
Abstract
Lnk, an intracellular adapter protein, is expressed in hematopoietic cell lineages, which has recently been proved as an essential inhibitory signaling molecule for stem cell self-renewal in the stem cell factor-c-Kit signaling pathway with enhanced hematopoietic and osteogenic reconstitution in Lnk-deficient mice. Moreover, the therapeutic potential of hematopoietic stem/endothelial progenitor cells (EPCs) for fracture healing has been demonstrated with mechanistic insight into vasculogenesis/angiogenesis and osteogenesis enhancement in the fracture sites. We report here, Lnk siRNA-transfected endothelial commitment of c-kit+/Sca-1+/lineage− subpopulations of bone marrow cells have high EPC colony-forming capacity exhibiting endothelial markers, VE-Cad, VEGF and Ang-1. Lnk siRNA-transfected osteoblasts also show highly osteoblastic capacity. In vivo, locally transfected Lnk siRNA could successfully downregulate the expression of Lnk at the fracture site up to 1 week, and radiological and histological examination showed extremely accelerated fracture healing in Lnk siRNA-transfected mice. Moreover, Lnk siRNA-transfected mice exhibited sufficient therapeutic outcomes with intrinstic enhancement of angiogenesis and osteogenesis, specifically, the mice demonstrated better blood flow recovery in the sites of fracture. In our series of experiments, we clarified that a negatively regulated Lnk system contributed to a favorable circumstance for fracture healing by enhancing vasculogenesis/angiogenesis and osteogenesis. These findings suggest that downregulation of Lnk system may have the clinical potential for faster fracture healing, which contributes to the reduction of delayed unions or non-unions.
Similar content being viewed by others


Main
Lnk shares a pleckstin homology domain, a Src homology 2 domain and potential tyrosine phosphorylation sites with APS and SH-2B. It belongs to a family of adaptor proteins implicated in the integration and regulation of multiple signaling events1, 2, 3, 4, 5 and has also been suggested to act as a negative regulator in the stem cell factor (SCF)-c-Kit signaling pathway.6 Takaki et al7 reported that Lnk expressed in hematopoietic cell lineages, and bone marrow (BM) cells of Lnk-deficient mice are competitively superior in hematopoietic population to those of wild-type mice. Ema et al8 also clarified that not only hematopoietic stem cell/progenitor cell (HSC/HPC) numbers but also the self-renewal capacity of some HSCs/HPCs were markedly increased in Lnk-deficient mice. In addition, the functional domains of Lnk were identified, and dominant-negative Lnk mutant inhibits the functions of Lnk endogenously expressed in the HSCs/HPCs and thereby potentiates the HPCs for engraftment.9
In previous studies, we demonstrated that Lnk deletion reinforces the function of the BM progenitor, promoting repair of an injured spinal cord through the acceleration of angiogenesis and astrogenesis.10 Another report suggested that Lnk regulated BM endothelial progenitor cells (EPCs) kinetics in vascular regeneration. Lnk-deficient EPCs are more potent actors than resident cells in hindlimb perfusion recovery and ischemic neovasucularization, mainly via the activity of BM EPCs.11 Moreover, our group have recently demonstrated that a lack of Lnk signaling, dependent on SCF-cKit signaling pathway, enhanced a regenerative response via vasculogenesis and osteogenesis in fracture healing by HSC/EPC mobilization and recruitment to the fracture sites in Lnk-deficient mice.12
Although most fractures heal appropriately in a certain period with callus formation, which bridges the fracture gap while bone repair takes place, a large number of patients with fractures waste a lot of time due to disability or confinement, leading to a tremendous loss of productivity. Moreover, a significant proportion (5–10%) of fractures fails to heal and results in delayed union or persistent non-union causing serious problems for a patient’s quality of life. Therefore, establishment of a novel therapeutic strategy for non-union healing is clinically warranted.13, 14, 15, 16 Among various causes of failed bone formation and remodeling, inappropriate neoangiogenesis is considered to be a crucial factor.13, 16, 17, 18 The majority of tissues require appropriate vascularization and blood supply to support proper function. Without such vascularization, cells suffer from hypoxia, lack of nutrients, accumulation of waste products and disruption of biomechanical signaling pathways, affecting tissue homeostasis and preventing tissue regeneration.19 Notably, appropriate vasculogenesis, by BM EPCs,20, 21 is emerging as a prerequisite for bone development and regeneration, and indeed there appears to be a developmental reciprocity between endothelial cells (ECs) and osteoblasts (OBs).22 We have recently proved the pathophysiological role and contribution of murine BM-derived Sca1+Lineage− cells, HSC/EPC-enriched fraction, for bone healing.23
Therefore, the concept of enhanced angiogenesis/osteogenesis by HSCs/EPCs is emerging, and one of the novel factors responsible for stem/progenitor cell mobilization from BM, Lnk, seems to be attractive for developing a therapeutic strategy utilizing circulating EPCs for skeletal medicine. Aiming for the induction of neovascularization at the fracture sites, local downregulation of Lnk may provide a novel therapeutic strategy for modifying and improving the fracture healing process and preventing non-union.
In the present study, we examined the in vitro and in vivo properties of Lnk small interfering RNA (siRNA) and investigated the hypothesis that downregulation of Lnk signaling enhances a regenerative response via vasculogenesis/angiogenesis and osteogenesis in fracture healing.
MATERIALS AND METHODS
siRNA Preparation
The expression of Lnk was suppressed by RNA interference (RNAi) in mice c-kit+/Sca-1+/Lineage− (KSL) cells, OBs and the tissues of a peri-fracture site. siRNAs were purchased from Dharmacon (Thermo Scientific, CO, USA) and HAYASHI KASEI. (Osaka, Japan) in deprotected, desalted and annealed forms. The sense strand sequences of the RNA duplexes were as follows: for mLnk No. 1, 5′-CGAGUUACCUCUUUCCUUA-3′, and for mLnk No. 2, 5′-GAGGUCGUAUUGCGCUAUA-3′. We used both the No.1 and No.2 siRNA for in vitro study and the No.1 siRNA for in vivo study after confirming that both the No.1 and No.2 siRNAs regulated Lnk in an in vitro study. The siRNAs were delivered into mouse KSL cells and OBs by lipofection according to the manufacturer’s instructions. Twenty-four hours after the transfection, cells were used for further experiment.
Preparation of Mouse KSLs (mKSLs) for In-Vitro Study
To investigate the effect of Lnk siRNA on mouse ECs in vitro, we isolated the mouse endothelial commitment of KSL subpopulations of BM cells from 10- to 12-week-old mouse by a modification of a method reported previously.24 BM cells were obtained from C57BL/6 mice (wild-type mice), and lineage-positive cells were removed using a magnetic cell separation system (BD IMag Hematopoietic Progenitor Cell Enrichment Set-DM; BD Biosciences, San Jose, CA, USA, http://www.bdbiosciences.com). In brief, cells isolated by centrifugation with a low-density solution (<1.077 g/ml) were stained with a mixture of biotinylated mouse lineage antibodies to CD3e, CD11b, CD45R/B220, Ly-6G and Ly-6C (Gr-1) and TER-119/erythroid cells (Ly-76). The lineage-positive cells were depleted with streptavidin-magnetic beads and the use of neodymium magnets (BD IMagnet; BD Biosciences). The remaining cells were collected and further stained with phycoerythrin-conjugated anti-Sca-1 and allophycocyanin-conjugated anti c-Kit antibodies. All antibodies were purchased from BD Biosciences. After washing, the cells were resuspended in staining medium supplemented with 7-amino-actinomycin D (7-AAD). Stained cells were analyzed by fluorescence-activated cell sorting using a BD FACSAria cell-sorting system (BD Biosciences), and KSL cells were sorted. Dead cells stained with 7-AAD were excluded from analysis and sorting.
Preparation of Mouse Osteoblasts (mOBs) for In-Vitro Study
To investigate the effect of Lnk siRNA on mouse OBs in vitro, we isolated mouse calvarial OBs from 3–5-day-old C57BL/6 mouse calvarial bone using a modification of the method described previously.25, 26 Briefly, after removal of sutures, calvaliae were subjected to 15-min digestions in an enzyme mixture containing 0.05% trypsin (Gibco, Life Technologies, CA, USA) and 1.5 U/ml collagenase P (Boehringer Mannheim, Mannheim, Germany) at 37 °C on a rocking platform. The enzyme activity was then neutralized by the addition of media containing FBS. The isolated cells were plated at a density of 1.5 × 105/mm2 in 35-mm culture dish with 10% FBS/Dulbecco’s modified Eagles’ medium (DMEM). The cells were harvested and used for in vitro studies as mOBs after 2–3 weeks in culture.
Sodium Dodecyl Sulfate–Polyacrylamide Gel Electrophoresis (SDS-PAGE) and Western Blotting
Cell pellets and minced tissues were resuspended in 600 μl of RIPA lysis buffer with 1% sodium orthovanadate, 1% PMSF and 1% protease inhibitor cocktail. The tissues were homogenized by microtube pestle before protein lysis. The proteins were lysed for 10 min on ice. The supernatants were recovered by centrifugation at 15 000 g for 10 min to remove insoluble materials. The samples were mixed with 4 × SDS sample buffer (0.25 M Tris-HCl (pH 6.8), 40% glycerol, 8% sodium dodecyl sulfate and 16% 2-mercaptoethanol), boiled for 5 min and loaded onto 0.1% SDS-10% polyacrylamide gel and run for 1 h at 150 V. After electrophoresis, proteins were transferred to nitrocellulose membranes in a Bio-Rad Electroblotter Apparatus. Membranes were blocked in PBST (PBS, 0.1% Tween-20) containing 2% blocking reagent for 30 min at room temperature and then reacted overnight at 4 °C with each antibodies: goat anti-Lnk antibody (M-20, sc-7222, Santa Cruz biotechnology, Santa Cruz, CA, USA), rabbit anti-BMP-2 antibody (ab82511, Abcam, Cambridge, MA, USA), rabbit anti-VE Cadherin antibody (ab33168, Abcam) and rabbit anti-β-tubulin antibody (Sigma, St Louis, MO, USA) as an internal control diluted 1:10 000 in PBST-blocking reagent. After three washes in PBST, the membranes were treated with affinity-purified horseradish peroxidase-conjugated donkey anti-goat IgG (Promega, Madison, WI, USA) and HRP-conjugated goat anti-rabbit IgG (GE Healthcare UK, UK) diluted 1:100 000. Membranes were washed three times in PBST and then developed with ECL Advance Kit (GE Healthcare UK). The images were obtained by luminescent image analyzer LAS-3000 (FUJIFILM). The densities of the blots were quantified using an image analysis software, ImageJ version (NIH Image). All values were normalized relative to Tublin expressions.
Immunoprecipitation
The proteins from the cell or tissue homogenate were prepared as described in western blotting method. The supernatants were pre-incubated with Protein G-agarose beads (Sigma) at 4 °C for 30 min to avoid non-specific protein absorption against agarose beads. The pre-incubated supernatants were mixed with goat anti-Lnk antibody for 1 h at 4 °C. The immune complex was then captured on a solid support by incubation with Protein G-agarose beads for 1 h at 4 °C followed by centrifugation and washing the pellets five times with PBS. Immunoprecipitated proteins were dissolved into SDS sample buffer and further analyzed by SDS-PAGE and immunoblotting to determine their molecular weights and to determine the presence and quantity of proteins.
EPC Colony-Forming Assay of Lnk and Control siRNA-Transfected KSL Cells
EPC colony-forming assay established in our laboratory was performed as reported previously.27, 28 The number of EPC colonies was assessed 16 days after seeding of Lnk siRNA and control siRNA-transfected KSL cells (1 × 103cells/well) in a six-well plate with methyl cellulose-containing medium M3236 (Stem Cell Technologies, Vancouver, BC, Canada) supplemented with 20 ng/ml SCF (Kirin, Tokyo, Japan), 50 ng/ml VEGF (R&D Systems, Minneapolis, MN), 20 ng/ml interleukin-3 (Kirin) and 50 ng/ml basic fibroblast growth factor (Wako, Osaka, Japan). Two different types of attaching cell colonies made of small EPCs and large EPCs were counted separately.
Induction of Osteogenic Differentiation in vitro
To induce osteogenic differentiation, 100 000 Lnk siRNA and control siRNA-transfected OBs were plated in six-well plates and cultured for 3 weeks under specific osteogenic conditions in alpha-MEM supplemented with 10% FBS, 2 mM L-glutamine, 60 μM ascorbic acid (Sigma-Aldrich, St Louis, MO, USA), 10 mM beta-glycerophosphate (Sigma-Aldrich) and 0.1 μM dexamethasone (Sigma-Aldrich). Cells were also cultured with alpha-MEM supplemented with 10% FBS and 2 mM L-glutamine as a negative control.
Proliferation and Migration Assays for Cultured Mouse KSLs and OBs
The proliferation activity of mEPCs and mOBs in the presence of Lnk siRNA and control siRNA was examined using a Cell Counting Kit-8 (Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer’s instructions. Briefly, Lnk and control siRNA were transfected into mouse KSL cells and OBs by lipofection according to the manufacturer’s instructions. Twenty-four hours after transfection, the siRNA-transfected KSL cells or OBs were seeded onto 96-well culture plates at a density of 5 × 103 cells per well and cultured in EBM2 or DMEM with 10% FBS for 48 h at 37 °C in 20% O2/5% CO2. Optical density was measured using a plate reader at 490 nm wavelength.
Migration activity was evaluated with a modified Boyden’s chamber assay as described previously.29 Mouse KSLs or OBs (5 × 104 cells per well) were seeded onto upper chambers in 24-well Transwell culture plates (Corning Incorporated Life Science, MA, USA) and lower chambers were filled with DMEM with 10% FBS containing 50 ng/ml of SDF-1a (Minneapolis, MN, USA) followed by incubation for 6 h at 37 °C in 5% CO2. Migrated cells were stained with 4’,6-diamidino-2-phenylindole (DAPI) and counted under a fluorescent microscope. All groups were studied at least in triplicate.
Histochemical Analysis of Cell Cultures
Mineralization was assessed by alizarin red staining method. Briefly, the cultures were rinsed twice with PBS, fixed in 100% ethanol for 30 min and stained with 1% Alizarin Red S (Hartman Leddon, Philadelphia, PA, USA) in 0.28% ammonia water for 10 min at room temperature. The stained cell layers were washed twice with distilled water and air-dried for micro/macroscopic observation. For quantification, Alizarin Red dye was extracted from the stained cell layer with 5% formic acid solution, and the optical density was measured by a photometer (Microplate leader Model 680, Bio-Rad, Tokyo, Japan) at 415 nm wavelength.
Mice Animal Model of Bone Fracture
Male mice (C57BL/6, CLEA Japan, Tokyo, Japan) aged 10–12 weeks and weighing 25–27 g were used in this study. The mice were fed a standard maintenance diet and provided water ad libitum. All animal procedures were performed in accordance with the Japanese Physiological Society Guidelines for the Care and Use of Laboratory Animals, and the study protocol was approved by the Ethical Committee of the RIKEN Center for Developmental Biology.
All surgical procedures were performed under anesthesia and normal sterile conditions. Anesthesia was performed with ketamine hydrochloride (60 mg/kg) and xylazine hydrochloride (10 mg/kg) administered intraperitoneally. A reproducible bone fracture model was created in the femur of the mice, according to a modification of Manigrasso’s mode.30 A lateral parapatellar knee incision on the right limb was made to expose the distal femoral chondyle. A 2-mm wedge was made using a 27-gauge needle on the interchondyle of the femur and then a 0.5-mm diameter, stainless wire was inserted in a retrograde fashion. The wire was advanced until its proximal end was positioned stable to the greater trochanter, and the distal end was cut close to the articular surface of the knee. A transverse femoral shaft fracture was then created in the right femur of each mouse. The wound was then irrigated with 10 cc of sterile saline, and the skin was closed in layers with 5-0 nylone sutures. Unprotected weight bearing was allowed immediately post-operatively. The left non-fractured femur served as a control.
Local Transplantation of siRNA and the Experimental Groups
Immediately after creation of fracture, mice received local administration of the following materials mixed in 100 μl of AteloGene atelocollagen gel (KOKEN, Tokyo, Japan), which was used as a bio-absorbable scaffold to retain the siRNA at the transplantation site:31, 32 (1) 10 μM Lnk siRNA (Lnk group), and (2) 10 μM control scramble siRNA (control group). Twenty animals were assigned in each group for radiological assessment and every additional three or five animals in each group were assigned in each study. The contralateral intact femurs were used as controls for histological and functional analyses.
Tissue Harvesting
Mice were euthanized with an overdose of ketamine and xylazine for biomechanical and histological analyses at the indicated time course described below. The femurs were directly frozen for biomechanical analysis, embedded in OCT compound (Sakura Finetek Japan) for histological analysis and snap frozen in liquid nitrogen and stored at −80 °C for real-time RT-PCR analysis. Three mice were randomly selected and euthanized in each group at weeks 1, 2 and 3 after surgery. The remaining rats were killed at week 4. Bilateral femurs were harvested and quickly treated for the above mentioned analyses as described previously.33, 34
Quantitative Real-Time RT-PCR Analysis with KSL Cells and OBs in Peri-Fracture site
Total RNA was obtained from KSL cells and OBs using Rneasy Mini Kit (QIAGEN KK, Tokyo, Japan) according to the manufacturer’s protocol. Total RNA was obtained from granulation tissues and callus tissues at the peri-fracture site at day 7 after transplantation using Tri-zol (Life Technologies, CA, USA; Gaithersburg, MD) according to the manufacturer’s instructions. For quantitative real-time RT-PCR, after the first-strand cDNA was synthesized using a RNA LA PCR Kit Ver1.1 (TAKARA BIO, Shiga, Japan), the converted cDNA samples (2 μl) were amplified in triplicate by real-time PCR (ABI PRISM 7700, Applied Biosystems, Foster City, CA, USA) in a final volume of 20 μl using SYBR Green Master Mix reagent (Applied Biosystems) with gene-specific primers listed in Table 1. Melting curve analysis was performed with Dissociation Curves Software (Applied Biosystems). Results were obtained using sequence detection software (ABI PRISM 7700) and the mean cycle threshold (Ct) values were used to calculate gene expressions with normalization to human or rat glyceraldehydes-3-phosphate (GAPDH).
Morphometric Evaluation of Capillary Density and OB Density
To evaluate capillary density and OB density at the peri-fracture sites, immunohistochemistry was performed with the following anti-mouse rat CD31 (1:50 dilution, Santa Cruz Biotechnology) as a sensitive marker for mice ECs, or anti-mouse goat OC (1:250 dilution, Biogenesis) a marker for mice OBs. The secondary antibodies for each immunostaining are as follows: Alexa-Fluor 488-conjugated goat anti-rat IgG (1:1000, Invitrogen, CA, USA) for CD31 and Alexa-Fluor 594-conjugated donkey anti-goat IgG (1:1000, Invitrogen, CA, USA) for OC. DAPI solution (1:5000, Sigma-Aldrich) was applied for 5 min for nuclear staining.
The capillary and OB density was morphometrically evaluated as average values in five randomly selected fields in the soft tissue of peri-fracture sites under a fluorescent microscope. Capillaries were recognized as tubular structures positive for CD31. OB-like cells were recognized as lining or floating cells positive for OC on new bone surface. All morphometric studies were performed by two examiners who were blinded to treatment.
Double Immunofluorescent Staining
To detect Lnk expressing BM progenitor cells in the mouse peri-fracture sites, immunohistochemistry was performed with the following antibodies: Lnk (C-20) (1:50 dilution, Santa Cruz Biotechnology) and anti-mouse CD117, cKit (1:100 dilution, Chemicon, Millipore, Burlingame, Billerica, MA) as a sensitive marker for immature BM progenitor cells. The used secondary antibodies for each immunostaining were as follows: Alexa Fluor 594-conjugated donkey anti-goat IgG1 (1:500, Invitrogen, CA, USA) for mLnk, Alexa Fluor 488-conjugated goat anti-rat IgG1 (1:500, Molecular Probes, Invitrogen Japan K.K., Tokyo, Japan) for mCD117. DAPI solution (1:500, Sigma-Aldrich) was applied for 5 min for nuclear staining.
Physiological Assessment for Blood Perfusion by Laser Doppler Imaging (LDPI)
LDPI system (Moor Instrument, Wilmington, DE, USA)35, 36 was used to measure serial blood flow in both hindlimbs at 0, 1, 2 and 3 weeks post fracture according to the manufacturer’s instruction. The ratio of fractured/intact contralateral blood flow was calculated to evaluate the serial blood flow recovery after surgery.
In these digital color-coded images, a red hue indicated regions with maximum perfusion, medium perfusion values were shown in yellow and the lowest perfusion values were represented as blue. The measurement was performed under anesthesia with animals supine and both limbs fully extended. At first, we palpated the femoral fracture site under the skin and pasted a small stainless steel marker on the skin above the fracture site. The steel marker was scanned as a defect spot in the LDPI image, and was recognized as the center of the region of interest (ROI). The steel marker was removed and scanned again. Then, the ROI squares on the second scanned image referring to the first image were set with the marker’s defect spot. The blood flow recovery following fracture was evaluated as the ratio of mean flux within the ROI in the fractured hindlimb to the same size ROI in the contralateral intact hindlimb.
Radiographic Assessment for Fracture Healing
Radiographs of the fractured hindlimbs were serially taken at weeks 0, 1, 2, 3 and 4 following creation of fractures under anesthesia with the animals supine and both limbs fully extended. Fracture union was identified by the presence of a bridging callus on two cortices. Radiographs in each animal were examined by three blinded observers.
For quantification of callus/trabecular bone formation, micro-computed tomography (CT) imaging analysis was performed 3 weeks after fracture creation using LaTheta (ALOKA, Tokyo, Japan). To evaluate the fracture healing process, relative callus areas around the fracture sites in scanned radiographs at week 3 were quantified using LaTheta. Bone densities in the callus areas were calculated with CT intensity in scanned images by single-energy X-ray absorptiometry method and averaged using LaTheta. To recognize fracture healing more clearly, three-dimensional CT images were reconstructed using Volume Graphics GmbH Studio MAX software (Heidelberg, Germany).
Histological Assessment of Fracture Healing
Histological evaluations (n=3 in each group) were performed with toluidine blue staining to address the process of endochondral ossification at weeks 1, 2 and 3. The degree of fracture healing was evaluated using a five-point scale (grade 0–4) proposed by Allen et al.37 Samples were sectioned at 6-μm thickness on slides followed by fixation with 4% paraformaldehyde at 4 °C for 5 min. All morphometric studies were performed by two orthopedic surgeons blinded to the treatment.
Biomechanical Analysis of Fracture Union
Three mice in each treatment group were used for biomechanical evaluation at week 4 post fracture. Biomechanical evaluation was performed in Kureha Special Laboratory (KSL, Tokyo, Japan). Fractured femurs and contralateral non-fractured femurs were prepared and intramedullar fixation pins were removed before the bending test. The standardized three-point bending test was performed using a load torsion and bending tester ‘MZ-500S’ (Maruto Instruments, Tokyo, Japan). The bending force was applied with cross-head at a speed of 2 mm/min until rupture occurred. The extrinsic stiffness (N/mm) and the ultimate stress (N) were interpreted and calculated from the load deflection curve. The relative ratio of the fractured (right) femur to non-fractured (left) femur was calculated in each group and averaged.
Statistical Analysis
The results were statistically analyzed using a software package (Graph Pad Prism, MDF software). All values were expressed as mean±s.e.m. Paired t-test were performed for comparison before and after fracture. Unpaired t-tests (Mann–Whitney U-test) were performed for comparison between the two groups. The comparison of radiological results was performed with a χ2-test. A probability value of <0.05 was considered to denote statistical significance.
RESULTS
Lnk siRNA-Transfected KSL Cells Show High Capacity of Colony Formation
We first confirmed that Lnk expression was significantly decreased in Lnk siRNA-transfected KSL cells (Supplementary Figures S1A and B) and OBs (Supplementary Figures S1C and D) by western blotting analysis. We found that the expression of Lnk was suppressed by Lnk siRNA in mouse KSL cells and in mouse OBs.
To evaluate the function of Lnk siRNA-transfected KSL cells as EPCs in vitro, cells were analyzed by EPC colony-forming assay. After 16 days in culture with a methylcellulose-based medium, the average number of small and large EPC colonies per dish in the Lnk group was significantly greater than that in the control group. (small colony: Lnk, 39.2±0.49 vs control, 28.7±1.87, P<0.05, n=10; large colony: Lnk, 7.00±0.45 vs control, 3.21±0.32, P<0.05, n=10; Figures 1a and b) Each EPC colony-formation unit showed a differentiation hierarchy from small EPC to large EPC colonies, indicating a primitive EPC stage with high proliferative activity and a definitive EPC stage with vasculogenic properties, respectively. These findings suggest that silencing Lnk gene increases the function of KSL cells as EPCs in vitro.

Colony-formation assay and endothelial abilities of Lnk siRNA-transfected mouse KSL cells in vitro. (a) Representative images of a small colony and a large colony. (b) The number of EPC colonies was assessed after Lnk and control siRNA transfection. Average number of EPC colonies per dish of Lnk siRNA-transfected KSL cells was significantly greater than that of control siRNA-transfected KSL cells. (c) mRNA expressions of mVE-Cad, mVEGF and mAng-1 were normalized to GAPDH. (d) Proliferation and migration activities of mouse BM-derived cultured KSLs in response to Lnk and control siRNA. The proliferation activity is expressed as optical density (490 nm). The migration activity is expressed as the number of migrated cells to SDF-1. *P<0.05 and **P<0.001 vs control siRNA.
Lnk siRNA Transfection Promotes the Differentiation Capacity of KSL Cells and OBs
Endothelial marker expressions were examined in KSL cells by real-time RT-PCR after 7 days in culture. The mRNA expression of each marker (VE-Cad, VEGF and Ang-1) was significantly upregulated by Lnk siRNA transfection. (VE-Cad: Lnk, 1294±20.1 vs control, 816.3±51.6, P<0.001; VEGF: Lnk 739.8±30.7 vs control 495.7±68.2, P<0.001; Ang-1: Lnk 1649±117.0 vs control 823.2±155.4, P<0.001, n=5; Figure 1c).
Following osteogenic induction, Lnk siRNA-transfected OBs exhibited remarkable matrix mineralizations as assessed by Alizarin Red staining for calcium detection (Figure 2a). Lnk siRNA-transfected OBs showed a significant high concentration of Alizarin Red dye compared with the control siRNA-transfected OBs (Figure 2b). In addition, mRNAs of mOC, mBMP-2, mCol1A1, mRunx2 and mALP were highly expressed significantly in Lnk siRNA group than the control siRNA group after osteogenic induction. (OC: Lnk, 1134±167.9 vs control, 861.8±160.0, P<0.05; BMP-2: Lnk 2183±17.97 vs control 1945±57.75, P<0.05; Col1A1: Lnk 938.2±57.59 vs control, 574.3±72.95, P<0.001; Rux2: Lnk, 725.3±51.8 vs control, 509.5±32.4, P<0.05; Osx: Lnk, 228.0±15.2 vs control, 185.5±9.83, n.s, ALP: Lnk, 2340±207.3 vs control, 1594±67.3, P<0.05, n=5; Figure 2c).

Osteogenic abilities of Lnk siRNA-transfected mouse osteoblasts in vitro. (a) In the wells of Lnk siRNA-transfected mOBs, matrix mineralization was more clearly demonstrated by alizarin red staining, indicating the presence of calcium. Right upper panels in each image indicate whole well images. (b) Quantification of alizarin red staining by photometric analysis. The extracted alizarin red dye was measured with an absorption spectrometer at 415 nm wavelength. The amount of alizarin red dye was expressed as an optical density (OD) value. (c) The mRNA expressions of mOC, mCol1A1, mRunx2, mOsx, mALP and mBMP-2 were normalized to GAPDH. (d) Proliferation and migration activities of mouse cultured OBs with Lnk siRNA transfection. The proliferation activity is expressed as optical density (490 nm). The migration activity is expressed as the number of migrated cells to SDF-1. *P<0.05 and **P<0.001 vs control siRNA.
These results suggest that Lnk gene silencing leads to differentiation of KSL cells into endothelial lineage (EPCs) and osteogenic induction of OBs.
Lnk siRNA Transfection Promotes Proliferation and Migration Activities of KSL Cells and OBs
The proliferative activities of Lnk siRNA or control siRNA-transfected KSL cells and OBs were expressed as a mean optical density value at a wavelength of 490 nm. Lnk siRNA transfection significantly promoted KSL cell proliferation (Figure 1d, left panel) and OB proliferation (Figure 2d, left panel) activities. (mKSLs: Lnk, 0.420±0.0221 vs control, 0.265±0.0141, P<0.001; mOBs: Lnk, 1.061±0.0857 vs control, 0.899±0.1128, P<0.05, n=5) The migration of KSL cells and OBs were evaluated using a Transwell culture plate, and the activity was expressed as the number of migrated cells toward SDF-1α-containing medium. Lnk siRNA transfection significantly increased migration activities of KSL cells (Figure 1d, right panel) and OBs (Figure 2d, right panel) (mEPCs: Lnk, 163±21.9 vs control, 132±13.5, P<0.05; mOBs: Lnk, 313±15.7 vs control, 271±30.7, P<0.05, n=5).
These results suggest that KSL cells and OBs were functionally activated by Lnk gene deletion.
Fracture Upregulates Lnk gene Expression at Peri-Fracture site.
We first compared the frequency of Lnk mRNA expression by real-time RT-PCR analysis in the sites of peri-fracture and BM at the indicated time course (Figure 3a). We collected BM tissue from box1 and peri-fractuer site tissue from box2. The relative expressions of Lnk mRNA to GAPDH mRNA were significantly upregulated in both the BM (Figure 3b) and peri-fracture site (Figure 3c) after fracture (BM; pre-fracture: 184.0±37.52, post fracture: day 1; 262.7±32.07, day 3; 305.7±9.545, day 5; 174.3±38.87, day 7; 182.8±28.47, respectively, peri-fracture site; pre-fracture: 77.26±25.67, post fracture: day 1; 296.0±85.97, day 3; 318.1±161.1, day 5; 335.3±105.1, day 7; 171.2±36.00, respectively, P<0.05 for pre-fracture vs post fracture day 1 in both the BM and peri-fracture site and day 7 in peri-fracture site, P<0.001 for pre-fracture vs post fracture day 3 in the BM and days 3 and 5 in the peri-fracture site, n=5), suggesting that fracture-induced Lnk gene upregulation in the BM, including the peri-fracture site. However, Lnk gene was not expressed in mid-late term (after 10 days) after fracture creation (Supplementary Figure S1E).

Phenotypic characterization with Lnk expression in bone marrow (BM) and peri-fracture site. (a) Representative tissue collection site of BM (1) and peri-fracture site (2). Arrowheads show fracture sites. (b, c) The expression level of Lnk was assessed by qPCR in the BM (b) and peri-fracture site (c) at pre-fracture (fx) stage and 1, 3 and 7 days post-fx stage. *P<0.05 and **P<0.001 vs pre-fx. (d, e) Representative double immunostaining for Lnk (red) and cKit (green) with tissue samples of the fracture site on day 5. (d) White dotted lines indicate the edge of the bone. Arrowheads show mouse femur fracture sites. White closed square is the region of interest (ROI) observed by fluorescent Lnk and cKit stainings for high magnification image. (e) High magnification image of the ROI. Arrows show representative double positive cells for Lnk (red) and cKit (green). (original magnification, × 40 in (d), × 400 in (e)). (f–i) Immunoblotting for the expression of Lnk protein at the peri-fracture sites after Lnk siRNA transplantation. Lnk siRNA transfection resulted in the decreased expression of Lnk protein for 7 days after the treatment. **P<0.001 vs control siRNA.
Next, to determine what specific type of cells express Lnk in the sites of fracture, we performed double immunostaining of c-Kit for immature progenitors and Lnk with the samples 5 days after fracture creation. We detected Lnk-expressing immature c-Kit+ cells at the peri-fracture site 1 week after fracture creation (Figures 3d and e); however, no Lnk expressed cells were observed in the samples obtained 2 and 4 weeks after surgery.
Lnk Gene Deletion Accelerates Fracture Healing
The results of western blotting with tissue samples collected on days 3, 5 and 7 post fracture demonstrated that Lnk expression was significantly decreased in the Lnk siRNA-transfected mice. We also confirmed that Lnk expression was suppressed in the peri-fracture site up to 1 week after Lnk siRNA-conjugated atelocollagen transplantation. (Figures 3f–i).
Next, to evaluate fracture repair morphologically, radiographical and histological examinations were performed. Fractures were united radiographically with bridging callus formation in 60% (12 out of 20) of the Lnk group mice at week 2 and 90% (18 out of 20) at week 3. In contrast, fracture sites in all the animals of the control group showed only 30% (6 out of 20) fracture healing at week 2 and 60% (12 out of 20) at week 3 (P<0.05 in week 2 and 3). The time course of union rate in each group is described in Supplementary Figure S2A. Morphological and functional fracture healing in each group was further evaluated by micro-CT 3 weeks after fracture surgery. The results of micro-CT exhibited striking trabecular bone formation in the Lnk group compared with the control group (Figure 4a, Supplementary Figure S2B). Quantitative analysis for bone formation was performed using micro-CT images and expressed as callus volume and bone density, trabecular width and number of trabeculae. Both parameters in the Lnk group exhibited significantly greater values than those in the control group. (callus volume: Lnk, 13.68±0.7127 vs control, 11.52±0.4497 mm2, P<0.05; bone density: Lnk, 607.7±11.91vs control, 536.6±19.86, P<0.05; number of trabecula: Lnk, 2.73±0.22 vs control, 2.00±0.18/mm, P<0.05; width of trabecular: Lnk, 42.79±1.55 vs control, 37.87±0.63 μm, P<0.05, n=3; Figures 4b–e). Fracture healing was also histologically evaluated by toluidine blue staining. In animals receiving Lnk siRNA, bridging callus formation was observed at week 2, and finally fractures were united completely at week 3. In the control group, although callus formation was observed, bridging callus formation was rarely found at week 2 (Figure 4h). The score of fracture healing assessed by Allen’s classification38 was significantly higher in the Lnk group compared with control group at weeks 2 and 3. week 2: Lnk 2.60±0.245 vs control 1.40±0.245, P<0.05; week 3: Lnk 3.80±0.20 vs 2.60±0.400 P<0.05, n=3; Figure 4i).


Radiographical, functional and histological assessments for fracture healing following Lnk siRNA transfection. (a) Representative micro-CT 3D reconstruction images and axial images of fracture sites in both the groups at week 3. Arrowheads show rat femur fracture sites. (b–e) Quantitative micro-CT analysis of fracture healing. Callus volume, bone density, width and number of trabecula were quantified in the micro-CT images and averaged. *P<0.05 and **P<0.001 vs control siRNA. (f, g) Functional recovery after fracture was assessed by biomechanical three-point bending test at week 4. The percentage of each parameter for ultimate stress (f) and extrinsic stiffness (g) indicates the ratio of each value in the fracture site to the contralateral intact femur (n=3 in each group and each parameter). *P<0.05 vs Control siRNA. (h and i) Histological evaluation of endochondral ossification by toluidine blue staining. White dotted line marks the edge of the callus formation. Arrowheads show the sites of femur fracture. Images are shown as sets of lower magnification figures (above) and higher ones (below) of the area surrounded by yellow square in the lower ones. bc, bridging callus; cb, cortical bone; ft, fibrous tissue; gt, granulation tissue; tb, trabecular bone (h). The extent of fracture healing was assessed by Allen’s classification (n=3 in each group and at each time point). *P<0.05 vs control siRNA (i).
These results clearly indicate that the femoral fracture could be quickly repaired by local Lnk gene silencing.
Lnk siRNA Transplantation Leads to Functional Bone Healing after Fractures
To confirm functional recovery of the fractured bones, biomechanical evaluation by a three-point bending test was performed at week 4 in both the groups (n=3 in each group). There was no significant difference of the specimen length between the Lnk group and control group. (19.8±0.90 vs 20.7±0.78 mm, NS) The percentage ratios of extrinsic stiffness and ultimate stress in the fractured femur vs the contra-lateral intact femur were significantly superior in the Lnk group than in the control group. (percentage of ultimate stress: Lnk, 78.8±3.40 vs control, 66.2±3.70, P<0.05; percentage of extrinsic stiffness: Lnk, 72.40±3.172 vs control, 62.80±3.707, P<0.05) (Figures 4f and g). These findings indicate that fractures are not only radiographically but also functionally repaired by local Lnk gene silencing.
Lnk siRNA Transfection Enhanced Intrinsic Angiogenesis and Osteogenesis in the Sites of Fracture
We confirmed enhanced angiogenesis and osteogenesis by Lnk siRNA transfection at the peri-fracture sites’ tissues by immunostaining with specific markers for EC and OB. Immunohistochemical vascular staining with CD31, a marker for EC, in tissue samples collected 1 week post fracture, demonstrated enhancement of intrinsic neovascularization around the endochondral ossification area in animals treated with Lnk siRNA (Figure 5a). Neovascularization assessed by capillary density was significantly greater in the Lnk siRNA group compared with the control siRNA group. (Lnk, 250.2±10.97 vs control, 162.2±9.98/mm2, P<0.001, n=3; Figure 5b).

Enhancement of angiogenesis and osteogenesis in fractured bone following Lnk siRNA transfection. (a, d, g), Representative vascular staining with mCD31 (green) and DAPI (blue) using tissue samples at the peri-fracture sites collected at week 1. (a) Representative osteoblast staining with anti-mice osteocalcin (red) and DAPI (blue) using tissue samples at the peri-fracture sites (d) and perioeteal site (g) collected at week 1. (d, g) Arrowheads show femur fracture sites. White dotted line marks the edge of the callus formation. (b, e, h), Quantification of capillaries (b) and osteoblasts (f, h). CD31-positive capillaries and OC-positive osteoblasts were counted in five randomly selected high-power fields and averaged (n=5 in each group). **P<0.001 vs control siRNA. (c, f) Gene expressions of intrinsic cytokine for angiogenesis and osteogenesis in sites of fracture. RNA was extracted from peri-fracture tissue 1 week after surgery and examined by quantitative real-time RT-PCR analysis. The mRNA expressions of CD31 (c) and OC (f) were normalized to GAPDH. *P<0.05 vs control siRNA (n=5 in each group).
OB staining with anti-mouse OC on tissue samples collected 1 week post fracture revealed the augmentation of osteogenesis in a newly formed bone area in the Lnk group compared with the control group (Figure 5d). Osteogenesis assessed by OB density was significantly greater in the Lnk siRNA group than in the control siRNA group. (Lnk, 426.6±10.05 vs control, 243.2±18.49/mm2, P<0.001, n=3; Figure 5e). As shown in Figure 5g, the number of osteocalcin-positive lining cells on the periosteal surface of the fractured bone was increased in the Lnk siRNA group at day 7. (number of cells: Lnk, 31.0±1.05 cells/mm; control, 20.2±1.83 cells/mm, P<0.001, n=5; Figure 5h).
These results indicate that the morphological enhancement of angiogenesis and osteogenesis were attributed to Lnk gene deficiency in fracture-induced environment.
Lnk siRNA Transfection Promotes Blood Flow Recovery at the Sites of Fracture
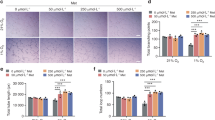
To evaluate blood flow recovery via neovascularization at the fracture sites, LDPI was serially performed after fracture creation (Figure 6a). In both the groups, severe reduction of blood flow was observed 1 h after fracture. There was no significant difference in the blood flow ratio of fractured to intact hindlimbs 1 h after fracture between the two groups. Although the blood perfusion ratios were significantly higher in the Lnk group than in the control group at week 1 (Lnk, 1.30±0.04 vs control, 1.12±0.04/mm2, P<0.05, n=3), a similar trend was observed until 2 weeks after surgery (Lnk, 1.45±0.06 vs control, 1.10±0.03/mm2, P<0.05, n=3). At week 3, the blood flow ratio exhibited no significant difference between the two groups (Figure 6b).

Improvement of blood flow recovery at the fracture sites following Lnk siRNA transfection. (a) Representative laser doppler perfusion imaging (LDPI) at weeks 0 (1 h after fracture), 1, 2 and 3 are shown. In these digital color-coded images, maximum perfusion values are indicated in white to red, medium values in green to yellow and the lowest values in dark blue. (b) Quantitative analyses of local blood perfusion. The blood flow within the fracture site (red square) and intact contralateral site (black square) were evaluated as mean flux, and the ratio of the mean flux in the fractured site to that in the contralateral site (mean flux ratio) was expressed as relative blood perfusion of fracture site. *P<0.05 vs control siRNA.
These results indicate that local Lnk gene silencing by transfection of Lnk siRNA contributes to early improvement of blood perfusion, which is consistent with the results of capillary density increase at the fracture sites.
Angiogenesis and Osteogenesis at the Sites of Fracture were Promoted in Lnk siRNA-Transfected Mice
To explore the underlying mechanism by which intrinsic angiogenesis and osteogenesis were enhanced following Lnk siRNA transfection therapy, we further assessed mRNA expressions of pro-angiogenic and -osteogenic cytokines in the peri-fracture sites 1 week after surgery by real-time RT-PCR, quantifying the expression of mVE-cad (mouse vascular endothelial cadherin (mVE-cad), mCD31, mouse angiopoietin 1 (mAng1), mouse vascular endothelial growth factor (mVEGF), mOC: mouse osteocalcin (mOC), mouse collagen 1 alpha 1 (mCol1A1), mBMP-2: mouse bone morphogenetic protein-2 (mBMP-2) and mouse bone morphogenetic protein 4 (mBMP-4) around the fracture sites. We also confirmed the protein expressions of CD31 and OC by immunohistochemistry and those of VE-Cadherin and BMP-2 by western blotting.
The results demonstrated a significantly high mRNA expressions of EC marker (CD31, VE-cad, Ang-1) in the Lnk group compared with the control group. However, there was no significant difference in VEGF between the Lnk group and the control group (VE-cad: Lnk, 501.8±43.28 vs control 257.7±34.00, P<0.001, CD31: Lnk, 254.9±33.30 vs control, 174.1±45.976, P<0.05, Ang-1: Lnk, 1001.0±51.46 vs control, 614.1±72.67, P<0.001,VEGF: Lnk, 194.4±20.89 vs control, 200.7±29.63, n.s., n=5; Figures 5c and 7c, Supplementary Figures S3A and B). The relative mRNA expressions of bone-related marker (OC, Col1A1, BMP-2 and BMP-4) to mGAPDH were significantly enhanced in the Lnk group compared with the control group (OC: Lnk, 837.2±120.1 vs control, 355.5±92.21, P<0.05, Col1A1: Lnk, 5855±1028 vs control, 2524±304.3, P<0.05, BMP-2: Lnk, 471.6137.73 vs control, 206.2±19.84, P<0.001, BMP-4: Lnk,185.6520.58 vs control, 102.9±5.699, P<0.05, n=5; Figures 5f and 7e, Supplementary Figures S3C and D). We also checked the above gene expresssions not only in the peri-fracture site but also in the BM; however, there were no statistical differences between the groups (data not shown), perhaps because Lnk gene expression was absent in the BM 7 days aftrer fracture (Figure 3f). The results of immunohistochemistry and western blot analysis also demonstrated that agniogneic (CD31 and VE-Cad; Figures 5a, b, 7a and b) and osteogenic (OC and BMP-2; Figures 5d, e and 7a, d) protein expressions were significantly increased in the Lnk siRNA group.

Immunoblotting for the expression of angiogenic and osteogenic protein in the peri-fracture site. (a) Representative immunoblotting for the expression of VE-Cadherin and BMP-2 protein in the peri-fracture sites 1 week after Lnk siRNA transfection. The immunoblots for VE-Cadherin normalized to Tublin (b) and for BMP-2 normalized to Tublin (d) in each group were quantified and shown as bar graphs. P<0.05 vs control siRNA. (c, d) Gene expressions of intrinsic cytokine for angiogenesis and osteogenesis in the sites of fracture. The relative expression levels of mouse specific VE-Cadherin (c) and BMP-2 (e) to mGAPDH. *P<0.05 and **P<0.001 vs control siRNA (n=5 in each group).
These results suggest that Lnk siRNA might enhance not only intrinsic angiogenesis but also osteogenesis at the fracture sites paracrinely via upregulation of pro-angiogenic and -osteogenic cytokines, leading to accelerated fracture healing with blood perfusion recovery.
DISCUSSION
In our series of experiments, we clarified that a negatively controlled Lnk system, using an atelocollagen-based siRNA transfection technique, contributed to a favorable environment for fracture healing by promoting vasculogenesis and osteogenesis at fracture sites, leading to rapid recovery from fracture. Moreover, our data indicated that Lnk siRNA-transfected KSL cells and OBs showed high capacities of vasculogenic and osteogenic terminal differentiations for fast fracture healing. Therefore, a negatively regulating Lnk system has important implications for the development of new therapeutic strategies in bone regeneration.
Lnk deletion has been shown to not only increase cell numbers but also reinforce self-renewal function of HSC/HPC and neural stem cells.8, 10, 38 In this study, we have demonstrated that Lnk was also expressed in KSL cells and OBs, and Lnk siRNA-transfected KSL cells and OBs showed increased proliferation activity and high vasculogenic and osteogenic activity. In vivo study, we demonstrated that c-Kit positive BM-derived progenitor cells co-expressed Lnk in a fracture-induced environment at the initial phase in the fracture repair process. Moreover, we have also demonstrated that local downregulation of Lnk enhanced bone healing at the fracture site in the in vivo study. These findings are thought to be caused by the following two mechanisms. First, osteogenesis and vasculogenesis closely regulate each other in terms of microenvironmental interaction for regenerative activity in BM. It has been reported that BM side population cells, which contain hematopoietic repopulating cells, can also engraft in the bone after transplantation39 and that the non-adherent population of BM cells, including KSL cells, contains primitive cells able to generate both hematopoietic and osteocytic lineage cells.40 Kwon et al11 reported that Lnk-deficient EPCs (KSLs) are more potent in BM-EPC kinetics, including the ability of cell growth, endothelial commitment, mobilization and recruitment for vascular regeneration. Calvi et al41 also found a parallel expansion of HSCs when the number of OBs was increased by parathyroid hormone infusion. Second, BMP-2-involved SCF-cKit signaling pathway is thought to be involved in the promotion of OB activity by silencing Lnk gene. Indeed, Matsumoto et al12 previously demonstrated that upregulation of BMP-2 mRNA expression was greater in the Lnk knockout mice than that in the wild-type mice and that a lack of Lnk or SCF supplement in OBs induced terminal differentiation of OBs and mineralized matrix formation with increased BMP-2 gene expression at the sites of fracture that were blocked by SCF antagonist. In the current study, the favorable effects of Lnk siRNA transfection on fractures were evidenced by a series of assessments in fracture healing. In the Lnk siRNA-transfected mice, quantitative histochemical analysis for ECs and OBs revealed the enhancement of angiogenesis and osteogenesis at the sites of fracture, leading to superiority of functional blood flow recovery and callus formation, and quantitative real-time RT-PCR analyses also exhibited upregulation of pro-angiogenic/-osteogenic gene expressions which is consistent with increased blood perfusion and callus formation at the peri-fracture sites. Regarding pro-angiogenic gene expression, the discrepancy between significant upregulation of mVEGF mRNA expression in KSL cells (Figure 1b) and no significant upregulation of mVEGF mRNA expression in peri-fracture tissue (Figure 7d) is attributed by sampling time course difference of 24 h after Lnk siRNA transfection in KSL cells vs 7 days after Lnk siRNA transfection. The series of findings could support the evidence that fractures promptly heal with sufficient mineralization and stiffness in the Lnk siRNA-transfected mice compared with control mice.
Silencing gene expression by siRNA is rapidly becoming a powerful tool for the genetic analysis of mammalian cells. However, the rapid degradation of siRNA and the limitation of its action call for an efficient delivery technology. In the past studies, several groups have described the use of viral vectors for siRNA delivery.42, 43, 44 However, viral vectors suffer from a problem of promoting side effects. Although the ‘hydrodynamic transfection method’ and a liposome transfection method were recently reported for siRNA delivery into animals,45, 46 more practical methods are needed for clinical use. Therefore, the development of safe and effective non-vector-based siRNA delivery systems is critical for the future of siRNA-based therapies. Recent reports described that atelocollagen complexed with siRNA is resistant to nucleases and is efficiently transduced into cells, thereby allowing long-term gene silencing.31, 47 Here, we used an atelocollagen-mediated siRNA transfer in an in vivo fracture model and could negatively control Lnk at the fracture site. Because atelocollagen allowed increased cellular uptake, nuclease resistance and prolonged release of siRNAs, atelocollagen complexed with siRNA rather than siRNA alone resulted in stronger gene silencing effects compared with other methods. As atelocollagen has an ability to transfer genes to both dividing and non-dividing cells, the atelocollagen-based siRNA transfer system represents an ideal method for achieving maximal outcome of siRNA-based gene silencing in vivo.
Despite the promising initial results obtained with Lnk siRNA transfection for fracture repair, there is a certain issue that needs to be solved before we move to clinical application. For instance, although recent reports described that atelocollagen complexed with siRNA was efficiently transduced into cells allowing long-term gene silencing not only in vivo but also in vitro,31, 47 we have a persistent concern that unexpected outcome will occur in other type of cells by long-term Lnk deletion. Indeed, previous reports suggest that Lnk is a negative regulator of brain neural stem cell proliferation and Lnk deficiency partially mitigates hematopoietic stem cell aging.38, 48 Nevertheless, our data shown in this study clearly indicate that downregulation of Lnk system promoted fracture healing.
In our series of experiments, we clarified that a negatively controlled Lnk system contributed to a favorable environment for fracture healing by enhancing vasculogenesis/angiogenesis and osteogenesis, leading to rapid bone reconstitution from fracture. The current study revealed the therapeutic potential of Lnk siRNA when transfected with a bioabsorbable scaffold, atelocollagen, at the sites of fracture in a mouse model. Therefore, downregulation of Lnk system may have a clinical impact for fast fracture healing that contributes to the reduction of delayed union or non-union fracture.
References
Huang X, Li Y, Tanaka K et al. Cloning and characterization of Lnk, a signal transduction protein that links T-cell receptor activation signal to phospholipase C gamma 1, Grb2, and phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA 1995;92:11618–11622.
Takaki S, Watts JD, Forbush KA et al. Characterization of Lnk. An adaptor protein expressed in lymphocytes. J Biol Chem 1997;272:14562–14570.
Yokouchi M, Suzuki R, Masuhara M et al. Cloning and characterization of APS, an adaptor molecule containing PH and SH2 domains that is tyrosine phosphorylated upon B-cell receptor stimulation. Oncogene 1997;15:7–15.
Li Y, He X, Schembri-King J et al. Cloning and characterization of human Lnk, an adaptor protein with pleckstrin homology and Src homology 2 domains that can inhibit T cell activation. J Immunol 2000;164:5199–5206.
Ahmed Z, Pillay TS . Adapter protein with a pleckstrin homology (PH) and an Src homology 2 (SH2) domain (APS) and SH2-B enhance insulin-receptor autophosphorylation, extracellular-signal-regulated kinase and phosphoinositide 3-kinase-dependent signalling. Biochem J 2003;371:405–412.
Takaki S, Sauer K, Iritani BM et al. Control of B cell production by the adaptor protein lnk. Definition of a conserved family of signal-modulating proteins. Immunity 2000;13:599–609.
Takaki S, Morita H, Tezuka Y et al. Enhanced hematopoiesis by hematopoietic progenitor cells lacking intracellular adaptor protein, Lnk. J Exp Med 2002;195:151–160.
Ema H, Sudo K, Seita J et al. Quantification of self-renewal capacity in single hematopoietic stem cells from normal and Lnk-deficient mice. Dev Cell 2005;8:907–914.
Takizawa H, Kubo-Akashi C, Nobuhisa I et al. Enhanced engraftment of hematopoietic stem/progenitor cells by the transient inhibition of an adaptor protein, Lnk. Blood 2006;107:2968–2975.
Kamei N, Kwon SM, Alev C et al. Lnk deletion reinforces the function of bone marrow progenitors in promoting neovascularization and astrogliosis following spinal cord injury. Stem Cells 2010;28:365–375.
Kwon SM, Suzuki T, Kawamoto A et al. Pivotal role of lnk adaptor protein in endothelial progenitor cell biology for vascular regeneration. Circ Res 2009;104:969–977.
Matsumoto T, Ii M, Nishimura H et al. Lnk-dependent axis of SCF-cKit signal for osteogenesis in bone fracture healing. J Exp Med 2010;207:2207–2223.
Colnot CI, Helms JA . A molecular analysis of matrix remodeling and angiogenesis during long bone development. Mech Dev 2001;100:245–250.
Gerstenfeld LC, Cullinane DM, Barnes GL et al. Fracture healing as a post-natal developmental process: molecular, spatial, and temporal aspects of its regulation. J Cell Biochem 2003;88:873–884.
Rodriguez-Merchan EC, Forriol F . Nonunion: general principles and experimental data. Clin Orthop Relat Res 2004;419:4–12.
Marsh D . Concepts of fracture union, delayed union, and nonunion. Clin Orthop Relat Res 1998;355:S22–S30.
Lu C, Miclau T, Hu D et al. Ischemia leads to delayed union during fracture healing: a mouse model. J Orthop Res 2007;25:51–61.
Matsumoto T, Kawamoto A, Kuroda R et al. Therapeutic potential of vasculogenesis and osteogenesis promoted by peripheral blood CD34-positive cells for functional bone healing. Am J Pathol 2006;169:1440–1457.
Folkman J . Angiogenesis. Annu Rev Med 2006;57:1–18.
Asahara T, Murohara T, Sullivan A et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997;275:964–967.
Akita T, Murohara T, Ikeda H et al. Hypoxic preconditioning augments efficacy of human endothelial progenitor cells for therapeutic neovascularization. Lab Invest 2003;83:65–73.
Karsenty G, Wagner EF . Reaching a genetic and molecular understanding of skeletal development. Dev Cell 2002;2:389–406.
Matsumoto T, Mifune Y, Kawamoto A et al. Fracture induced mobilization and incorporation of bone marrow-derived endothelial progenitor cells for bone healing. J Cell Physiol 2008;215:234–242.
Osawa M, Hanada K, Hamada H et al. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science 1996;273:242–245.
Li H, Marijanovic I, Kronenberg MS et al. Expression and function of Dlx genes in the osteoblast lineage. Dev Biol 2008;316:458–470.
Wong GL, Cohn DV . Target cells in bone for parathormone and calcitonin are different: enrichment for each cell type by sequential digestion of mouse calvaria and selective adhesion to polymeric surfaces. Proc Natl Acad Sci USA 1975;72:3167–3171.
Kwon SM, Eguchi M, Wada M et al. Specific Jagged-1 signal from bone marrow microenvironment is required for endothelial progenitor cell development for neovascularization. Circulation 2008;118:157–165.
Masuda H, Alev C, Akimaru H et al. Methodological development of a clonogenic assay to determine endothelial progenitor cell potential. Circ Res 2011;109:20–37.
Shoji T, Ii M, Mifune Y et al. Local transplantation of human multipotent adipose-derived stem cells accelerates fracture healing via enhanced osteogenesis and angiogenesis. Lab Invest 2010;90:637–649.
Manigrasso MB, O’Connor JP . Characterization of a closed femur fracture model in mice. J Orthop Trauma 2004;18:687–695.
Minakuchi Y, Takeshita F, Kosaka N et al. Atelocollagen-mediated synthetic small interfering RNA delivery for effective gene silencing in vitro and in vivo. Nucleic Acids Res 2004;32:e109.
Kinouchi N, Ohsawa Y, Ishimaru N et al. Atelocollagen-mediated local and systemic applications of myostatin-targeting siRNA increase skeletal muscle mass. Gene Ther 2008;15:1126–1130.
Matsumoto T, Kuroda R, Mifune Y et al. Circulating endothelial/skeletal progenitor cells for bone regeneration and healing. Bone 2008;43:434–439.
Iwasaki H, Kawamoto A, Ishikawa M et al. Dose-dependent contribution of CD34-positive cell transplantation to concurrent vasculogenesis and cardiomyogenesis for functional regenerative recovery after myocardial infarction. Circulation 2006;113:1311–1325.
Linden M, Sirsjo A, Lindbom L et al. Laser-Doppler perfusion imaging of microvascular blood flow in rabbit tenuissimus muscle. Am J Physiol 1995;269:H1496–H1500.
Wardell K, Jakobsson A, Nilsson GE . Laser Doppler perfusion imaging by dynamic light scattering. IEEE Trans Biomed Eng 1993;40:309–316.
Allen HL, Wase A, Bear WT . Indomethacin and aspirin: effect of nonsteroidal anti-inflammatory agents on the rate of fracture repair in the rat. Acta Orthop Scand 1980;51:595–600.
Ahlenius H, Devaraju K, Monni E et al. Adaptor protein LNK is a negative regulator of brain neural stem cell proliferation after stroke. J Neurosci 2012;32:5151–5164.
Dominici M, Pritchard C, Garlits JE et al. Hematopoietic cells and osteoblasts are derived from a common marrow progenitor after bone marrow transplantation. Proc Natl Acad Sci USA 2004;101:11761–11766.
Olmsted-Davis EA, Gugala Z, Camargo F et al. Primitive adult hematopoietic stem cells can function as osteoblast precursors. Proc Natl Acad Sci USA 2003;100:15877–15882.
Calvi LM, Adams GB, Weibrecht KW et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003;425:841–846.
Xia H, Mao Q, Paulson HL et al. siRNA-mediated gene silencing in vitro and in vivo. Nat Biotechnol 2002;20:1006–1010.
Hemann MT, Fridman JS, Zilfou JT et al. An epi-allelic series of p53 hypomorphs created by stable RNAi produces distinct tumor phenotypes in vivo. Nat Genet 2003;33:396–400.
Rubinson DA, Dillon CP, Kwiatkowski AV et al. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet 2003;33:401–406.
Sorensen DR, Leirdal M, Sioud M . Gene silencing by systemic delivery of synthetic siRNAs in adult mice. J Mol Biol 2003;327:761–766.
Song E, Lee SK, Wang J et al. RNA interference targeting Fas protects mice from fulminant hepatitis. Nat Med 2003;9:347–351.
Yamada A, Ishimaru N, Arakaki R et al. Cathepsin L inhibition prevents murine autoimmune diabetes via suppression of CD8(+) T cell activity. PLoS One 2010;5:e12894.
Bersenev A, Rozenova K, Balcerek J et al. Lnk deficiency partially mitigates hematopoietic stem cell aging. Aging Cell 2012;11:949–959.
Acknowledgements
We thank Ms Janina Tubby for her editing assistance in manuscript preparation.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflicts of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
Lnk is an intracellular adaptor protein expressed in hematopoietic cell lineages, and Lnk-deficient mice demonstrate enhanced hematopoietic reconstitution. A new role for this negative regulator of hematopoietic cytokine signaling is described in this study: Down-regulation of Lnk contributes to a favorable environment for healing bone fractures by enhancing vasculogenesis and osteogenesis.
Rights and permissions
About this article
Cite this article
Kawakami, Y., Ii, M., Matsumoto, T. et al. A small interfering RNA targeting Lnk accelerates bone fracture healing with early neovascularization. Lab Invest 93, 1036–1053 (2013). https://doi.org/10.1038/labinvest.2013.93
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2013.93

{kind=link}
{kind=link}
{kind=link}
{kind=link}