
Abstract
A new methodology has been developed to amplify RNA from minute amounts of starting material. Specifically, an efficient means of second-strand (ss) cDNA synthesis using a sequence-specific ‘terminal continuation’ (TC) method is demonstrated. An RNA synthesis promoter is attached to the 3′ and/or 5′ region of cDNA utilizing the TC mechanism. The orientation of amplified RNAs is ‘antisense’ or a novel ‘sense’ orientation. TC RNA amplification is utilized for many downstream applications including gene expression profiling, cDNA microarray analysis, and cDNA library/subtraction library construction. Synthesized sense TC-amplified RNA can also be used as a template for in vitro protein translations and downstream proteomic applications. The TC RNA amplification methodology offers high sensitivity, flexibility, and throughput capabilities. A likely mechanism is that the TC primer binds preferentially to GC-rich CpG islands flanking 5′ regions of DNA that contain promoter sequences. Following TC RNA amplification, a large proportion of genes can be assessed quantitatively as evidenced by bioanalysis and cDNA microarray analysis in mouse and human postmortem brain tissues.
Similar content being viewed by others


Main
Contemporary gene expression profiling is performed using cDNA array technology.1, 2, 3, 4, 5 An array platform allows investigation of multiple genes (eg, hundreds to thousands) simultaneously. However, fairly large tissue quantities are needed for RNA extraction. Unfortunately, sources of tissues may be limited and a heterogeneous cell population may be analyzed. Single-cell and/or population-cell analysis is highly desirable, especially as it relates to the pathophysiology of disease and pharmacogenomic drug discovery.6, 7, 8 Amplified antisense RNA (aRNA) synthesis amplifies genetic signals from limited amounts of fresh, frozen, or fixed tissues and cells.9, 10, 11, 12 aRNA is effective, yet can be a relatively difficult protocol to perform. The principal obstacle for increasing efficiency is the problematic second-strand (ss) cDNA synthesis. This impediment is not specific to the aRNA protocol. Rather, this issue is endemic to all current RNA amplification methods. Two procedures utilized for ss cDNA synthesis include self-priming and replacement synthesis. Self-priming uses a hairpin at the 3′ end of first-strand (fs) cDNA to self-prime ss cDNA synthesis. However, the loop formed at the end must be removed using S1 nuclease digestion in a poorly controlled reaction and invariably leads to the loss of the 5′ signal.13 Replacement synthesis employs multiple enzymes to synthesize ss cDNA including RNase H, DNA polymerase I, and T4 DNA ligase.14, 15 Replacement synthesis suffers from low efficiency, likely caused by the multiple enzymatic steps involved.
Current methods of RNA amplification attach a bacteriophage transcriptional promoter sequence (eg, T7, T3, SP6) to the 3′ end of fs cDNA, resulting in transcripts with antisense orientation. Key factors to improving RNA amplification include increasing the efficiency of ss cDNA synthesis and allowing for flexibility in the placement of bacteriophage transcriptional promoter sequences.
A new experimental procedure is described that utilizes a method of terminal continuation (TC) to attach an oligonucleotide primer of known sequence to the 3′ region of fs cDNA. Therefore, ss cDNA synthesis can be initiated by annealing a second oligonucleotide primer complementary to the attached oligonucleotide. By providing a known sequence at the 3′ region of fs cDNA and a primer complementary to it, hairpin loops will not form, avoiding use of the destructive S1 nuclease digestion step. ss cDNA synthesis can be performed with robust DNA polymerases, such as Taq, and the TC reaction is highly efficient. One round of amplification is sufficient for downstream genetic analyses. Furthermore, TC RNA transcription can be driven using a promoter sequence attached to either the 3′ or 5′ oligonucleotide primers, therefore transcript orientation can be ‘antisense’ or ‘sense’ (or both if using two separate promoter sequences), depending upon the design of the experimental paradigm.
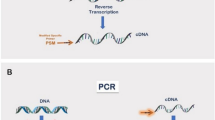
Amplification of genetic signals includes synthesizing fs cDNA complementary to the RNA template, subsequently generating ss cDNA complementary to the fs cDNA, and finally in vitro RNA transcription using the double-stranded (ds) cDNA as template. Synthesis of the fs cDNA complementary to template mRNA entails the use of two oligonucleotide primers, a poly-d(T) primer and a TC primer. The poly-d(T) primer is similar to conventional primers that exploit the poly-A+ sequence present on most mRNAs (Table 1). The TC primer consists of an oligonucleotide sequence at the 5′ terminus and a short span of three cytidine triphosphates (CTPs) at the 3′ terminus (Table 1). For antisense RNA amplification (similar to conventional aRNA methods), the bacteriophage promoter sequence is placed on the poly-d(T) primer. For the novel sense orientation, the promoter sequence is attached to the TC primer (Figure 1a).

Overview and analysis of the TC RNA amplification method. (a) A TC primer (containing a bacteriophage promoter sequence for sense orientation) and a poly-d(T) primer are added to the mRNA population to be amplified (green rippled line). First strand (blue line) synthesis occurs as an mRNA–cDNA hybrid is formed after reverse transcription and terminal continuation of the oligonucleotide primers. Following RNase H digestion to remove the original mRNA template strand, ss (red line) synthesis is performed using Taq polymerase. The resultant ds product is utilized as template for in vitro transcription, yielding high fidelity, linear RNA amplification of sense orientation (green rippled lines). (b) Schematic similar to (a) illustrating the TC RNA amplification procedure amplifying RNA in the antisense orientation (yellow rippled lines). (c)Bioanalysis of TC RNA amplification products to illustrate relative abundance and distribution. Note the long-range representation of transcripts from high (greater than 7.5 kb) to low (100 bp) on the electropherogram gel. Lane 1 depicts the molecular weight standards. Lane 2 is a control lane with a commercially available purified mRNA (arrowhead) used for amplification in lanes 3–5. Highly consistent amplification is observed in representative TC RNA amplification reactions shown in triplicate (lanes 3–5). Note the relative paucity of signal variation from the three separate reactions. Amplification efficiency of approximately 2500–3000-fold is estimated using the control poly-A+ mRNA as a starting template.
Materials and methods
RNA Preparation
RNAs, either total or mRNAs, are extracted from tissues, single cells, or bodily fluids from C57BL/6 mice and postmortem human brains as described in detail elsewhere.4, 10, 11 Tissue accession and accrual procedures are in accordance with IRB and IACUC standards at the Nathan Kline Institute and NYU School of Medicine. The TC RNA amplification method is especially useful when employed in conjunction with single-cell (or population-cell) laser capture microdissection (LCM) or microaspiration.4, 8, 16 For optimal extraction from fixed tissues (eg, formaldehyde or ethanol-based fixatives), single cells or populations are incubated in 250 μl of Proteinase K solution (Ambion, 50 μg/ml) for 12 hours at 37°C prior to extraction. RNA can be extracted using conventional organic methods (eg, Trizol reagent, Invitrogen) or semiautomated magnetic mRNA extraction methods (eg, KingFisher, ThermoLabsystems).17
RNA Amplification
Extracted RNAs are obtained from single cells/populations using microaspiration or LCM techniques.4, 8, 16 In addition to the microaspirated samples, a 1:20 × dilution series (sampling at 1:1, 1:20, 1:400, 1:8000, 1:160 000, and 1:3 200 000) of RNA extracted from mouse hippocampus was performed to assess signal recovery using the same TC RNA amplification procedures. Extracted RNAs are reverse transcribed in the presence of the poly-d(T) primer (10 ng/μl) and TC primer (10 ng/μl) in 1 × fs buffer (Invitrogen) 1 mM dNTPs, 5 mM DTT, 20 U of RNase inhibitor (Ambion) and 5 U reverse transcriptase (Superscript III; Invitrogen) in a final volume of 20 μl. Synthesized single-stranded cDNAs are converted into ds cDNAs by adding into the reverse transcription reaction the following: 10 mM Tris (pH 8.3), 50 mM KCl, 1.5 mM MgCl2, and 0.5 U Rnase H (Invitrogen) in a total volume of 99 μl. Samples are placed in a thermal cycler and ss synthesis proceeds as follows: Rnase H digestion step 37°C, 10 min.; denaturation step 95°C, 3 min., annealing step 50°C, 3 min.; elongation step 75°C, 30 min. 5 U (in one microliter) Taq polymerase (PE Biosystems) is added to the reaction at the initiation of the denaturation step (ie, hot start). Phenol:chloroform extracted and ethanol precipitated cDNAs are resuspended and drop dialyzed on 0.025 μm filter membranes (Millipore) against 50 ml of RNase-free H2O for 2 h. The sample is collected off the dialysis membrane and hybridization probes are synthesized by in vitro transcription using radiolabel, fluorescent, or biotin incorporation. For example, radiolabeling with 33P occurs in the following solution: 40 mM Tris (pH 7.5), 7 mM MgCl2, 10 mM NaCl, 2 mM spermidine, 5 mM of DTT, 0.5 mM of ATP, GTP, and CTP, 10 μM of cold UTP, 20 U of RNase inhibitor, and 40 μCi of 33P-UTP (Amersham Biosciences). The reaction is performed at 37°C for 4 h. Synthesized radioisotope-labeled RNA probes are added into the prehybridization solution directly without further purification.
cDNA Array Analysis
Labeled probes can be used for a variety of downstream applications including expression profiling in combination with a myriad of cDNA array platforms. We typically utilize single-cell microdissection in conjunction with TC RNA amplification to hybridize to custom-designed cDNA arrays consisting of 220–480 cDNAs and expressed sequence-tagged cDNAs (ESTs) for the analysis of neurodegeneration-related paradigms in mouse brain and human postmortem brain tissues.4, 8, 9, 10, 11, 16, 18 Specifically, 1 μg of linearized cDNA purified from plasmid preparations is adhered to arrays using high-density nitrocellulose (Amersham Biosciences). Each cDNA/EST on the custom-deigned cDNA arrays is verified by restriction digestion and sequence analysis. Mouse, rat, and human clones are employed successfully on the arrays. Arrays are prehybridized (4 h) and hybridized (12–24 h) in a solution consisting of 6 × SSPE, 5 × Denhardt's solution, 50% formamide, 0.1% sodium dodecyl sulfate (SDS), and denatured salmon sperm DNA (200 μg/ml) at 42°C in a rotisserie oven.4, 8, 9, 10, 11, 16, 18 Following hybridization, arrays are washed sequentially with 2 × SSC/0.1% SDS, 1 × SSC/0.1% SDS, and 0.5 × SSC/0.1% SDS for 20 min each at 42°C. TC hybridization signal intensity is detected by phosphor imaging. Specific signal intensity (minus background using the empty vector pBs) of TC-amplified RNA bound to each linearized cDNA is expressed as a ratio of the total hybridization signal intensity of the array, thereby minimizing variations due to differences in the specific activity of the probe and the absolute quantity of probe present.8, 10, 16, 18 Data analyzed in this manner do not allow the absolute quantitation of mRNA levels, but generate an expression profile of the relative changes in mRNA levels.2, 4, 9, 10, 11, 12, 18 Relative changes in individual mRNAs are analyzed using an ANOVA with post hoc analysis (Newmann–Keuls test) for individual comparisons.4, 10, 11 Expression levels are analyzed and clustered using a bioinformatics and graphics software package (GeneLinker Gold, Predictive Patterns Inc.). Differentially expressed genes are also clustered into functional protein categories for multivariate coordinate gene expression analysis.
Results and discussion
To evaluate the ability of the TC method to amplify RNA species, yield and size distribution profiles are estimated by bioanalysis (2100 Bioanalyzer and RNA 6000 Lab Chip, Agilent Technologies). This bioanalysis assay utilizes a capillary device and a sensitive fluorescent RNA dye for electrophoretic separation and detection of RNA profiles. Bioanalysis is more sensitive than denaturing gel electrophoresis or spectrophotometric analysis of minute quantities (eg, picogram to nanogram) of RNA species. However, variability in detection of extremely small quantities of RNA species does exist (see the Bioanalyzer 2100 website specifications at www.chem.agilent.com). Using a purified control poly-A+ (Invitrogen), highly reproducible, robust linear amplification is demonstrated (Figure 1c). Concordance analysis (n=6; control poly-A+ as starting template; series run twice in triplicate) is r2=0.97. Amplification efficiency (as estimated using bioanalysis) of approximately 2500–3000- fold is demonstrated with the control poly-A+ mRNA and approximately 1000–1500-fold is demonstrated using biological samples of RNA extracted from a variety of brain sources including post mortem hippocampus and basal forebrain (Figure 2a). The efficiency of RNA amplification appears independent of the method of RNA extraction, as both conventional organic extraction and magnetic bead extraction both yield high-quality transcripts for TC RNA amplification (Figure 2b). A dendrogram is presented for gene expression level data acquired using three independent samples of individual CA1 pyramidal neurons (Figure 3a). Results indicate a high degree of expression level similarity for high, moderate, and low expressed genes using the TC RNA amplification method (Figure 3a). Scatter plots demonstrate a linear relationship between the TC RNA input concentration and mean hybridization signal intensity as well as for individual clones (Figure 3b). These observations are strikingly similar to linearity data obtained using an aRNA amplification methodology.10 Furthermore, a serial dilution experiment using RNA extracted from mouse hippocampus illustrates high fidelity and recovery of the TC RNA amplification reaction, as the mean hybridization signal intensity was preserved across four out of five 20-fold serial dilutions before the TC RNA amplification reaction reached the limit of detection on the array platform (Figure 3c). Recovery of signal obtained from serial dilutions was also successful using individual cDNAs including CAMKII and CREB (data not shown). A caveat of the dilution experiment is that serially titrating down RNA extracted from a region enriched with RNA does not necessarily approximate the experimental conditions of extracting RNAs from minute tissue sources such as microaspirating a single cell or population of identified cells from fixed and/or frozen tissues.

TC RNA amplification of single cells and discrete regions in the human hippocampus. (a) Bioanalysis illustrating the robust effect of TC RNA amplification. Note the strong signal intensity from a range of high to low molecular weights (>7.5 kb to <100 bp), indicating amplification of large and small transcripts and the lack of a ‘blob’ of short oligonucleotides at the lower molecular weights that often occurs with conventional aRNA methodologies. Lanes 1 and 2 depict TC RNA amplification from paraffin-embedded hippocampus, whereas lanes 3 and 4 are single-cell amplifications of individual neurofilament-immunoreactive CA1 pyramidal neurons from the same human brain, microdissected from a 70% ethanol fixed, paraffin-embedded tissue section of the hippocampus by microaspiration. Lane 5 illustrates a p75 (low-affinity nerve growth factor receptor)-immunoreactive cholinergic basal forebrain (CBF) neuron microaspirated from a paraformaldehyde fixed, frozen section of the basal forebrain. (b) Two identical custom-designed arrays hybridized with 33P-UTP labeled probes generated using TC RNA amplification from adjacent samples from the same case (corresponding lanes 1 and 2 of panel (a)) using organic (top panel) and magnetic extraction methods (bottom panel). No quantitative differences have been observed between extraction conditions.

Expression profile distribution and linearity following TC RNA amplification. (a) Dendrogram illustrating expression levels for >230 genes from individual CA1 pyramidal neurons aspirated from the hippocampus of postmortem human brain. TC1a–TC1c represent TC RNA amplifications run in triplicate from adjacent cells. Note the wide range of color-coded relative expression levels and the high degree of similarity of expression profiles for the three samples. (b) Scatter plots demonstrating a linear relationship between TC-amplified RNA input concentration and mean hybridization signal intensity of 96 cDNAs/ESTs on a custom-designed array (r2=0.98) as well as for an individual clone (CREB is depicted; r2=0.96). (c) Histogram illustrating mean hybridization signal intensity with standard deviations from mouse hippocampus (run three to four times per dilution) to illustrate the high level of signal recovery following serial 20-fold dilutions. Average calculated recoveries from serial dilutions: 1:1; 1:20 (actual recovery 1:26.4±6.8), 1:400 (actual recovery 1:457.4±44.6), 1:8000 (actual recovery 1:5864.2±478.3), 1:160 000 (actual recovery 1:126 139±1788.6), and 1:3 200 000 (N/D). Note the logarithmic scale and limit of detection at the final dilution.
TC RNA amplification produces robust and reproducible hybridization signal intensity after one round of amplification. The threshold of detection of genes with low hybridization signal intensity is also greatly increased. For example, several genes that are at the limit of detection using conventional aRNA can be readily observed with the TC method (Figure 4a). An approximate 3.5–4-fold increase in total, normalized hybridization signal intensity is observed on custom-designed cDNA arrays (Figure 4b). Importantly, the increased sensitivity appears greatest for genes with relatively low abundance (Figure 4a). Moreover, background hybridization (ie, hybridization to empty vector) is significantly greater using aRNA than the TC RNA amplification. Additionally, we observed that ‘hot start’ ss synthesis significantly reduced background. No overall quantitative differences have been detected in total hybridization signal intensity between 3′ (antisense orientation) and 5′ (sense orientation) TC RNA amplification reactions (Figure 4b). However, individual genes have been identified that are expressed differentially. For example, the neurofilament genes NF-M and NF-H display a relative increase in the 3′ TC amplification protocol as compared to the 5′ TC RNA amplification protocol using single neurofilament-immunoreactive CA1 pyramidal neurons from normal human hippocampus (Table 2). In contrast, the nicotinic acetylcholine receptor subunits nAchr α1 (CHRNa4) and nAchr α7 (CHRNa7) display a relative increase in the 5′ TC amplification vs the 3′ TC RNA amplification. Therefore, hybridization signal intensity of individual genes and/or cDNAs can vary between the 3′ and 5′ TC RNA amplification, yet total populations of mRNAs have similar expression levels, indicating relatively equivalent signal detection efficiency for TC RNA antisense and sense amplification.

Comparison of TC RNA amplification hybridization signal intensity with conventional RNA amplification. The left panel illustrates a comparison of two adjacent regions from the same hippocampal tissue section amplified by TC RNA amplification (top panel) and by conventional RNA amplification (bottom panel). The relative hybridization signal intensity of several low, moderate, and higher expressing genes is depicted. All steps in the procedures are performed identically, including hybridization time, identical washing regimens, and source of the custom-designed arrays. Significant increases in gene expression levels are detected (* denotes P<0.05 and **denotes P<0.01) for β-actin (β-act*), β-tubulin (β-tub*), neurofilament subunits (NF-L*, NF-M**, and NF-H**), 4-repeat tau (tau44*), nestin (nest*), utrophin (utro**), AMPA glutamate receptors (GluR1*, GluR3*), and kainite receptors (GluR5**, GluR6**, and GluR7**). Other clones represented on the arrays include: γ-actin (g-act), α-tubulin (a-tub), microtubule-associated proteins 2 and 5 (MAP2 and MAP5), largest isoform tau and three-repeat tau (tau40 and tau43), internexin (INX), AMPA glutamate receptors (GluR2 and GluR4), and kainate receptors (KA1 and KA2). The right panel depicts a histogram illustrating mean hybridization signal intensity for custom-designed cDNA arrays (containing 230 cDNAs/ESTs) using 5′ TC RNA amplification (sense orientation), 3′ TC RNA amplification (antisense orientation), and aRNA amplification from the same hippocampal tissue sections. A total of 12 reactions from three brains (four RNA amplification reactions per brain) are performed for each method and reported as normalized hybridization signal intensity±s.e.m. A 3.5–4-fold increase in signal intensity is observed for both 5′ and 3′ TC RNA amplification methods (see Table 2 for further details).
A mechanism for the TC primer to anneal preferentially to 5′ regions of transcripts is being investigated. Based upon sequence analysis of TC RNA-amplified products from human and mouse brains, we hypothesize that the TC primer, with its span of CTPs (guanosine triphosphates (GTPs) work as well; data not shown) anneals preferentially within CpG islands. CpG islands are nonmethylated GC-rich regions of the genome that tend to include the 5′ end of genes and are found at a significantly less frequency (CpGs are 25% less frequent than predicted) throughout the rest of the genome.19, 20 Estimates suggest that above 60% of all human genes are located near CpG islands.19, 20 It is apparent that the TC primer has to base pair with the complementary CTPs or GTPs at the termination site of the reverse transcription reaction in order to provide a short template for DNA synthesis to continue. The reverse transcriptase reaction has been reported to add a few dCTPs nonspecifically at the end of mRNA template.21, 22 Essential structural requirements of the TC primer include a short stretch of CTPs or GTPs at the 3′ end. Replacement of the CTPs/GTPs with adenosine triphosphates (ATPs) or thymidine triphosphates (TTPs) vastly diminishes the TC reaction.8 In addition, random base pairing of ATPs and TTPs with complementary T's and A's in mRNAs may interrupt a proper reverse transcription process that is essential for generation of the fs cDNA.
The development of techniques such as LCM23, 24 and single-cell microaspiration4, 9, 10, 11, 16, 18 enables accession of minute amounts of starting materials. However, an RNA amplification procedure is requisite to generate significant hybridization signal intensity for cDNA microarray platforms. PCR is not suitable for this application because exponential amplification cannot preserve optimally the quantitative relationships between the expressed genes,5, 25 a parameter critical for expression profiling. Hybrid protocols using PCR-based technology combined with aRNA have yielded positive results.26 The TC RNA amplification method is a protocol that meets both requirements of amplifying genetic signals and preserving the quantitative relationships between expressed genes. Compared to conventional RNA amplification methodologies, the TC method is more robust (approximately 3.5–4-fold stronger signal intensity) with less background hybridization and significantly less laborious (the procedure takes approximately 2 days to complete). Bioanalysis is used as an analytical technique to estimate quantity and relative distribution of RNA species amplified by the TC method. Although its use has been validated for larger amounts of input RNAs that undergo subsequent amplification, single-cell RNA amplification approaches the limit of sensitivity of the assay.2, 4, 27 Other analytical methods such as denaturing gel electrophoresis, spectrophotometry, and fluorimetric analysis are either less sensitive, or approach limits of sensitivity as well.
The present series of results are primarily from brain tissues accrued from post mortem human samples and animal models of neurodegeneration. The brain is an obvious site for single-cell RNA exploratory studies, due to the plethora of cell types and intricate connectivity of regions. The TC RNA amplification methodology, however, has much broader applications. Virtually, any in vivo or in vitro paradigm can be employed for TC RNA amplification. Disciplines that will benefit from this technology include, but are not restricted to, cancer biology, development, drug discovery, and a myriad applications where input sources of RNA are limited and/or cell-type specific. Current tissue sources include human, monkey, rat, and mouse tissues, and other sources are being investigated. The requirement appears to be polyadenylation on the 3′ end (identical to conventional RNA amplification), and a stretch of C's or G's at the 5′ region (either through CpG islands or other structures).
In summary, TC RNA amplification provides a novel technical advance to amplify minute amounts of mRNAs for subsequent microarray or proteomic-based analyses. Conceivably, the downstream applications of synthesized RNA are expanded, and the direction of RNA can be chosen according to the need of the end-user. Thus, the TC RNA amplification methodology provides high fidelity, linear RNA amplification that may become an essential, relatively straightforward laboratory technique to amplify antisense or sense RNA transcripts.
References
Brown PO, Botstein D . Exploring the new world of the genome with DNA microarrays. Nat Genet 1999;21(Suppl):33–37.
Eberwine J, Kacharmina JE, Andrews C, et al. mRNA expression analysis of tissue sections and single cells. J Neurosci 2001;21:8310–8314.
Geschwind DH . Mice, microarrays, and the genetic diversity of the brain. Proc Natl Acad Sci USA 2002;97:10676–10678.
Ginsberg SD . Gene expression profiling using single cell microdissection combined with cDNA microarrays, In: Geschwind DH (ed). DNA Microarrays: The New Frontier in Gene Discovery and Gene Expression Analysis. Society for Neuroscience Press: Washington, DC, 2001, pp 61–70.
Van Deerlin VMD, Ginsberg SD, Lee VM-Y, et al. The use of fixed human post mortem brain tissue to study mRNA expression in neurodegenerative diseases: applications of microdissection and mRNA amplification, In: Geschwind DH, Gregg JP (Eds). Microarrays for the Neurosciences: An Essential Guide, MIT Press: Boston, MA 2002, pp 201–235.
Dixon AK, Lee K, Richardson PJ, et al. Single cell expression analysis — pharmacogenomic potential. Pharmacogenomics 2002;3:809–822.
Freeman TC, Lee K, Richardson PJ . Analysis of gene expression in single cells. Curr Opin Biotechnol 1999;10:579–582.
Ginsberg SD, Che S . RNA amplification in brain tissues. Neurochem Res 2002;27:981–992.
Hemby SE, Ginsberg SD, Brunk B, et al. Gene expression profile for schizophrenia: discrete neuron transcription patterns in the entorhinal cortex. Arch Gen Psychiatry 2002;59:631–640.
Ginsberg SD, Hemby SE, Lee VM-Y, Eberwine JH, and Trojanowski JQ . Expression profile of transcripts in Alzheimer's disease tangle-bearing CA1 neurons. Ann Neurol 2000;48:77–87.
Ginsberg SD, Crino PB, Hemby SE, et al. Predominance of neuronal mRNAs in individual Alzheimer's disease senile plaques. Ann Neurol 1999;45:174–181.
Eberwine J, Yeh H, Miyashiro K, et al. Analysis of gene expression in single live neurons. Proc Natl Acad Sci USA 1992;89:3010–3014.
Sambrook J, Fritsch E, Maniatis T . Molecular Cloning: A Laboratory Manual, 2nd edn. Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, 1989.
Luzzi V, Holtschlag V, Watson MA . Expression profiling of ductal carcinoma in situ by laser capture microdissection and high-density oligonucleotide arrays. Am J Pathol 2001;158:2005–2010.
Gubler U, Hoffman BJ . A simple and very efficient method for generating cDNA libraries. Gene 1983;25:263–269.
Mufson EJ, Counts SE, Ginsberg SD . Single cell gene expression profiles of nucleus basalis cholinergic neurons in Alzheimer's disease. Neurochem Res 2002;27:1035–1048.
Ginsberg SD . High quality mRNA extraction with KingFisher for molecular fingerprinting. Focus 2002;1:5.
Hemby SE, Trojanowski JQ, Ginsberg SD . Neuron-specific age-related decreases in dopamine receptor subtype mRNAs. J Comp Neurol 2003;456:176–183.
Antequera F, Bird A . Number of CpG islands and genes in human and mouse. Proc Natl Acad Sci USA 1993;90:11995–11999.
Cross SH, Clark VH, Bird AP . Isolation of CpG islands from large genomic clones. Nucleic Acids Res 1999;27:2099–2107.
SMART technology overview. Clontechniques 2002, Jan: 22–23.
Matz M, Shagin D, Bogdanova E, et al. Amplification of cDNA ends based on template-switching effect and step-out PCR. Nucleic Acids Res 1999;27:1558–1560.
Bonner RF, Emmert-Buck M, Cole K, et al. Laser capture microdissection: molecular analysis of tissue. Science 1997;278:1481–1483.
Emmert-Buck MR, Bonner RF, Smith PD, et al. Laser capture microdissection. Science 1996;274:998–1001.
Brail LH, Jang A, Billia F, et al. Gene expression in individual cells: analysis using global single cell reverse transcription polymerase chain reaction (GSC RT-PCR). Mutat Res 1999;406:45–54.
Wang E, Miller LD, Ohnmacht GA, et al. High-fidelity mRNA amplification for gene profiling. Nat Biotechnol 2000;18:457–459.
Polacek DC, Passerini AG, Shi C, et al. Fidelity and enhanced sensitivity of differential transcription profiles following linear amplification of nanogram amounts of endothelial mRNA. Physiol Genomics 2003;13:147–156.
Acknowledgements
We thank Mr John T Le, and Ms Irina Elarova for expert technical assistance. Support for this project comes from the NIH (NS43939 and CA94520; SDG) and the Alzheimer's Association (SDG). We also express our appreciation to the families of the patients studied here who made this research possible.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Che, S., Ginsberg, S. Amplification of RNA transcripts using terminal continuation. Lab Invest 84, 131–137 (2004). https://doi.org/10.1038/labinvest.3700005
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700005
Keywords
This article is cited by
-
Frontal cortex and striatal cellular and molecular pathobiology in individuals with Down syndrome with and without dementia
Acta Neuropathologica (2019)
-
Locus coeruleus cellular and molecular pathology during the progression of Alzheimer’s disease
Acta Neuropathologica Communications (2017)
-
Expression profile analysis of hippocampal CA1 pyramidal neurons in aged Ts65Dn mice, a model of Down syndrome (DS) and Alzheimer’s disease (AD)
Brain Structure and Function (2015)
-
Alterations in discrete glutamate receptor subunits in adult mouse dentate gyrus granule cells following perforant path transection
Analytical and Bioanalytical Chemistry (2010)
-
An alternative method to amplify RNA without loss of signal conservation for expression analysis with a proteinase DNA microarray in the ArrayTube® format
BMC Genomics (2006)
