
Abstract
Hepatic myofibroblasts (MFB) show increased proliferation, migration and collagen production, which are crucial for hepatic fibrogenesis. Atorvastatin treatment inhibits proliferation, apoptosis and cytokine production of MFB in bile duct-ligated (BDL) rats in vivo. Here, we have further investigated the underlying mechanisms. Primary rat hepatic stellate cells (HSC) were isolated and culture-activated to hepatic MFB. Following 3 days of incubation with atorvastatin (10−4, 10−5 and 10−6 M), transcription levels of profibrotic cytokines (transforming growth factor-β1, connective tissue growth factor and TIMP1) and procollagen Ia were analyzed by real time PCR. Proliferation was investigated by 5'-bromo-2'-deoxyuridine assays. α-Smooth muscle actin protein expression was examined by western blotting. Fluorescence-activated cell sorting analysis of Annexin V and propidium iodide were used to measure apoptosis. Furthermore, p21 western blotting and β-galactosidase staining were investigated in MFB as senescence markers. Subsequently, hepatic expression of desmin and senescence markers were analyzed in the livers of rats receiving atorvastatin (15 mg/kg*d) for 1 week starting 3 and 5 weeks after BDL. Atorvastatin inhibited the activation of HSC to MFB and decreased cytokine and collagen production in MFB in vitro. In addition, proliferation, cytokine and collagen production of MFB were reduced by atorvastatin. Atorvastatin initiated apoptosis at 10−4 M and attenuated it at 10−5 M. Atorvastatin induced p21 protein expression and β-galactosidase staining of MFB in vitro and in vivo. Atorvastatin elicits similiar effects on MFB as previously seen in vivo: it decreases MFB turnover and fibrogenesis. We suggest that a further mechanism explaining these effects is senescence of cells.
Similar content being viewed by others


Main
During hepatic fibrosis, the desmin-positive hepatic stellate cells (HSC) and other myofibroblastic cells (MFB) are activated and show profibrotic activity.1, 2, 3 These profibrotic MFB produce high amounts of extracellular matrix proteins (eg, collagen) and profibrotic cytokines (transforming growth factor-β, TGF-β; connective tissue growth factor, CTGF; platelet-derived growth factor, PDGF), supporting progression of fibrosis in an autocrine and paracrine manner.4, 5 MFB express α-smooth muscle actin (α-SMA), which is used as a marker of their activation.2, 6, 7
Statins have shown beneficial effects on hepatic fibrosis and portal hypertension in cirrhosis, most likely because the inhibition of HMG-CoA-reductase impedes small GTPases (RhoA and Ras).8, 9, 10, 11, 12, 13 The portal pressure-lowering effect was attributed to the inhibition of RhoA translocation to the membrane of myofibroblastic HSC and thereby disruption of RhoA/Rho-kinase signaling, resulting in decreased contraction of these cells and reduced intrahepatic resistance.9 In a model of progressive biliary fibrosis using bile duct ligation (BDL) in rats,14 prophylactic and early atorvastatin treatment attenuated activation of MFB and subsequent collagen deposition.8 In fully established biliary fibrosis, atorvastatin treatment reduced MFB turnover by reducing apoptosis and proliferation of MFB.8 These effects, without a simultaneous reduction of cell number, may be explained by growth arrest and decreased fibrotic activity. Such phenomena have previously been observed in senescent myofibroblastic HSC.15, 16, 17 Statins might induce senescence, as shown for prostate cancer cells.18 After cellular stress many cells show increased β-galactosidase expression predominantly in lysosomes, reflecting senescence.19, 20, 21 Further markers of senescence are increased expression of p21,22, 23 as well as activation of the Wnt pathway.24 WNT1-inducible-signaling pathway protein 2 (WISP-2) (CCN5), a component of the Wnt pathway, mediates growth arrest and blunts TGF-β-induced effects.25, 26
The present study addresses the cellular effects of atorvastatin on hepatic MFB in vitro and in vivo with special emphasis on proliferation, apoptosis and senescence.
MATERIALS AND METHODS
Isolation and Culture of Primary Rat HSC
HSC were isolated as described previously.9 Briefly, after sequential in situ perfusion of the liver with collagenase type IV (Sigma, St. Louis, USA) and pronase E (Merck, Darmstadt, Germany) solution, dispersed cells were fractionated by density gradient centrifugation using Optiprep (Nycomed, Sweden). Cells were harvested at densities <1.053 (9% Optiprep). Viability and purity were systematically over 95%, as determined by trypan-blue exclusion and morphological characterization (vitamin A autofluorescence). Cells were seeded on uncoated plastic culture dishes and cultured in DMEM medium supplemented with 10% fetal calf serum, 0.6 IU/ml insulin, 2 mM glutamine and 1% antibiotic–antimycotic solution (Invitrogen, Karlsruhe, Germany). Contact with plastic leads to activation and subsequent transdifferentiation of HSC.27 The medium was renewed every 48–72 h.
Incubation of Primary Rat HSC With Atorvastatin
Atorvastatin was added to the culture medium of HSC at different concentrations (10−4, 10−5 and 10−6 M) starting at day 1 or day 3 after isolation. Culture-activated HSC receiving medium without atorvastatin served as controls. Cytological features (lipid droplets, cell shape and spreading) were evaluated by light microscopy. Cells were used for experiments 7 days after isolation. After the first passage, myofibroblastic HSC were fully activated. For these experiments, atorvastatin was added to the culture medium of these cells at different concentrations (10−4, 10−5 and 10−6 M) for 3 days, or cells remained untreated. Experiments were performed using at least three independent isolations.
5'-Bromo-2'-Deoxyuridine (BrdU) Proliferation Assay of HSC
The effect of atorvastatin on proliferation rates of rat HSCs was determined via a colorimetric BrdU ELISA kit (Roche Diagnostic GmbH, Penzberg, Germany). Briefly, cells were plated at a density of 6 × 105 cells/ml in flat-bottomed 96-well plates. During the last 24 h of the experiment, cells were labeled with 10 μM BrdU and its incorporation was measured according to manufacturers’ guidelines (Roche Diagnostic GmbH).
Detection of Apoptosis by Annexin V Fluorescence-activated Cell Sorting (FACS) Analysis
Analysis of apoptotic rat hepatic myofibroblastic HSC (MFB) was performed using a commercially available Annexin V Apoptosis Detection Kit (BD Biosciences, Heidelberg, Germany). Briefly, 105 rat MFB were washed two times with Annexin V-binding buffer. Cells were resuspended in 100 μl Annexin V-binding buffer and incubated with 5 μl FITC-conjugated Annexin V and 5 μl propidium iodide (PI) staining solution for 15 min at room temperature in the dark. After adding 400 μl Annexin V-binding buffer, the cell suspension was analyzed by flow cytometry within 30 min. Apoptotic cells were defined as PI-negative and Annexin V-positive.
Detection of Apoptosis/necrosis by Identification of subG1 Cells Using Cycle Analysis
105 MFB were washed with cold phosphate-buffered saline (PBS) and resuspended gently in 500 μl PI-hypotonic lysis buffer (0.1% (w/v) sodium citrate, 0.1% (w/v) Triton X-100, 100 μg/ml RNAse and 50 μg/ml PI in deionizied H2O). After an incubation time for at least 20 min, free nuclei were analyzed flow cytometrically within 2 h of hypotonic treatment. Flow cytometric analysis was performed using the cell cycle tool of FlowJo software (Tristar, Ashland, USA) that includes automatic identification of cell cycle phases.28
Western Blotting
Snap-frozen cells as well as liver samples were homogenized in a buffer containing 25 mM Tris/HCl, 5 mM ethylenediamine tetraacetic acid, 10 μM phenylmethanesulfonyl fluoride, 1 mM benzamidine and 10 μg/ml leupeptin. Samples were diluted with sample buffer as previously described.9 Protein concentration of the homogenates was determined using the DC-assay kit (Biorad, Munich, Germany). Samples (20 μg of protein per lane) were subjected to SDS-polyacrylamide gel electrophoresis (10% gels), and proteins were blotted to nitrocellulose membranes. Ponceau-S staining was performed to assure equal protein loading, and GAPDH served as endogenous control. Membranes were blocked, incubated with primary antibodies for α-SMA (Abcam plc, Cambridge, UK), GAPDH, p21 (sc-25778, sc-397, Santa Cruz Biotechnology, Santa Cruz, CA, USA) or desmin (GeneTex, Irvine, CA, USA), respectively. Subsequently, membranes were incubated with corresponding secondary peroxidase-coupled antibody (Calbiochem, San Diego, USA). Blots were developed with enhanced chemiluminescence (Amersham, UK). Intensities of the digitally detected bands were evaluated densitometrically using Chemi-Smart (PeqLab Biotechnologies, Erlangen, Germany).
Quantitative Real-Time PCR (RT-PCR)
RNA isolation, reverse transcription with 0.5 μg total RNA and detection by RT-PCR were performed as described previously.9 Primers and probes for RT-PCR were obtained as a ready-to-use mix (TGF-β-1, TIMP1, CTGF, procollagen Ia and PDGF-β receptor (PDGFβ-R)) from Applied Biosystems (Foster City, USA). 18 S rRNA served as endogenous control (primers and probes ready-to-use mix by Applied Biosystems). RT-PCR (ABI 7300 sequence detector) and PCR reaction (2 × TaqMan-PCR-mastermix, Applied Biosystems) were performed as described previously.9 For each of the genes, a validation experiment was performed. Efficiencies of RT-PCR for the target gene and the endogenous control were approximately equal. −ΔCT expresses the difference between the number of cycles (CT) of the target genes and the endogenous control. For better understanding, we normalized all −ΔCT values to those of controls. Results of liver samples were expressed as 2−ΔΔCt, and express the x-fold increase of gene expression compared with the control group. Experiments were performed using at least three independent isolations.
β-galactosidase Staining of Cells
MFB were fixed in 2% formaldehyde and 0.2% glutaraldehyde. After washing with PBS, the cells were incubated overnight at 37 °C with 0.1% (w/v) X-Gal (Peqlab) in 5 mM potassium hexacyanoferrate(III), 5 mM potassium hexacyanidoferrate(II), 2 mM magnesium chloride, 0.01% (w/v) sodium deoxycholate and 0.02% (v/v) Nonidet P-40 in PBS. Visualization was performed using a Nikon Eclipse TS100 microscope equipped with the NIS-Elements D3.2 software (Nikon, Düsseldorf, Germany).
Animals
We used male Sprague-Dawley rats with an initial body weight of 180 to 200 g. Sixteen rats underwent BDL as previously described.9 Eight animals were killed 4 weeks after BDL (4 W), and eight animals were killed 6 weeks after BDL (6 W). At 3 and 5 weeks after BDL, half of the 4 W BDL and 6 W BDL rats, respectively, received orally 15 mg atorvastatin per kg body weight per day for 1 week before killing as described previously.8, 9 Dissected livers were cut into fragments and either snap-frozen in liquid nitrogen (stored at −80°C) or stored in formaldehyde as previously described.9 The study was approved by the local committee for animal studies (Bezirksregierung Köln, 50.203.2-BN22,46/05).
Double Staining for β-Galactosidase, α-SMA and Desmin
Cryosections from liver tissue (20 and 7 μm) were fixed in 2% formaldehyde and 0.2% glutaraldehyde. After washing with PBS, the sections were incubated overnight at 37 °C with x-Gal-solution (Peqlab). For immunohistochemical staining of α-SMA, 20 μm slides with sections were incubated with a mouse-anti-SMA antibody (clone 1A4; Sigma-Aldrich) diluted 1:600 in Tris-buffered saline for 60 min. A biotinylated rabbit-anti-mouse secondary antibody absorbed with rat serum (Dako, Glostrup, Denmark) was applied (1:200, 45 min) and complexed with strepdavidin-conjugated alkaline phosphatase (1:200, 45 min; Dako). Stain was developed in fresh fuchsin-naphtol AS-BI solution (Sigma-Aldrich). For the morphometric analysis of stained specimen at least 10 mm2 of liver tissue were analyzed by means of computional analysis (Histoquant, 3DHistech, Budapest, Hungary) as described.29, 30 Large bile ducts and vessels were excluded.
Seven micometer cryosections were analyzed by confocal microscopy using an LSM710 confocal microscope (Zeiss, Jena, Germany). In order to combine X-Gal staining with anti-α-SMA or anti-desmin immunohistochemistry and DAPI, the slides were additionally incubated with anti-SMA-antibody (1:100; A2547, Sigma-Aldrich) or anti-desmin-antibody (1:100; GTX 28592, GeneTex) and with the respective secondary cy5-labeled antibody or Dylight 649 antibody (Dianova, Hamburg, Germany). Sections were counterstained with DAPI (Sigma-Aldrich) after X-Gal staining. Specimens were monitored by fluorescence microscopy and evaluated using AxioVision 4.8 (Carl Zeiss AG, Jena, Deutschland).
Statistical Analysis
Data are presented as mean±s.e.m. Student’s t-test was used for comparison where appropriate; Mann–Whitney U-test was used for comparison between groups. P-values <0.05 were considered statistically significant.
RESULTS
Atorvastatin Inhibits Activation and Proliferation of Primary Rat HSC In Vitro
Recently, we showed that atorvastatin treatment attenuates liver fibrosis in BDL rats.8 To analyze the effect of atorvastatin on the activation of HSC, primary quiescent rat HSC were either treated with atorvastatin from the first (d1) or third day (d3) after isolation, or they remained untreated. Analysis were performed at day 7 after isolation (Figure 1a). The proliferation of these HSC in vitro, as assessed by the BrdU assay, was significantly decreased in a dose-dependent manner by higher doses of atorvastatin (10−4 and 10−5 M), regardless of the incubation time point (d1, d3), whereas the lowest dose (10−6 M) did not reveal changes of BrdU uptake in comparison with controls (Figure 1b). Similarly, PDGFβ-R mRNA levels, used as marker for HSC activation, were significantly lower in d1 and d3 HSC after incubation with the higher atorvastatin doses (10−4 and 10−5 M). Again, atorvastatin at 10−6 M did not influence the steady-state level of PDGFβ-R mRNA (Figure 1c). Atorvastatin at the highest concentration (10−4 M) reduced α-SMA protein expression (Figure 1d). Similarly, atorvastatin at 10−5 M markedly decreased α-SMA protein expression of HSC as a marker of activation, whereas 10−6 M had essentially no effect on activation of HSC (Figure 1d). In summary, these results demonstrate that atorvastatin attenuates activation of quiescent HSC and inhibits proliferation of myofibroblastic HSC.

Proliferation and activation of HSC 7 days after isolation. Experimental design using primary rat HSC (a). Incubation of quiescent HSC with atorvastatin (10−4, 10−5 and 10−6 M) started on day 1 (d1) or day 3 (d3) after isolation. Control HSC remained untreated. HSC experiments were performed 7 days after their isolation from healthy rat livers. Cell proliferation was assessed by BrdU incorporation using ELISA (b). Relative mRNA expression of PDGFβ-R (c) as a proliferation marker was determined in HSC lysates by quantitative RT-PCR, corrected to 18 S rRNA as housekeeping gene and compared with HSC controls (expressed as 2−ΔΔCt). Atorvastatin dose dependently inhibited proliferation of HSC. Protein expression of α-SMA in the atorvastatin-treated and control HSC was measured by western blotting (d). Atorvastatin dose dependently inhibited the activation of primary rat HSC. Experiments were performed using three independent isolations.
Atorvastatin Incubation Decreased Transcription of Profibrotic Cytokines and Collagen in Quiescent HSC
In subsequent experiments, we assessed the role of atorvastatin on the profibrotic activity of HSC, as reflected by mRNA levels of profibrotic cytokines and collagen. Incubation of primary HSC with atorvastatin at higher concentrations (10−4 and 10−5 M) resulted in significantly decreased mRNA levels of the profibrotic cytokines TGF-β-1 and CTGF in comparison to untreated HSC, whereas no effect was measured at the lowest dose of atorvastatin (Figure 2a and b). Conversely, the expression of the matrix metalloprotease inhibitor TIMP1 was blocked at all atorvastatin concentrations and time points, suggesting a direct, dose- and time-independent effect (Figure 2c). In accordance with the anti-fibrotic effect of atorvastatin, the mRNA levels of procollagen Ia were significantly decreased at both time points and at all concentrations (Figure 2d). The highest concentration of atorvastatin reduced procollagen Ia expression levels almost to zero (Figure 2d). Thus, atorvastatin apparently prevented the activation of primary rat HSC, and thereby their proliferation and profibrotic activity.

Profibrotic cytokine and collagen expression of quiescent HSC. HSC were incubated with atorvastatin (10−4, 10−5 and 10−6 M) starting on day 1 (d1) or day 3 (d3) after isolation. The results were compared with fully activated HSC controls 7 days after isolation. Relative mRNA expression of TGF-β-1 (a), CTGF (b), TIMP1 (c) and procollagen type Ia (d) were determined in HSC lysates by quantitative RT-PCR and corrected to 18 S rRNA as housekeeping gene and compared with HSC controls (expressed as 2−ΔΔCt). Experiments are performed using three independent isolations.
Atorvastatin Incubation Decreased Proliferation of MFB
In a previous study, we could demonstrate that treatment with atorvastatin of BDL rats in established liver fibrosis decreased the proliferation of MFB.8 However, the effect of atorvastatin on the already activated MFB has remained unclear. We therefore used culture-activated primary rat HSC after the first passage, a stage at which HSC are myofibroblast-like cells (MFB). These cells were incubated with atorvastatin for 3 days or remained untreated (Figure 3a). In contrast to quiescent HSC (Figure 1), atorvastatin incubation of MFB did not change their α-SMA expression (Figure 3b). Interestingly, proliferation of MFB assessed by BrdU assay was attenuated significantly and in a dose-dependent manner by higher doses of atorvastatin (10−4 and 10−5 M), whereas the lowest dose (10−6 M) did not affect BrdU uptake in the MFB in comparison with controls (Figure 3c). Similarly, PDGFβ-R mRNA levels in MFB were markedly decreased after incubation with atorvastatin at all doses (Figure 3d). Again, atorvastatin at 10−6 M had less influence on PDGFβ-R expression than the higher concentrations of atorvastatin, but the effect was still significant (Figure 3d). This set of experiments showed that although atorvastatin decreased the proliferation of MFB, it did not change the expression of α-SMA, a marker for their activation.

Proliferation and activation of MFB. Experimental design in MFB (a). Incubation of MFB with atorvastatin (10−4, 10−5 and 10−6 M) started after the first passage for 3 days. Control MFB remained untreated. Protein expression of α-SMA (b) in the atorvastatin-treated and control MFB was measured by western blot, but did not differ between groups. Cell proliferation was assessed by BrdU incorporation using ELISA (c). Relative mRNA expression of PDGFβ-R (d) in MFB lysates was determined by quantitative RT-PCR and corrected to 18 S rRNA as housekeeping gene and compared with MFB controls (expressed as 2−ΔΔCt). Atorvastatin dose dependently inhibited proliferation of MFB. Experiments are performed using three independent isolations.
Atorvastatin Incubation Blunted the Expression Levels of Profibrotic Cytokines and Collagen in MFB
Atorvastatin treatment of BDL rats in established liver fibrosis decreased the cytokine expression at the tissue level, but not hepatic collagen contents.8 Here, we analyzed in detail the expression of profibrotic cytokines and collagen of MFB after incubation with atorvastatin. mRNA levels of profibrotic cytokines TGF-β-1 and CTGF were also significantly decreased in MFB after incubated with atorvastatin compared with untreated MFB (Figure 4a and b). Similarly, transcription of TIMP1 and procollagen Ia were also significantly decreased at all concentrations (Figure 4c and d). The highest concentration of atorvastatin showed the most pronounced effect on the transcription of CTGF and procollagen Ia, whereas the lowest concentration of atorvastatin resulted in the mildest but still significant effect on TGF-β-1 and TIMP1 mRNA levels (Figure 4). These results show that atorvastatin inhibits the profibrotic activity of MFB by reducing the transcription of profibrotic cytokines and collagen.

Profibrotic cytokine and collagen expression of MFB. Incubation of MFB with atorvastatin (10−4, 10−5 and 10−6 M) started after the first passage for 3 days. Control MFB remained untreated. Relative mRNA expression of TGF-β-1 (a), CTGF (b), TIMP1 (c) and procollagen type Ia (d) in MFB lysates was determined by quantitative RT-PCR and corrected to 18 S rRNA as housekeeping gene and compared with MFB controls (expressed as 2−ΔΔCt). Atorvastatin dose dependently inhibited proliferation of MFB. Experiments are performed using three independent isolations.
Analysis of Apoptosis and Cell Death in MFB After Atorvastatin Incubation
Atorvastatin reduces the proliferation of MFB without changing the expression of α-SMA (Figure 3). To rule out that these effects are simply due to induction of apoptosis, as shown by others for HSC (but not for MFB),31 a FACS analysis of Annexin V was used to detect apoptosis in MFB after atorvastatin incubation in comparison with MFB controls. The highest concentration of atorvastatin (10− 4 M) increased the number of Annexin V-positive, ie, apoptotic, MFB (Figure 5a and b). The intermediate concentration applied (10−5 M atorvastatin) did not, however, increase the number of Annexin V-positive MFB, and showed significantly less apoptotic MFB than the higher dose. There was a tendency towards a decrease in apoptosis of MFB, yet this was not statistically significant (Figure 5a and b). The lowest concentration of atorvastatin did not induce significant alterations in levels of apoptosis in comparison with MFB controls, but significantly more apoptotic MFB as compared with 10−5 M and significantly less apoptosis in comparison with the highest concentration of atorvastatin (Figure 5a and b).

Apoptosis of MFB in vitro. Incubation of MFB with atorvastatin (10−4, 10−5 and 10−6 M) started after the first passage for 3 days. Control MFB remained untreated. FACS analysis of annexin V binding was used to detect apoptosis in MFB after atorvastatin incubation and compared with MFB controls (a). Representative experiments are shown in b. The subG1 cell fractions (c) were detected by FACS analysis using the method described by Watson 28 and compared with MFB controls. The highest concentration of atorvastatin (10−4 M) induced apoptosis in MFB, whereas atorvastatin at 10−5 M shows a trend to protect from apoptosis. Experiments are performed using three independent isolations.
The necrotic/apoptotic cells (subG1 cells) were detected in FACS using the method described by Watson.28 Similarly to Annexin V assay, the highest dose of atorvastatin (10−4 M) induced massive cell death, whereas atorvastatin at 10−5 M seemed to act protective (Figure 5c). Again, atorvastatin at 10−6 M did not increase cell death when compared with controls, but showed significantly more cell death than at a concentration of 10−5 M (Figure 5c).
Summarizing these findings, atorvastatin incubation of MFB for 3 days at a concentration of 10−5 M seemed to protect against apoptosis and cell death. In contrast, higher concentrations of atorvastatin resulted in significantly higher levels of apoptosis and necrosis of MFB than the intermediate concentration.
Atorvastatin Incubation Induced Senescence in MFB
Our results suggest that atorvastatin decreased proliferation, profibrotic activity and modified apoptosis of MFB. The MFB phenotype as analyzed by α-SMA expression remained unchanged, but the cells seem to be more quiescent. Therefore, we analyzed the expression of the established senescence marker p2122, 23 in order to explain this state of MFB. The highest concentration of atorvastatin (10−4 M) failed to induce an increase in p21 expression in MFB when compared with untreated control cells (Figure 6a). In contrast, incubation with atorvastatin at 10−5 and 10−6 M for 3 days markedly increased the expression of p21 in MFB (Figure 6a).

Senescence of MFB in vitro. Incubation of MFB with atorvastatin (10−4, 10−5 and 10−6 M) started after the first passage for 3 days. Control MFB remained untreated. Protein expression of p21, a senescence marker, was measured by western blot in the atorvastatin-treated MFB and compared with controls (a). Representative sections of β-galactosidase (blue) stainings of MFB (b) detected senescent MFB. The quantification of the staining is shown in c. Relative mRNA expression of Wnt1 (d) and WISP-2 (e) in MFB lysates was determined by quantitative RT-PCR and corrected to 18 S rRNA as housekeeping gene and compared with MFB controls (expressed as 2−ΔΔCt). Senescence was induced by atorvastatin at 10−5 M in MFB. Experiments are performed using three independent isolations.
To strengthen this finding, we used β-galactosidase staining as a complementary tool to detect senescence.19, 20 Again, atorvastatin markedly increased β-galactosidase staining of MFB (10−4 and 10−5 M) in comparison with control cells (Figure 6b and c). However, incubation of MFB with atorvastatin at 10− 6 M did not result in β-galactosidase staining (data not shown).
A further hint for the presence of senescence in mesenchymal cells is activation of the Wnt pathway.24 We investigated this as a third line of evidence for increased senescence of MFB upon incubation with atorvastatin. Indeed at a concentration of 10− 4 M, Wnt1 expression was undetectable, but mRNA levels of Wnt1 were highly increased at concentrations of 10− 5 and 10− 6 M (Figure 6d). Downstream of the Wnt-signaling pathway, the expression of WISP-2 (CCN5), which has important roles in growth arrest and the repression of TGFβ pathways,25, 26 is induced. Again, a strong increase in WISP-2 mRNA levels was detected in MFB after incubation with atorvastatin at 10− 5 M (Figure 6e). Taken together, our results demonstrate that a concentration of 10− 5 M atorvastatin is capable of inducing sencescence in myofibroblastic HSC in vitro (Figure 6).
Analysis of Senescence in Livers of BDL Rats
Previously, we have shown that atorvastatin treatment of rats for 1 week reduces proliferation and apoptosis of hepatic α-SMA-positive MFB without changing their number.8 As our in vitro experiments indicate senescence in activated MFB, we investigated senescence in livers from these rats using the markes described above, p21 and β-galactosidase. BDL rats were treated with atorvastatin for 1 week after either 3 or 5 weeks of BDL, or they remained untreated (Figures 7a and b). We have previously described that atorvastatin treatment resulted in significantly reduced cytokine expression, proliferation and apoptosis of hepatic MFB, without influencing their total number or the accumulation of collagen in both treatment regimes.8

p21 and desmin protein expression in the BDL rat model in vivo. Experimental design in BDL rats in vivo (a, b). Rats received fibrosis induction by bile duct ligation and were kept for 4 weeks (a) and 6 weeks, respectively, with atorvastatin treatment only during the last week in both groups (each group n=4). Hepatic protein expression of p21 (c, d) and desmin (e, f) was analyzed by western blot analysis. Panels c, d, e and f show representative western blots and densitometric quantifications in means±s.e.m. relative densitometric units (d.u.) compared with control BDL rats (100 d.u.). Atorvastatin induced hepatic p21 protein expression in 6 W BDL rats in vivo (d) and reduced hepatic desmin protein expression in these rats (f).
Treatment with atorvastatin for 1 week, after 3 weeks of BDL (Figures 7a), did not change significantly hepatic p21 or desmin expression in these rats (Figures 7c and e). By contrast, atorvastatin treatment initiated after 5 weeks of BDL (Figure 7 b) led to increased hepatic expression of p21 as well as decreased levels of desmin protein expression in comparison with controls (Figures 7d and f).
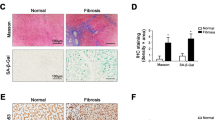
In addition to these findings, our experiments also revealed that atorvastatin treatment significantly increased the number of β-galactosidase-positive cells in both treatment regimes (Figure 8a and b). Immunostainings showed perinuclear localization of β-galactosidase expression—possibly in lysosomes—as described for senescence-associated β-galactosidase expression.19, 20, 21 These cells express α-SMA or desmin in their cytoplasm, as shown by double staining and confocal microscopy (Figure 8c and d). These data suggest the induction of senescence in activated HSC and hepatic MFB (Figure 8c and d).

Localization of senescent cells in the liver of BDL rats in vivo. The original hepatic β-galactosidase expression (blue, arrow) was found in atorvastatin-treated rats when treatment was started 3 weeks (4 W) and 5 weeks (6 W) after BDL, as shown in representative sections (a). Quantification of β-galactosidase stainings is given in b as means±s.e.m. Panel c shows representative confocal section of immunofluorescence staining for α-SMA (upper panel) or desmin (lower panel) in red, nuclear staining DAPI (blue) and perinuclear lysosomal β-galactosidase staining (black) in the liver sections of 4 W BDL+atorvastatin. Panel d gives representative confocal sections of immunofluorescence stainings for α-SMA (upper panel) or desmin (lower panel) in red, nuclear staining DAPI (blue) and perinuclear lysosomal β-galactosidase staining (black) in liver sections of 6 W BDL+atorvastatin. In the overlayed confocal sections (staining for DAPI, βGal and desmin/αSMA), the perinuclear lysosomal β-galactosidase staining is co-localized between cytoplasmatic α-SMA and DAPI-positive nuclei, suggesting an induction of senescence in activated HSC and MFB cells in vivo in both atorvastatin-treated groups.
DISCUSSION
In this study, we have investigated the cellular effects of atorvastatin on quiescent HSC in vitro, as well as on activated MFB in vitro and in vivo. We found that incubation with atorvastatin inhibits activation of cultured quiescent rat primary HSC. In addition, atorvastatin induced senescence in MFB, both in vitro and in vivo. The induction of senescence might at least partially explain why MFB show a decrease in proliferation, apoptosis and profibrotic activity after treatment with atorvastatin.
Hepatic MFB have a central role in promoting injury-induced liver fibrosis, regardless of its origin.2, 3, 4, 5 Hepatic injury activates MFB characterized by increased cytokine (TGF-β, PDGF and CTGF) and collagen expression, altered receptor formation (upregulation of PDGFβR) and increased proliferation.2, 4, 5 In vitro-activated HSC have also been demonstrated to change their morphology with loss of lipid droplets and increased expression of α-SMA.32, 33, 34
In our previous study, prophylactic treatment starting directly after the BDL resulted in a lower number of MFB in the liver and less collagen accumulation, but without significant decrease in cytokine production or inflammation.8 If atorvastatin was administrated later, but still before established fibrosis, collagen formation was reduced.8 These in vivo findings have now been underlined substantially by our cell culture experiments. In particular, atorvastatin dose-dependently inhibited proliferation of quiescent HSC (Figure 1) without induction of apoptosis as described previously by others.31 As the expression of PDGFβR (a proliferation marker) and α-SMA (a MFB marker) were significantly decreased (Figure 1), the most likely explanation is inhibition of HSC activation.4, 6 Supporting this assumption, atorvastatin-incubated MFB expressed less profibrotic cytokines and collagen (Figure 2).
In contrast, the effect of atorvastatin on MFB remains enigmatic. In fully established fibrosis in vivo, atorvastatin treatment decreased cytokine expression without causing a detectable reduction in inflammation or in hepatic cellular injury, as shown in our previously published data.8 Furthermore, the proliferation of MFB was reduced, suggesting a direct effect of atorvastatin on MFB in fibrotic livers.8 These effects were now analyzed in more detail in vitro, and we found that both proliferation and the expression of profibrotic mediators were decreased by atorvastatin incubation in a dose-dependent manner (Figures 3 and 4). Interestingly, this effect was not associated with changes in α-SMA protein expression (Figure 3b), suggesting that—though the MFB remained activated—their profibrotic activity was blunted by atorvastatin.
Thus, atorvastatin dose dependently decreased proliferation of MFB, as well as transcription of profibrotic factors, in these activated and fully transdifferentiated MFB.
Previously, it has been shown that culture-activated HSC show a dose-dependent increase in apoptosis after incubation with atorvastatin for 22 hours.31 Aprigliano et al31 attributed this effect to cell cycle arrest and caspase induction, and these effects might indicate early toxic effects of atorvastatin in cell culture. In contrast to these in vitro data, in our hands chronic atorvastatin treatment for 1 week did not change caspase activity; it even reduced apoptosis of MFB in cells with fully established fibrosis as shown previously in rats.8 Therefore, in the present study, we used fully transdifferentiated MFB (after first passage) and incubated these with atorvastatin for a longer time (3 days) than Aprigliano et al did.31 In this situation, apoptosis of MFB increased only after incubation with the highest dose (10−4 M) of atorvastatin, but it decreased at 10−5 M (Figure 5). The effect after 3 days of incubation with 10−5 M was rather stable, without a possible toxic overlap that could have been the case for the high dose (10−4 M). Toxic effects might also explain the observed short-term effects as described previously.31
Taken together, atorvastatin reduced (at least for 10−5 M) proliferation, profibrotic activity and apoptosis in MFB. We suggest senescence of MFB, previously described for HSC,15, 16, 17 as one explanation for this finding. However, the effects of atorvastatin towards induction of apoptosis or senescence are dose-dependent. The high dose of atorvastatin (10−4 M) induced senescence (β-galactosidase staining), which was followed by apoptosis (Figures 5 and 6). In contrast, the lower dose (10−5 M) predominantly induced expression of p21 and β-galactosidase as markers of senescence, without increasing apoptosis of these cells. Recently, it has also been shown that Wnt1 might induce senescence in mesenchymal cells.24 Interestingly, atorvastatin at 10−5 M induced the expression of Wnt1 and its downstream effector WISP-2 (Figure 6), a repressor of the TGF-β pathway.26
We also investigated senescence in vivo using p21 and β-galactosidase staining in rats treated for 1 week with atorvastatin 3 and 5 weeks after BDL (Figures 7 and 8).8 In these rats, atorvastatin did not affect collagen and MFB accumulation, but decreased proliferation, transcription of profibrotic factors and apoptosis of MFB.8 At the same time our present work revealed an induction of senescence, as assessed by β-galactosidase stainings (Figure 8a and b). This was more pronounced in more advanced stages of fibrosis (6 W BDL, Figure 8b), and might explain why p21 expression, a further senescence marker, was only increased in the 6-week BDL group treated with atorvastatin. The higher rate of senescent cells is probably caused by the fact that livers with more advanced fibrosis harbor more MFB. Co-localization microscopy (Figure 8c and d) identified MFB (α-SMA-positive cells) and HSC (desmin-positive cells) as the senescent cells (β-galactosidase-positive cells). This senescence of MFB we found is currently believed to be a stage between their profibrotic active state and apoptosis.15, 16, 17
Thus, our in vivo and in vitro findings suggest that atorvastatin drives MFB to senescent, inactive cells, which are not yet apoptotic.
References
Friedman S . The hepatic stellate cell. In: PD. B (ed) Semin Liver Dis. Thieme: New York, 2001, pp 307–452.
Friedman SL . Mechanisms of hepatic fibrogenesis. Gastroenterology 2008;134:1655–1669.
Ramadori G, Saile B . Mesenchymal cells in the liver--one cell type or two? Liver 2002;22:283–294.
Pinzani M . PDGF and signal transduction in hepatic stellate cells. Front Biosci 2002;7:d1720–d1726.
Bataller R, Brenner DA . Liver fibrosis. J Clin Invest 2005;115:209–218.
Gressner AM, Weiskirchen R . Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-beta as major players and therapeutic targets. J Cell Mol Med 2006;10:76–99.
Parola M, Marra F, Pinzani M . Myofibroblast—like cells and liver fibrogenesis: emerging concepts in a rapidly moving scenario. Mol Aspects Med 2008;29:58–66.
Trebicka J, Hennenberg M, Odenthal M et al. Atorvastatin attenuates hepatic fibrosis in rats after bile duct ligation via decreased turnover of hepatic stellate cells. J Hepatol 2010;53:702–712.
Trebicka J, Hennenberg M, Laleman W et al. Atorvastatin lowers portal pressure in cirrhotic rats by inhibition of RhoA/Rho-kinase and activation of endothelial nitric oxide synthase. Hepatology 2007;46:242–253.
Abraldes JG, Albillos A, Banares R et al. Simvastatin lowers portal pressure in patients with cirrhosis and portal hypertension: a randomized controlled trial. Gastroenterology 2009;136:1651–1658.
Laufs U, Liao JK . Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem 1998;273:24266–24271.
Laufs U, Marra D, Node K et al. 3-Hydroxy-3-methylglutaryl-CoA reductase inhibitors attenuate vascular smooth muscle proliferation by preventing rho GTPase-induced down-regulation of p27(Kip1). J Biol Chem 1999;274:21926–21931.
Rikitake Y, Liao JK . Rho GTPases statins, and nitric oxide. Circ Res 2005;97:1232–1235.
Schuppan D, Strobel D, Hahn EG. . Hepatic fibrosis - therapeutic strategies. Digestion 1998;59:385–390.
Krizhanovsky V, Yon M, Dickins RA et al. Senescence of activated stellate cells limits liver fibrosis. Cell 2008;134:657–667.
Green MR . Senescence: not just for tumor suppression. Cell 2008;134:562–564.
Schrader J, Fallowfield J, Iredale JP . Senescence of activated stellate cells: not just early retirement. Hepatology 2009;49:1045–1047.
Lee J, Lee I, Park C et al. Lovastatin-induced RhoA modulation and its effect on senescence in prostate cancer cells. Biochem Biophys Res Commun 2006;339:748–754.
Debacq-Chainiaux F, Erusalimsky JD, Campisi J et al. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc 2009;4:1798–1806.
Dimri GP, Lee X, Basile G et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 1995;92:9363–9367.
Lee BY, Han JA, Im JS et al. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 2006;5:187–195.
Chen Z, Trotman LC, Shaffer D et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005;436:725–730.
Kuilman T, Michaloglou C, Mooi WJ et al. The essence of senescence. Genes Dev 2010;24:2463–2479.
Zhang DY, Wang HJ, Tan YZ . Wnt/beta-catenin signaling induces the aging of mesenchymal stem cells through the DNA damage response and the p53/p21 pathway. PLoS One 2011;6:e21397.
Mason HR, Lake AC, Wubben JE et al. The growth arrest-specific gene CCN5 is deficient in human leiomyomas and inhibits the proliferation and motility of cultured human uterine smooth muscle cells. Mol Hum Reprod 2004;10:181–187.
Sabbah M, Prunier C, Ferrand N et al. CCN5, a novel transcriptional repressor of the transforming growth factor beta signaling pathway. Mol Cell Biol; 31:1459–1469.
Weiskirchen R, Gressner AM . Isolation and culture of hepatic stellate cells. Methods Mol Med 2005;117:99–113.
Watson JV, Chambers SH, Smith PJ . A pragmatic approach to the analysis of DNA histograms with a definable G1 peak. Cytometry 1987;8:1–8.
Huss S, Schmitz J, Goltz D et al. Development and evaluation of an open source Delphi-based software for morphometric quantification of liver fibrosis. Fibrogenesis Tissue Repair 2011;3:10.
Trebicka J, Racz I, Siegmund SV et al. Role of cannabinoid receptors in alcoholic hepatic injury: steatosis and fibrogenesis are increased in CB(2) receptor-deficient mice and decreased in CB(1) receptor knockouts. Liver Int 2011;31:862–872.
Aprigliano I, Dudas J, Ramadori G et al. Atorvastatin induces apoptosis by a caspase-9-dependent pathway: an in vitro study on activated rat hepatic stellate cells. Liver Int 2008;28:546–557.
Kato M, Iwamoto H, Higashi N et al. Role of Rho small GTP binding protein in the regulation of actin cytoskeleton in hepatic stellate cells. J Hepatol 1999;31:91–99.
Rombouts K, Kisanga E, Hellemans K et al. Effect of HMG-CoA reductase inhibitors on proliferation and protein synthesis by rat hepatic stellate cells. J Hepatol 2003;38:564–572.
Sohail MA, Hashmi AZ, Hakim W et al. Adenosine induces loss of actin stress fibers and inhibits contraction in hepatic stellate cells via Rho inhibition. Hepatology 2009;49:185–194.
Acknowledgements
The study was supported by grants from the Deutsche Forschungsgemeinschaft (SFB TRR57 projects 1, 12, 18, Q1) and the Bonfor-Stiftung (O-107.0084 and O-107.0103). We thank G Hack and S Bellinghausen for excellent technical assistance, and S Dentler for critical reading.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Atorvastatin prevents liver fibrosis and is also a beneficial treatment for established fibrosis. The mechanisms are twofold: the prevention of hepatic stellate cell (HSC) activation, and decreased cytokine and collagen production in previously activated HSCs. Furthermore, atorvastatin decreases turnover and induces senescence of activated HSCs and hepatic myofibroblasts in injured livers.
Rights and permissions
About this article
Cite this article
Klein, S., Klösel, J., Schierwagen, R. et al. Atorvastatin inhibits proliferation and apoptosis, but induces senescence in hepatic myofibroblasts and thereby attenuates hepatic fibrosis in rats. Lab Invest 92, 1440–1450 (2012). https://doi.org/10.1038/labinvest.2012.106
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2012.106
Keywords
This article is cited by
-
Atorvastatin-induced senescence of hepatocellular carcinoma is mediated by downregulation of hTERT through the suppression of the IL-6/STAT3 pathway
Cell Death Discovery (2020)
-
Statine bei Lebererkrankungen: gut oder schlecht?
Gastro-News (2020)
-
Novel Targets and Drug Development in Portal Hypertension
Current Hepatology Reports (2019)
-
Loss of pericyte smoothened activity in mice with genetic deficiency of leptin
BMC Cell Biology (2017)
-
Liraglutide improves liver microvascular dysfunction in cirrhosis: Evidence from translational studies
Scientific Reports (2017)
