
Abstract
Mutational activation of KIT or PDGFRA is considered an early step in pathogenesis of gastrointestinal stromal tumors (GISTs); however, other nonrandom genetic changes have also been identified. At least three common regions of deletions on chromosome 22q, which may harbor putative tumor suppressor genes, have been defined. However, mapping of these regions has been inconsistent. It has also been speculated that GI autonomous nerve tumors (GANTs), GISTs with ultrastructural features suggestive of autonomic nerve differentiation, are characterized by a specific deletion involving 22q13 cytogenetic region. This study was undertaken to evaluate loss of heterozygosity (LOH) on chromosome 22q in 50 GISTs, including 10 GANTs. Four tumors were incidental minimal lesions ≤10 mm in diameter. LOH was evaluated using 20 PCR-based microsatellite markers and capillary gel electrophoresis. In all, 15 (30%) cases showed LOH of more than 75% of informative markers, suggesting loss of chromosome 22q. A total of 24 GISTs (50%) revealed LOH of one to seven informative markers clustered in different loci suggesting simultaneous involvement of different regions. The highest frequency of LOH was seen at D22S922 and D22S425, mapped to 22q13.33 and 22q11.22, respectively. However, LOH at other regions including IL2RB and NF2 locus was also found. No NF2 mutations were identified in four analyzed tumors. LOH on chromosome 22q was more frequent among intestinal than among gastric GISTs; however, there was no difference between LOH pattern seen in tumors defined by different histologic, ultrastructural (GANT) and molecular features (KIT and PDGFRA mutations). Although minimal GISTs revealed LOH on chromosome 22q, there was a higher LOH frequency in malignant than in benign tumors. An isolated LOH at D22S425 was equally found in both benign and malignant tumors. These observations may suggest that LOHs on chromosome 22q in GISTs play a role in early stages of tumor formation as well as in late tumor progression.
Similar content being viewed by others

Main
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors of the gastrointestinal (GI) tract, previously often diagnosed as benign or malignant smooth muscle tumors, GI autonomic nerve tumors (GANTs) and schwannomas.1 A great majority of GISTs express KIT and have gain-of-function KIT or PDGFRA mutations. Mutational alteration of KIT or PDGFRA leads to ligand-independent activation (phosphorylation) of these tyrosine kinase receptors, has a transforming effect in vitro and is considered to be an early molecular event leading to the development of GISTs.2, 3 Germline KIT or PDGFRA mutations similar to those identified in sporadic GISTs have been reported in human familial GIST syndromes.4, 5 Moreover, features of human familial GIST syndrome were reproduced in a mouse model by introduction of KIT activating mutation.6
Although mutational activation of KIT or PDGFRA plays an important role in GIST pathogenesis, other changes, mostly losses of genetic material, have been documented in primary tumors.7, 8, 9, 10, 11, 12, 13, 14, 15, 16 Total or partial loss of chromosome 22 has been found in benign and malignant GISTs indicating that this change might play a role in GIST tumorigenesis.9 However, some of the studies linked loss of heterozygozity (LOH) on chromosome 22q to tumor progression and malignant outcome.12, 15 A comparative genomic hybridization (CGH) study defined 22q12–qter as a common region of deletion in GISTs,9 while fluorescence in situ hybridization (FISH) study based on five GISTs with ultrustructural features suggestive of autonomic nerve differentiation (GISTs/GANTs) identified the common region of deletion at 22q13.16 Based on one LOH study, NF2 was indicated as a possible target of deletions on chromosome 22q in GISTs; however, no convincing evidence of ‘Knudson type’ two hit inactivation of NF2 has been found.11 Moreover, two frequent LOH regions on chromosome 22, separated from NF2 locus, were recently identified in malignant GISTs.15
This study was undertaken to address some of the above-mentioned controversies emerging from the previously published investigations.11, 15, 16 A group of 50 well-characterized GISTs including small incidentally detected tumors and GISTs/GANTs was evaluated for LOH on chromosome 22q to identify common region/regions of deletions, which may harbor genes important for GISTs pathogenesis.
Materials and methods
Tissue Material
Samples of the tumor and corresponding normal tissue, demographic and clinical data were obtained from the files of the Armed Forces Institute of Pathology, Washington, DC, USA; the Department of Pathology, New York University Medical Center, New York, NY, USA, the Haartman Institute of the University of Helsinki, Finland, from the Medical College of Georgia and Veterans Affairs Medical Center, Augusta, Georgia; the Collegium Medicum of the Jagiellonian University, Krakow Poland and the Otto-von-Guericke University, Magdeburg, Germany.
Tumors were diagnosed as GISTs using previously established histological, immunohistochemical and molecular genetic criteria.1 Also based on published criteria,17, 18 tumor size and mitotic activity were used to evaluate the likelihood of malignant behavior (Table 1). All GISTs were classified into six prognostic groups. Group 1 and 2 tumors were considered benign or of very low malignant potential. GISTs classified into groups 3–6 represented tumors of uncertain malignant potential to the high-grade sarcomas (group 6).
Genetic Studies
Tumor and normal tissue samples were microdissected from formalin-fixed paraffin embedded (FFPE) tissue blocks and evaluated for possible cross-contamination to ensure purity of DNA samples. DNA was extracted as previously described.19 LOH was evaluated by PCR amplification of 20 microsatellite markers mapped to the chromosome 22q. Primer sequences were obtained from human genome microsatellite marker databases linked to the webpage of the National Center for Biotechnology Information (NCBI) (www.ncbi.nlm.nih.gov).
PCR amplification was performed using standard conditions recommended by Applied Biosystems (www.appliedbiosystems.com). PCR products were analyzed on ABI PRISM® 310 Genetic Analyzer following the Applied Biosystems procedure. The LOH was defined as recommended by PE Biosystems and previously reported.20 A ratio of the peak high values (fluorescence intensity) between longer and shorter alleles was calculated for the normal and tumor tissues. To obtain LOH value, an allele ratio from normal tissue was divided by an allele ratio from tumor tissue. The values ≤0.5 and ≥1.5 were considered to represent loss of heterozygosity.
Marker positions were established based on Human Chromosome 22 Sequence Map, deCODE Map and Marshfield Map (www.ncbi.nlm.nih.gov). Table 2 shows the orders of chromosome 22q microsatellite and FISH markers used in this and other studies.11, 15, 16
In eight cases that were noninformative for D22S929 microsatellite marker mapped to the first NF2 intron, additional single nucleotide polymorphic (SNP) NF2 markers were evaluated by PCR amplification and direct sequencing as previously described.21 Moreover, NF2 coding sequences (exons 1–16) were screened for mutations by PCR amplification and direct sequencing in 4 cases, following previously published procedures.22
Genomic sequences of KIT exons 9, 11, 13 and 17 and PDGFRA exons 12 and 18 were evaluated for mutations by PCR amplification and direct sequencing as previously described.23, 24, 25
In one case, tumor tissue was enzymatically disaggregated and cultured for cytogenetic analysis as previously reported.26 Chromosomes and chromosomal abnormalities were identified using GTW banding and described according to the International System of Human Cytogenetic Nomenclature.27
Results
Demographic, Clinical and Pathologic Features
All demographic, clinical and pathologic data are summarized in Table 3. The patient age varied from 29 to 86 years with median age of 59 years. The male to female ratio was 32:18. There were 31 gastric, 10 small intestinal, two colonic, four rectal and two apparently retroperitoneal primary GISTs. In Case 42, primary small intestinal vs colonic localization of the tumor could not be clearly established. Tumor size, available in all cases, varied from 1 to 36 cm (median 6.5 cm). Four small GISTs approximately 1 cm in diameter represented incidental findings during unrelated surgery (n=2), endoscopy (n=1) and autopsy (n=1).
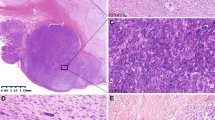
In all, 29 GISTs had pure spindle cell morphology, 13 were epithelioid and eight revealed mixed histology with both spindle and epithelioid components. Pleomorphic features were seen in one spindle cell tumor. KIT expression was documented immunohistochemically in all analyzed cases. Representative histological and immunohistochemical images are shown in Figure 1. In all, 10 previously reported GISTs28 showed ultrastructural features suggestive of autonomic nerve differentiation and were considered gastrointestinal autonomic nerve tumor variants of GIST (GIST/GANT).

Representative histological and immunohistochemical images of GISTs analyzed in this study. Case 4, gastric carcinoma (a, right) and minimal GIST (a, left) with epithelioid histology and KIT expression (b) with documented LOH of all analyzed 22q markers; Case 32, spindle cell benign intestinal GIST (c) and Case 9, malignant gastric GIST with pleomorphic features (d), both tumors with documented isolated LOH at D22S425; Case 30, epithelioid gastric GIST/GANT (e) with isolated losses of D22S425 (IGLV locus) and D22S929 (NF2 locus); Case 41, spindle component of intestinal GIST/GANT (f) with documented LOH at all informative loci on chromosome 22q.
Based on tumor size and mitotic activity, GISTs were assigned to six clinicopathologic groups according to the expected biologic potential. A total of 17 cases were classified as benign or of very low malignant potential (groups 1 and 2), 15 of uncertain malignant potential (group 3) and 18 as malignant (groups 5 and 6).
Clinical follow-up data were available in 37 cases. In all, 19 patients showed no evidence of disease with an average follow-up of 55 months. Nine patients died of disease or developed intra-abdominal metastases. Five patients died of unrelated causes and one patient died of unknown causes 312 months after surgery.
Genetic Studies
A total of 20 microsatellite markers were used to screen 50 tumors for LOH on chromosome 22q. Successful PCR amplification was obtained in 986 of 998 analyses (98.8%). LOH was calculated based on 754 heterozygous, informative markers and found in 295 analyses.
The pattern and frequency of LOH on chromosome 22q in 50 GISTs are shown in Figures 2 and 3. Representative examples of LOH analyses are shown in Figure 4.

Patterns of LOH on chromosome 22q identified in 50 GISTs. Single assay is represented by a circle. White, black and gray colors indicate constitutional heterozygosity with retention of both alleles, loss of heterozygosity and constitutional homozygosity (noninformative loci), respectively. Markers are listed in consecutive order from the centromeric to the telomeric part of chromosome arm 22q. ua: unsuccessful PCR amplification; nd: not done.

Frequencies of LOH at the 20 chromosome 22 loci examined in 50 GISTs.

Two examples of LOH on chromosome 22 identified in GISTs by PCR amplification of D22S425 and D22S928 microsatellite markers and capillary gel electrophoresis. Black arrows indicate lost alleles.
In 15 of 50 (30%) analyzed tumors, more than 75% of informative markers were lost indicating loss of the entire of chromosome 22q, however, in nine (18%) cases LOH was not found at any of the analyzed loci. In the remaining 26 (52%) cases, isolated losses of one or multiple markers often clustering in the distinctive regions of chromosome 22q were found. The highest frequency of LOH was seen at D22S922 (50%) and at D22S425 (47%) mapped to 22q13.31 and 22q11.22, respectively. However, LOH of other markers including IL2RB, D22S277 and markers flanking or mapped to NF2 locus were also found in 20–30% of the informative cases.
In addition, five NF2 intronic SNPs were analyzed in eight tumors noninformative for D22S929, the microsatellite marker mapped to the first NF2 intron. However, finding of LOH in two of six tumors informative for at least one SNP marker did not substantially increase LOH frequency at NF2 locus. The NF2 coding sequences (exons 1–16) were evaluated for mutations in four GISTs. Two tumors with LOH at NF2, one with intragenic deletion documented by SNP analysis and one with LOH of marker mapped immediately distal to NF2 locus, showed wild-type NF2 sequences.
KIT and PDGFRA mutations were found in 39 of 50 (78%) analyzed tumors. KIT juxtamembrane domain (exon 11) mutations consisted of one to several codon deletions (n=28) sometimes complicated by coexisting point mutations (DEL/DEL+PM), single PMs (n=9) affecting codons 557, 559, 560 and 576 and in two cases internal tandem duplications (ITDs). In two tumors, respectively, a rare but typical AY502 ITD in KIT extracellular domain and N822 K PM in KIT tyrosine kinase domain (exon 17) were found. All six PDGFRA mutations affected codon 842 in tyrosine kinase domain leading to either valin (D842 V) or tyrosine (D842Y) substitution for aspartic acid. KIT and PDGFRA mutational status of all analyzed GISTs is shown in Table 3.
Cytogenetic study was performed in Case 48 and the following karyotype (Figure 5) was identified 40–43,X, dic(X;1)(q12;p12),+7,−13,−15,der(17)t(1;17)(q23;q25),−22 [cp14].

Karyotype 40–43,X, dic(X;1)(q12;p12),+7,−13,−15,der(17)t(1;17)(q23;q25),−22 identified in the Case 48, rectal malignant GIST.
LOH on Chromosome 22q and Clinicopathological Features of GISTs
There were no substantial differences in LOH pattern and frequency among epithelioid and spindle cell tumors. GISTs assigned to groups 1 and 2 considered to be benign or of low malignant potential showed two times lower frequency of LOH on chromosome 22q than the highly malignant tumors asigned to group 6. Moreover, LOH of more than 75% of informative markers suggesting loss of the entire chromosome 22q were found in eight of 13 (61.5%) of GISTs assigned to group 6 compared with only two of 16 (12.5%) assigned to groups 1 and 2. However, isolated LOH at D22S425 mapped to 22q11.22 was equally found in both benign and malignant tumors. Intestinal GISTs revealed a higher LOH frequency (50.4%) than gastric (35.6%) tumors; however, GISTs assigned to groups 1 and 2 were two times more frequent among gastric (38.7%) than among intestinal (17.7%) tumors.
The patterns of LOH on chromosome 22q in GISTs/GANTs were similar to the ones seen in other GISTs. Three of 10 (33%) of these tumors showed loss of more than 75% of markers indicating possible loss of entire chromosome 22q. In five GISTs/GANTs, isolated LOH of single or multiple markers were found in the regions (including D22S425, NF2, IL2RB and 22q13.3) of chromosome 22q affected by deletions in other GISTs. In two GANTs, LOH was not found at any of the analyzed loci.
Three of four incidentally found, minimal GISTs (1 cm in diameter or less) showed LOH on chromosome 22q. In two tumors, all or the majority (63.3%) of informative markers were lost, indicating total or extended partial loss of chromosome 22q, respectively. In one minimal GIST, two isolated deletions were found. One of them involved D22S277 marker, shown in this study to be affected by LOH in 27% of all analyzed GISTs.
The patterns and frequencies of LOH on chromosome 22q were similar in the subcohorts of GISTs defined by KIT or PDGFRA mutational status.
Discussion
Gastrointestinal stromal tumors (GISTs) represent common mesenchymal tumors of the gastrointestinal tract driven by pathologic activation of KIT or PDGFRA tyrosine kinase receptors.1 KIT and PDGFRA gain-of-function mutations are considered the early if not the first step in GISTs pathogenesis;2, 3 however, other genetic changes, mainly losses and gains have also been reported.1 Losses of genetic material from chromosome 22 have been documented in GISTs by classical karyotyping, CGH, FISH and LOH studies.3, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16
In this study, we have evaluated LOH on chromosome 22q in 50 well-characterized GISTs using 20 PCR-based microsatellite markers and capillary gel electrophoresis. In 15 cases (30%), LOH of all or almost all markers suggested loss of the entire chromosome 22. Previously published LOH and CGH studies on GISTs, respectively, found loss of the entire chromosome 22 in 39% and 26% of analyzed tumors.9, 15
The frequency of loss of chromosome 22 in GISTs estimated by LOH or CGH studies differs substantially from the frequency of loss of chromosome 22 estimated by classical karyotyping. Two large, separate karyotyping studies of 19 and 52 GISTs reported loss of one or two copies of the chromosome 22, respectively, in 63 and 58% of analyzed cases.3, 14 The higher frequency of loss of chromosome 22 in GISTs detected by classic karyotyping compared to LOH or CGH studies could be an artifact related to the preferential culture of clones lacking chromosome 22. Recent report of primary GISTs with partial deletion of chromosome 22q and the recurrent tumor from the same patient with the loss of the entire chromosome 22 may support the notion of ongoing ‘in vivo’ clonal evolution of GISTs.16
Higher frequency of partial vs total loss of chromosome 22 in GISTs analyzed by LOH or CGH could also be related to the occult retention of the parts of chromosome 22 due to submicroscopic translocations. Genetic material from chromosome 22 was recently shown to be a part of the marker chromosome in GIST karyotype with monosomy of chromosome 22.29 In this study, discrepancy between karyotyping and LOH data was found in one analyzed tumor with the karyotype revealing monosomy of chromosome 22 and LOH analysis showing only partial loss of chromosome 22.
The highest frequency of LOH on chromosome 22q in GISTs was seen at D22S922 (50%), followed by D22S425 (47%) and D22S928 (37%) microsatellite markers. Two of three frequently deleted markers (D22S922, D22S928) are mapped to 22q13.3. This may suggest that deletion of 22q13.3 cytogenetic region plays a role in GISTs development. Recently published GIST karyotype with the specific deletion of 22q13 could partially support this notion3 as well as a study on GISTs/GANTs that reported exclusive loss of chromosome 22q13 cytogenetic region in one of five analyzed cases.16 Previous LOH studies on GISTS did not report significant LOH at 22q13.3;11, 12, 15 however, only one 22q13.3 microsatellite marker, D22S274, positioned proximal to D22S928, was evaluated and found to be deleted in some cases.11 Also in this study, markers mapped proximal to D22S274 at 22q13.2 were not commonly deleted. This again supports the mapping of the common deletion region in GISTs to 22q13.3–qter. However, further studies are required to define minimal regions of deletions and identified putative tumor suppressor gene/genes inactivated in GISTs. The cytogenetic region 22q13.3 has been shown to be affected by deletions in a spectrum of human cancers including breast, colon, ovarian and oral carcinomas as well as actrocytomas.30, 31, 32, 33
D22S425, mapped to 22q11.22 cytogenetic region, was the second most frequently lost marker in GISTs. 22q11.22 was not indicated as a common region of deletion in previously published GIST LOH studies.11, 15 However, LOH at D22S446, which currently is mapped relatively closed to D22S425 was found in 53.8% of analyzed GISTs.15 Considering the updated mapping of the markers (Table 2) and our and previously published data,15 the second most common region of deletion on chromosome 22q in GISTs might be defined by LOH at D22S425 and D22446. This region includes immunoglobulin lambda light chain variable region (IGLV) genes and other genes whose functions are not well known (www.ncbi.nlm.nih.gov).
Frequent LOH at NF2 locus (53%) was reported by Fukosawa et al11 in the first comprehensive study of LOH in GISTs and recently confirmed by Pylkkanen et al.15 In our study, LOH at NF2 locus was seen only in 27% of analyzed tumors, almost two times less than LOH at D22S922, the most commonly deleted marker. Although in some cases, NF2 was affected by an intragenic deletion as shown using the panel of SNP markers, no mutations were found in four GISTs with documented LOH at NF2 locus or its vicinity. Fukosawa et al11 reported one loss-of-function and one splicing mutation in 22 analyzed GISTs (9%); however, another study failed to find NF2 mutations in five primary GISTs.15 Considering all this, it appears that ‘Knudson type’ two hit inactivation of NF2 cannot be confirmed in GISTs.
An increased frequency of LOH was also seen at Interleukin 2 receptor beta (IL2RB) locus and D22S277 microsatellite marker, mapped to 22q12.3 cytogenetic region. This finding may correspond to previously reported increased frequency of LOH in the region defined by D22S683 and D22S445 microsatellite markers.15 This region overlaps with the common region of deletions reported in astrocytomas34 and breast carcinomas.35
Loss of genetic material from chromosome 22 was documented in benign and malignant, sometimes called low- and high-risk, GISTs.9, 11 Although some of the studies linked LOH on chromosome 22q to tumor progression and unfavorable outcome,12, 15 in our study, loss of genetic material from chromosome 22 was documented in three of four minimal GISTs incidentally detected due to other medical procedures. This suggests that LOH on chromosome 22 or loss of chromosome 22 occurs early at the tumor formation. However, frequency of LOH in malignant GISTs assigned to group 6 (>5 cm, >5 mitosis/50HPF) was higher than in benign tumors assigned to groups 1 and 2 (≤2 cm, <5 mitosis/50HPF and >2 cm <5 cm, ≤5 mitosis/50HPF). Of 13 tumors assigned to group 6, eight (61.5%) revealed deletions of at least 75% of chromosome 22q markers, suggesting loss of the entire chromosome 22. In contrast, only two of 16 (12.5%) tumors assigned to groups 1 and 2 with usually favorable outcome showed extensive losses on chromosome 22q. This may suggest that initial LOH on chromosome 22 at the early stage of tumor formation is followed by either accumulation of LOH on chromosome 22 due to genetic instability or ongoing clonal selection, giving growing advantage to the tumor cells with losses of chromosome 22.
In this study, isolated losses at IGLV locus (D22S425) were found in both benign and malignant tumors. The LOH studies on astrocytomas and breast carcinomas reported the same type of isolated losses on chromosome 22q in both early lesions and highest malignancy grade tumors. However, significant increase of LOH frequency was seen only in the latter.30, 33, 34 More tumors with isolated losses of specific chromosome 22q markers should be studied to confirm this phenomenon in GISTs.
Intestinal GISTs showed higher frequency of LOH on chromosome 22q than in gastric tumors. However, this may be related to the higher number of benign, group 1 and 2 tumors in the latter cohort. There was no significant difference in the LOH pattern on chromosome 22q between GISTs defined by morphological (epithelioid vs spindle cell) and molecular genetic features (KIT mutant vs PDGFRA mutant vs KIT and PDGFRA wild type).
GI autonomic nerve tumors (GANTs), which show ultrastructural features suggestive of autonomic nerve differentiation, are now considered GIST variants based on KIT expression and presence of GIST-specific KIT28 or PDGFRA-activating mutations. Recent FISH/CGH-based study reported loss of whole or part of chromosome 22q with the common overlapping area at 22q13 in five malignant intestinal GISTs/GANTs.16 It was also speculated that deletion of 22q13 may be a characteristic genetic feature of GANTs.16 In this study, we have evaluated 10 previously reported GANTs for the LOH on chromosome 22. The pattern of detected losses was similar to the one seen in other GISTs, giving no indication that GANTs would be distinctive in their involvement of chromosome 22.
In summary, we have shown that LOH on chromosome 22q in GISTs could be found at the early tumor formation and late stage of tumor progression. Although similar LOH patterns were seen in both early and late stages of tumor development, the frequency of total losses significantly increased in highly malignant tumors. Based on this study, at least three different regions harboring genes important for GISTs development can be defined and should be a subject of further molecular genetic studies.
References
Miettinen M, Lasota J . Gastrointestinal stromal tumors (GISTs): definition, occurrence, pathology, differential diagnosis and molecular genetics. Pol J Pathol 2003;54:3–24.
Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998;279:577–580.
Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003;299:708–710.
Nishida T, Hirota S, Taniguchi M, et al. Familial gastrointestinal stromal tumours with germline mutation of the KIT gene. Nat Genet 1998;19:323–324.
Chompret A, Kannengiesser C, Barrois M, et al. PDGFRA germline mutation in a family with multiple cases of gastrointestinal stromal tumor. Gastroenterology 2004;126:318–321.
Sommer G, Agosti V, Ehlers I, et al. Gastrointestinal stromal tumors in a mouse model by targeted mutation of the KIT receptor tyrosine kinase. Proc Natl Acad Sci USA 2003;100:6706–6711.
Dal Cin P, Boghosian J, Sandberg AA . Cytogenetic findings in leiomyosarcoma of the small bowel. Cancer Genet Cytogenet 1988;30:285–288.
Bardi G, Johansson B, Pandis N, et al. Recurrent chromosome aberrations in abdominal smooth muscle tumors. Cancer Genet Cytogenet 1992;62:43–46.
El-Rifai W, Sarlomo-Rikala M, Miettinen M, et al. DNA copy number losses in chromosome 14: An early change in gastrointestinal stromal tumors. Cancer Res 1996;56:3230–3233.
Breiner JA, Meis-Kindblom J, Kindblom L, et al. Loss of 14q and 22q in gastrointestinal stromal tumors (pacemaker cell tumors). Cancer Genet Cytogenet 2000;120:111–116.
Fukasawa T, Chong JM, Sakurai S, et al. Allelic loss of 14q and 22q, NF2 mutation, and genetic instability occur independently of c-kit mutation in gastrointestinal stromal tumor. Jpn J Cancer Res 2000;91:1241–1249.
Kim NG, Kim JJ, Ahn JY, et al. Putative chromosomal deletions on 9p, 9q and 22q occur preferentially in malignant gastrointestinal stromal tumors. Int J Cancer 2000;85:633–638.
Debiec-Rychter M, Lasota J, Sarlomo-Rikala M, et al. Chromosomal aberrations in malignant gastrointestinal stromal tumors: correlation with c-KIT gene mutation. Cancer Genet Cytogenet 2001;128:24–30.
Gunawan B, Bergmann F, Hoer J, et al. Biological and clinical significance of cytogenetic abnormalities in low-risk and high-risk gastrointestinal stromal tumors. Human Pathol 2002;33:316–321.
Pylkkanen L, Sarlomo-Rikala M, Wessman M, et al. Chromosome 22q alterations and expression of the NF2 gene product, merlin, in gastrointestinal stromal tumors. Human Pathol 2003;34:872–879.
Debiec-Rychter M, Pauwels P, Lasota J, et al. Complex genetic alterations in gastrointestinal stromal tumors with autonomic nerve differentiation (GANTs). Modern Pathol 2002;15:692–698.
Miettinen M, El-Rifai W, Lasota J, et al. Evaluation of malignancy and prognosis of gastrointestinal stromal tumors: a review. Hum Pathol 2002;33:478–483.
Miettinen M, Kopczynski J, Makhlouf HR, et al. Gastrointestinal stromal tumors, intramural leiomyomas, and leiomyosarcomas in duodenum. Am J Surg Pathol 2003;27:625–641.
Lasota J, Jasinski M, Sarlomo-Rikala M, et al. C-kit mutations occur preferentially in malignant vs. benign gastrointestinal stromal tumors and do not occur in leiomyomas and leiomyosarcomas. Am J Pathol 1999;154:53–60.
Canzian F, Salovaara R, Hemminki A, et al. Semiautomated assessment of loss of heterozygosity and replication error in tumors. Cancer Res 1996;56:3331–3337.
Lasota J, Fetsch JF, Wozniak A, et al. The neurofibromatosis type 2 (NF2) gene is mutated in perineurial cell tumors: a molecular genetic study of 8 cases. Am J Pathol 2001;158:1223–1229.
Lasota J, Wasag B, Dansonka-Mieszkowska A, et al. Evaluation of NF2 and NF1 tumor suppressor genes in distinctive gastrointestinal nerve sheath tumors traditionally diagnosed as benign schwannomas: a study of 20 cases. Lab Invest 2003;83:1361–1371.
Lasota J, Wozniak A, Sarlomo-Rikala M, et al. Mutations in exons 9 and 13 of KIT gene are rare events in gastrointestinal stromal tumors. A study of two hundred cases. Am J Pathol 2000;157:1091–1095.
Lasota J, Dansonka-Mieszkowska A, Sobin LH, et al. A great majority of GISTs with PDGFRA mutations represents gastric tumors of low or no malignant potential. Lab Invest 2004;84:874–883.
Miettinen M, Sarlomo-Rikala M, Lasota J . KIT expression in angiosarcomas and early fetal endothelial cells and lack of c-kit mutations in angiosarcoma. Mod Pathol 2000;13:536–541.
Limon J, Dal Cin P, Sandberg AA . Application of long-term collagenase disaggregation for the cytogenetic analysis of human solid tumors. Cancer Genet Cytogenet 1986;23:305–313.
Mitelman F . ISCN (1995): An International System for Human Cytogenetic Nomenclature. Karger: Basel, 1995.
Lee JR, Joshi V, Griffin Jr JW, et al. Gastrointestinal autonomic nerve tumor: immunohistochemical and molecular identity with gastrointestinal stromal tumor. Am J Surg Pathol 2001;25:979–987.
Gerresheim F, Kolin-Gerresheim I, Boltze C, et al. Complex karyotypes in two cases of gastrointestinal stromal tumors. Medizinische Genetik 2002;14:274.
Castells A, Gusella JF, Ramesh V, et al. A region of deletion on chromosome 22q13 is common to human breast and colorectal cancer. Cancer Res 2000;60:2836–2839.
Bryan EJ, Watson RH, Davis M, et al. Localization of an ovarian cancer tumor suppressor gene to a 0.5-cM region between D22284 and CYP2D, on chromosome 22q. Cancer Res 1996;56:719–721.
Miyakawa A, Wang XL, Nakanishi H, et al. Allelic loss on chromosome 22 in oral cancer: possibility of the existence of the tumor suppressor gene on 22q13. Int J Oncol 1998;13:705–709.
Oskam NT, Bijleveld EH, Hulsebos TJ . A region of common deletion in 22q13.3 in human glioma associated with astrocytoma progression. Int J Cancer 2000;85:336–339.
Ino Y, Silver JS, Blazejewski L, et al. Common regions of deletion on chromosome 22q12.3–q13.1 and 22q13.2 in human astrocytomas appear related to malignancy grade. J Neuropathol Exp Neurol 1999;58:881–885.
Iida A, Kurose K, Isobe R, et al. Mapping of new target region of allelic loss to a 2-cM interval at 22q13.1 in primary breast cancer. Genes Chromosomes Cancer 1998;21:108–112.
Acknowledgements
We thank Dr Brian West from the Department of Pathology, New York University Medical Center, New York, NY, for the contribution of cases for this study. The opinions and assertions contained herein are the expressed views of the authors and are not to be construed as official or reflecting the views of the Departments of the Army or Defense. This study was partially supported by the Grant from the Polish Committee for the Scientific Research (3PO5C05925) and by the American Registry of Pathology.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lasota, J., Wozniak, A., Kopczynski, J. et al. Loss of heterozygosity on chromosome 22q in gastrointestinal stromal tumors (GISTs): a study on 50 cases. Lab Invest 85, 237–247 (2005). https://doi.org/10.1038/labinvest.3700218
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700218
Keywords
This article is cited by
-
Metastatic Liver Disease Associated with Gastrointestinal Stromal Tumors: Controversies in Diagnostic and Therapeutic Approach
Journal of Gastrointestinal Cancer (2015)
-
Chromosome copy number changes carry prognostic information independent of KIT/PDGFRA point mutations in gastrointestinal stromal tumors
BMC Medicine (2010)
-
A molecular portrait of gastrointestinal stromal tumors: an integrative analysis of gene expression profiling and high-resolution genomic copy number
Laboratory Investigation (2010)
-
Impact of KIT and PDGFRA Gene Mutations on Prognosis of Patients with Gastrointestinal Stromal Tumors After Complete Primary Tumor Resection
Journal of Gastrointestinal Surgery (2009)
-
Neurofibromatosis type 1-related gastrointestinal stromal tumors: a special reference to loss of heterozygosity at 14q and 22q
Journal of Cancer Research and Clinical Oncology (2009)
