;19:e81–e88.
Wagoner J, Negash A, Kane OJ et al. Multiple effects of silymarin on the hepatitis C virus lifecycle. Hepatology 2010;51:1912–1921.
Crespo I, García-Mediavilla MV, Almar M et al. Differential effects of dietary flavonoids on reactive oxygen and nitrogen species generation and antioxidant enzymes in Chang Liver cells. Food Chem Toxicol 2008;46:1555–1569.
Benedicto I, Molina-Jiménez F, Barreiro O et al. Hepatitis C virus envelope components alter localization of hepatocyte tight junction associated proteins and promote occludin retention in the endoplasmic reticulum. Hepatology 2008;48:1044–1053.
Waris G, Felmlee DJ, Negro F et al. Hepatitis C virus induces proteolytic cleavage of sterol regulatory element binding proteins and stimulates their phosphorylation via oxidative stress. J Virol 2007;81:8122–8130.
Crespo I, García-Mediavilla MV, Gutiérrez B et al. A comparison of the effects of quercetin and kaempferol on cytokine-induced proinflammatory status of cultured human endothelial cells. Br J Nutr 2008;100:968–976.
Miquilena-Colina ME, Lima-Cabello E, Sánchez-Campos S et al. Hepatic fatty acid translocase CD36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non-alcoholic steatohepatitis and chronic hepatitis C. Gut 2011;60:1394–1402.
Gómez-Foix AM, Coats WS, Baqué S et al. Adenovirus mediated transfer of the muscle glycogen phosphorylase gene into hepatocytes confers altered regulation of glycogen metabolism. J Biol Chem 1992;267:25129–25134.
Huang H, Chen Y, Ye J . Inhibition of hepatitis C virus replication by peroxidation of arachidonate and restoration by vitamin E. Proc Natl Acad Sci USA 2007;107:18666–18670.
Gnoni GV, Paglialonga G, Siculella L . Quercetin inhibits fatty acid and triacylglycerol synthesis in rat-liver cells. Eur J Clin Invest 2009;39:761–768.
Yoshikawa T, Shimano H, Amemiya-Kudo M et al. Identification of liver X receptor-retinoid X receptor as an activator of the sterol regulatory element-binding protein 1c gene promoter. Mol Cell Biol 2001;21:2991–3000.
Tsubota A, Fujise K, Namiki Y et al. Peginterferon and ribavirin treatment for hepatitis C virus infection. World J Gastroenterol 2011;17:419–432.
Kim K, Kim KH, Kim HY et al. Curcumin inhibits hepatitis C virus replication via suppressing the Akt-SREBP-1 pathway. FEBS Lett 2010;584:707–712.
Feld JJ, Hoofnagle JH . Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature 2005;436:967–972.
Schindler C, Levy DE, Decker T . JAK–STAT signaling: from interferons to cytokines. J Biol Chem 2007;282:20059–20063.
Tai ZF, Zhang GL, Wang F . Identification of small molecule activators of the janus kinase/signal transducer and activator of transcription pathway using a cell-based screen. Biol Pharm Bull 2012;35:65–71.
Khachatoorian R, Arumugaswami V, Ruchala P et al. A cell-permeable hairpin peptide inhibits hepatitis C viral non-structural protein 5A-mediated translation and virus production. Hepatology 2012;55:1662–1672.
Higueras V, Raya A, Rodrigo JM et al. Interferon decreases serum lipid peroxidation products of hepatitis C patients. Free Radic Biol Med 1994;16:131–133.
Romero MJ, Bosch-Morell F, Romero B et al. Serum malondialdehyde: posible use for the clinical management of chronic hepatitis C patients. Free Radic Biol Med 1998;25:993–997.
Ando M, Korenaga M, Hino K et al. Mitochondrial electron transport inhibition in full genomic hepatitis C virus replicon cells is restored by reducing viral replication. Liver Int 2008;28:1158–1166.
Yang W, Hood BL, Chadwick SL et al. Fatty acid synthase is up-regulated during hepatitis C virus infection and regulates hepatitis C virus entry and production. Hepatology 2008;48:1396–1403.
Syed GH, Siddiqui A . Effects of hypolipidemic agent nordihydroguaiaretic acid on lipid droplets and hepatitis C virus. Hepatology 2011;54:1936–1946.
Brusselmans K, Vrolix R, Verhoeven G et al. Induction of cancer cell apoptosis by flavonoids is associated with their ability to inhibit fatty acid synthase activity. J Biol Chem 2005;280:5636–5645.
Odbayar TO, Badamhand D, Kimura T et al. Comparative studies of some phenolic compounds (quercetin, rutin, and ferulic acid) affecting hepatic fatty acid synthesis in mice. J Agric Food Chem 2006;54:8261–8265.
Jung CH, Cho I, Ahn J et al. Quercetin reduces high-fat diet-induced fat accumulation in the liver by regulating liver metabolism genes. Phytother Res 2013;27:139–143.
Harris C, Herker E, Farese RV et al. Hepatitis C virus core protein decreases lipid droplet turnover: a mechanism for core-induced steatosis. J Biol Chem 2011;49:42615–42625.
Neuschwander-Tetri BA . Nontriglyceride hepatic lipotoxicity: the new paradigm for the pathogenesis of NASH. Curr Gastroenterol Rep 2010;12:49–56.
Toyoda M, Kitaoka A, Machida K et al. Association between lipid accumulation and the cannabinoid system in Huh7 cells expressing HCV genes. Int J Mol Med 2011;27:619–624.
Konrad T, Zeuzem S, Vicini P et al. Evaluation of factors controlling glucose tolerance in patients with HCV infection before and after 4 months therapy with interferon-alpha. Eur J Clin Invest 2000;30:111–121.
Kaul D, Sikand K, Shukla AR . Effect of green tea polyphenols on the genes with atherosclerotic potential. Phytother Res 2004;18:177–179.
Moon HS, Chung CS, Lee HG et al. Inhibitory effect of (-)-epigallocatechin-3-gallate on lipid accumulation of 3T3-L1 cells. Obesity 2007;15:2571–2582.
Lee J, Jung E, Lee J et al. Isorhamnetin represses adipogenesis in 3R3-L1 cells. Obesity 2009;17:226–232.
Imai Y, Yoshimori M, Fukuda K et al. The PI3K/Akt inhibitor LY294002 reverses BCRP-mediated drug resistance without affecting BCRP translocation. Oncol Rep 2012;27:1703–1709.
Milenkovic D, Deval C, Gouranton E et al. Modulation of miRNA expression by dietary polyphenols in apoE deficient mice: a new mechanism of the action of polyphenols. PLoS ONE 2012;7:e29837.
Hwang MK, Song NR, Kang NJ et al. Activation of phosphatidylinositol 3-kinase is required for tumor necrosis factor-alpha-induced upregulation of matrix metalloproteinase-9: its direct inhibition by quercetin. Int J Biochem Cell Biol 2009;41:1592–1600.
Natsume Y, Ito S, Satsu H et al. Protective effect of quercetin on ER stress caused by calcium dynamics dysregulation in intestinal epithelial cells. Toxicology 2009;258:164–175.
Kaur S, Uddin S, Platanias LC . The PI3’ kinase pathway in interferon signaling. J Interferon Cytokine Res 2005;25:780–787.
Mannová P, Beretta L . Activation of the N-Ras-PI3K-Akt-mTOR pathway by hepatitis C virus: control of cell survival and viral replication. J Virol 2005;79:8742–8749.
Matsumoto A, Ichikawa T, Nakao K et al. Interferon-alpha-induced mTOR activation is an anti-hepatitis C virus signal via the phosphatidylinositol 3-kinase-Akt-independent pathway. J Gastroenterol 2009;44:856–863.
Acknowledgements
This work was supported by grants to Javier González-Gallego from Ministerio de Educación y Ciencia (BFU2010-15784), Francisco Jorquera from Junta de Castilla y León (GRS 482/A/10), Pedro L Majano from Ministerio de Ciencia e Innovación, Instituto de Salud Carlos III, FEDER (PI10/00101), and Ramiro Jover from Fondo de Investigación Sanitaria (FIS), Instituto de Salud Carlos III (PI10/00194). María V García-Mediavilla and Marta Benet were supported by CIBERehd contracts. CIBERehd is funded by the Instituto de Salud Carlos III, Spain. Sandra Pisonero-Vaquero was supported by the program ‘Formación del Profesorado Universitario’ (FPU, AP2009-4484) from the Ministry of Education (Spain).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
The authors demonstrate that the antioxidant flavonoid quercetin shows a marked anti- hepatitis C virus (HCV) activity when combined with interferon α. Quercetin decreases HCV-induced reactive oxygen and nitrogen species generation and lipoperoxidation, and inhibits liver X receptor (LXR)α-induced lipid accumulation through phosphatidylinositol 3-kinase/AKT pathway inactivation.
Supplementary information
Rights and permissions
About this article
Cite this article
Pisonero-Vaquero, S., García-Mediavilla, M., Jorquera, F. et al. Modulation of PI3K-LXRα-dependent lipogenesis mediated by oxidative/nitrosative stress contributes to inhibition of HCV replication by quercetin. Lab Invest 94, 262–274 (2014). https://doi.org/10.1038/labinvest.2013.156
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2013.156
Keywords
This article is cited by
-
Plant-derived antivirals against hepatitis c virus infection
Virology Journal (2018)
-
The role of PTEN - HCV core interaction in hepatitis C virus replication
Scientific Reports (2017)
-
Antioxidant, anti-inflammatory, and analgesic activities of Citrus reticulata Blanco leaves extracts: An in vivo and in vitro study
Phytothérapie (2017)
-
An extract from Taxodium distichum targets hemagglutinin- and neuraminidase-related activities of influenza virus in vitro
Scientific Reports (2016)
-
A comprehensive overview of hepatoprotective natural compounds: mechanism of action and clinical perspectives
Archives of Toxicology (2016)
