
Abstract
In mouse senile amyloidosis, apolipoprotein A-II polymerizes into amyloid fibrils (AApoAII) and deposits systemically. Peripheral injection of AApoAII fibrils into young mice induces systemic amyloidosis (Higuchi et al, 1998). We isolated AApoAII amyloid fibrils from the livers of old R1.P1-Apoa2c mice and injected them with feeding needles into the stomachs of young R1.P1-Apoa2c mice for 5 consecutive days. After 2 months, all mice had AApoAII deposits in the lamina propria of the small intestine. Amyloid deposition extended to the tongue, stomach, heart, and liver at 3 and 4 months after feeding. AApoAII suspended in drinking water also induced amyloidosis. Amyloid deposition was induced in young mice reared in the same cage for 3 months with old mice who had severe amyloidosis. Detection of AApoAII in feces of old mice and induction of amyloidosis by the injection of an amyloid fraction of feces suggested the propagation of amyloidosis by eating feces. Here, we substantiate the transmissibility of AApoAII amyloidosis and present a possible pathogenesis of amyloidosis, ie, oral transmission of amyloid fibril conformation, where we assert that exogenous amyloid fibrils act as templates and change the conformation of endogenous amyloid protein to polymerize into amyloid fibrils.
Similar content being viewed by others


Introduction
Amyloidosis refers to a group of protein misfolding diseases (Westermark, 1998). Various innocuous and soluble proteins in physiological conditions polymerize to insoluble amyloid fibrils in several serious diseases, including Alzheimer's disease, type II diabetes, amyloidosis associated with inflammation, chronic dialysis, prion diseases, and familial and sporadic amyloid diseases (Booth et al, 1997; Glenner, 1980). Although the diverse amyloid proteins have unrelated sequences, they can all form fibrils with a similar ultrastructural appearance and a similar core structure comprising β-pleated sheets. In mouse senile amyloidosis, apolipoprotein A-II (apoA-II), the second most abundant apolipoprotein in serum high-density lipoproteins (HDL), polymerizes to amyloid fibrils (AApoAII) and deposits systemically, which is associated with aging (Higuchi et al, 1986, 1999). Three alleles of the apoA-II gene (Apoa2a, Apoa2b, and Apoa2c) code three variants of apoA-II protein (types A, B, and C) with different amino acid substitutions at four positions among inbred strains of mice (Higuchi et al, 1991a). One amino acid substitution, proline to glutamine, at position 5 (P5Q) in type C apoA-II protein markedly accelerates age-associated deposition of AApoAII (Higuchi et al, 1991b, 1995, 1996; Naiki et al, 1993).
Nucleation-dependent polymerization is postulated to be a model that explains well the kinetics of fibrillizaton of amyloid proteins and the pathogenesis of amyloidosis. This model comprises two phases: nucleation and extension. Nucleus formation requires a series of monomer association steps, which are thermodynamically unfavorable. Thus, the nucleation phase is considered the rate-limiting step in the development of amyloidosis. Once the nucleus has been formed, further addition of monomers becomes thermodynamically favorable, resulting in the rapid extension of amyloid fibrils. The dramatically hastened fibril formation in vitro by the addition of amyloid fibrils (nucleus) into the solution of amyloid protein monomers represents nucleation-dependent polymerization (Horwich and Weissman, 1997; Jarrett and Lansbury, 1993; Kelly and Lansbury, 1994; Rochet and Lansbury, 2000; Weissmann, 1999). The addition of AApoAII amyloid fibrils into apoA-II solutions removed the nucleation phase and hastened the fibril extension in vitro (Naiki et al, 1991). Furthermore, intravenous or intraperitoneal injections of AApoAII amyloid fibrils accelerated dramatically the progression of amyloidosis in young mice with type C apoA-II protein (Higuchi et al, 1998).
Prion diseases are associated with the accumulation of a conformational isomer (PrPSc) of host-derived prion protein (PrPC). PrPSc forms amyloid fibrils. Prion transmission or propagation involves the conversion of cellular PrPC into PrPSc with an increase in its β-sheet secondary structure content. According to the protein-only hypothesis, introduction of the abnormal conformer PrPSc into the organism would result in the conversion of PrPC into a likeness of itself. The nucleation-dependent polymerization model provides a feasible mechanism for the conformational conversion of PrPC to PrPSc (Prusiner, 1997; Prusiner et al, 1998; Supattapone et al, 1999).
Here, we attempted to verify the transmission of non-prion amyloidosis using a mouse model and found new evidence for the transmissibility through digestive tracts in AApoAII amyloidosis. From these findings, we postulate that invasion of exogenous amyloid fibrils or amyloid fibril-like proteins might be a possible mechanism for initiation of mouse senile amyloidosis.
Results
Oral Transmission of AApoAII Amyloidosis
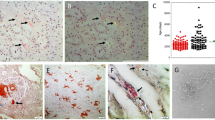
Sonicated AApoAII amyloid fibrils (20 μg) were injected once daily with feeding needles for 5 days into the stomachs of 2-month-old R1.P1-Apoa2c mice. At 2 months after injection, all mice (13/13) had AApoAII deposits in the lamina propria of the small intestine (Fig. 1A). The amyloid deposition was found mainly in the jejunum and ileum (Fig. 2A) and rarely in the duodenum and large intestine. After 2 months the intensity of the amyloid deposition increased. Amyloid deposition extended to the papillary layer of the tongue, the glandular portion and the squamous-glandular junction of the stomach, the collecting tubules in the papillae of the kidneys, the interstitium of the heart muscles, and to the Disse spaces of the periportal sinusoid of the liver (Fig. 2B and C). By contrast, no amyloid deposition was detected in the mice injected with saline into the stomach, except for in one mouse who had a slight amyloid deposition in the small intestine at 3 months after the injection (Fig. 1B). When 100-μg and 20-μg AApoAII fibril applications were injected into the stomachs at one try, half (6/12) and none (0/6) of the mice had amyloid deposition after 2 months, respectively (data not shown).

Oral transmission of AApoAII amyloidosis. A, Sonicated AApoAII fibrils were injected into the stomachs of 2-month-old male R1.P1-Apoa2c mice. A total of 37 mice (•) were injected with AApoAII fibrils, and 13, 20, and 4 mice were killed at 2, 3, and 4 months after the first injection. The intensity of the AApoAII amyloid deposition was determined using the amyloid index (AI) as a parameter. All 37 mice were amyloid-positive, and AI increased with time after the injection. B, A total of 27 mice (○ were injected with 0.2 ml saline, and 10, 12, and 5 mice were killed at 2, 3, and 4 months after the first injection, respectively. Only 1 mouse was amyloid-positive.

AApoAII deposition in the mouse tissues with intragastric injection of AApoAII amyloid fibrils. AApoAII deposits were immunohistochemically identified. A, AApoAII deposits (Grade 2) in the lamina propria of the jejunum at 2 months after injection. B, AApoAII deposits (Grade 3) in the lamina propria of the small intestine at 3 months after AApoAII injection. C, AApoAII deposits (Grade 2) in the papillary layer of the tongue at 3 months after injection. D, Intensive AApoAII deposits (Grade 4) in the lamina propria and submucosa of jejunum of 17-month-old R1.P1-Apoa2c mice reared with young mice. Scale bar equals 25 μm.
Amyloid deposits were not detected in any mice 1 month after drinking water containing sonicated AApoAII fibrils. AApoAII deposits were detected in the small intestine of all R1.P1-Apoa2c mice 2 months after drinking AApoAII fibrils suspended in water (Table 1). Amyloid deposits were observed in the lamina propria and submucosa of the small intestine, in the collecting tubules in the papillae of the kidneys, and in the squamous-glandular junction of the stomach. The intensity (amyloid index [AI] = 0.35 ± 0.14) and tissue distribution of amyloid deposition were almost the same at 2 months as those in mice injected with AApoAII into the stomach (AI = 0.41 ± 0.15 at 2 months). Spontaneous amyloid deposition was not observed in R1.P1-Apoa2c mice who drank distilled water (DW), except for in 2 mice at 4 months who had slight amyloid deposition in the small intestine, stomach, and tongue.
Transmission of Amyloidosis from Old Mice to Young Mice
Nine to 14 female 2-month-old R1.P1-Apoa2c mice were separated into two groups. Mice in Group B were kept in the same cage for 3 months with 2 old (12- to 19-month-old) female SAMR1; mice in Group C were kept with 2 old (12- to 19-month-old) R1.P1-Apoa2c mice. The transmissibility of amyloidosis was examined in five independent experiments (Fig. 3). Amyloid deposition was detected in 25 of 28 young R1.P1-Apoa2c mice reared with old R1.P1-Apoa2c mice. By contrast, amyloid deposition was detected in only 1 of 28 mice reared with old SAMR1 mice. Amyloid deposition in the small intestine, tongue, and stomach was observed in 25, 15, and 10 of 25 young R1.P1-Apoa2c mice, respectively. The intensity of amyloid deposition (AI = 0.35 ± 0.21) was significantly lower than that in mice at 3 months after injection of AApoAII into the stomach (AI = 0.79 ± 0.25, p < 0.05 by Mann-Whitney U-test). No old SAMR1 mice reared with young mice had amyloid deposition in any tissues, but severe and systemic amyloid deposition was confirmed in all old R1.P1-Apoa2c mice with AI of 3.28 ± 0.38 (Fig. 2D). These data suggest that amyloidosis in the old R1.P1-Apoa2c mice might be transmitted to young R1.P1-Apoa2c mice.

Transmission of amyloidosis to the young mice reared with old mice. Young female R1. P1-Apoa2c mice were separated into two groups. Group B mice (○ were reared in the same cage with two old female SAMR1 mice with type B apoA-II for 3 months (17- and 19-month-old SAMR1 mice in Experiment 1; 12- and 19-month old mice in Experiment 2; 14- and 17-month-old mice in Experiment 3; 14- and 15-month- old mice in Experiment 4; and 14- and 18-month-old mice in Experiment 5). Group C mice (•) were reared in the same cage with two old female R1.P1-Apoa2c mice (17- and 19-month-old R1.P1-Apoa2c mice in Experiment 1; 12- and 19-month- old mice in Experiment 2; 14- and 17-month-old mice in Experiment 3; 14- and 16-month-old mice in Experiment 4; and 17- and 19-month-old mice in Experiment 5). After 3 months, all young and old mice were killed and amyloid depositions were determined.
AApoAII in the Feces
Western blot analysis after one-dimensional Tris-Tricine-Urea/SDS PAGE of the amyloid fibril fractions isolated from old R1.P1-Apoa2c feces revealed two anti-apoA-II reactive bands with molecular weights of 6.8 and 30 kilodalton (KDa) (Fig. 4A). These molecular weights corresponded to the apoA-II monomer and its tetrameric form, respectively. These bands were not detected in the water suspensions of old SAMR1 feces. Western blot analysis after two-dimensional (2-D) PAGE revealed that the tetrameric form was almost completely depolymerized to the monomeric form. AApoAII in the feces comprised four isoforms: one major and three minors (Fig. 4B). The pattern of these isoforms was similar to those found in AApoAII isolated from liver of R1.P1-Apoa2c (data not shown).

A, Western blot of feces proteins. Two anti-apoA-II reactive bands were detected in amyloid fractions from feces of an old R1.P1-Apoa2c mouse (Lane 1). The molecular weight corresponded to apoA-II monomer (6.8 KDa) and its tetrameric form (30 KDa). No anti-apoA-II reactive bands were detected in water suspensions from old SAMR1 feces (Lane 2). B, Two-dimensional immunoblot of feces proteins from old R1.P1-Apoa2c mouse. The acidic part of the strip is shown here. Anti-apoA-II reactive spots were detected with molecular weights of 6.8 KDa and 30 KDa. ApoA-II monomer in feces comprised four isoforms: one major (arrow) and three minor (thin arrows). The isoelectric point (pI) measurements of the four spots were approximately 4.9. Two small faint spots of 30 KDa were detected (arrow heads).
Amyloid fractions isolated from old R1.P1-Apoa2c feces were pooled and injected intraperitoneally (0.1 mg) into 2-month-old R1.P1-Apoa2c mice. Amyloid deposition was observed in the small intestine and tongue in eight of nine injected mice (Fig. 5), although the degree of amyloid deposition was not high (AI = 0.43 ± 0.28). AApoAII deposition was not observed in mice injected with DW or feces amyloid fractions into the stomach.

AApoAII deposition in the mouse tissues with intraperitoneal injection of amyloid fibril fractions of feces. Two groups of 2-month-old male R1.P1-Apoa2c mice were injected with sonicated amyloid fibril fractions of feces (0.1 mg/0.1 ml) into the abdominal cavities (IP) (n = 9) and into the stomachs (Stom) (n = 6). Distilled water (DW) was injected intraperitoneally into six R1.P1-Apoa2c mice as a control. Mice were killed at 2 months after the first injection. The intensity of the AApoAII deposition is represented as AI.
Discussion
Although the etiology and pathogenesis of amyloid disease are not fully understood, drastic structural changes of the amyloid protein from the normal and soluble forms to the unique β-sheet fibrils may be the most important event in amyloid diseases. However, the mechanism of the structural changes is poorly understood. Acceleration of amyloid fibril formation in test tubes by pre-existing amyloid fibrils was shown in many amyloid proteins, including human Aβ peptides (Naiki and Nakakuki, 1996); β2-microglobulin, (Hashimoto et al, 1999); immunoglobulin light chain (Ionescu-Zanetti et al, 1999); Huntingtin (Scherzinger et al, 1999); yeast prion protein (Sup35p and Ure2p) (DePace et al, 1998; Santoso et al, 2000); and mouse apoA-II protein (Naiki et al, 1991). In vivo, we showed that intravenous and intraperitoneal injection of AApoAII amyloid fibrils markedly accelerated mouse senile amyloidosis (Higuchi et al, 1998). Further, it has recently been reported that intravenous injection of amyloid-like fibrils made from synthetic peptides of transthyretin (Johan et al, 1998) and denatured silk (Kisilevsky et al, 1999) accelerates mouse AA amyloidosis. These findings have led us to the proposal that the invasion of exogenous amyloid fibrils or amyloid fibril-like proteins may act as a template or seed for fibril formation, subsequently inducing amyloidosis. Here, we verified this proposal using mouse senile (AApoAII) amyloidosis. AApoAII is found in the mouse but not in the human. However, whole molecules of apoA-II (78 amino acids) polymerize into AApoAII amyloid fibrils without proteolysis and deposit systemically, which provides a relatively simple and important model with which to study the common mechanism of amyloid diseases.
A series of oral injections of AApoAII amyloid fibrils (20 μg/day for 5 days) induced amyloidosis. Previously, we showed that a single intravenous injection of 100 μg AApoAII hastens amyloid deposition rapidly in the whole body, and the AI increased to 2.70 at 3 months after injection (Higuchi et al, 1998). Compared with intravenous injection, intragastric injection induced amyloidosis more slowly (AI = 0.75 at 3 months). AApoAII amyloid deposition characteristically started from the lamina propria in the jejunum. This suggested that small amounts of AApoAII may be absorbed through the villi of the jejunum and act as a template there. A single injection of a relatively large amount of AApoAII fibrils (100 μg) induced amyloidosis in only half of the injected mice. Repeated oral injection of tissue homogenates containing prion protein was reported to propagate prion disease more effectively than a single injection (Diringer et al, 1998).
It was previously thought that induction of amyloidosis by oral injection of amyloid fibrils (transmission of amyloidosis) does not occur in any kind of amyloidosis other than prion diseases (Caughey, 2000; Chesebro, 1998). Here, we demonstrate for the first time the transmissibility of amyloid fibrils by means other than prion proteins. It has been reported that oral injection of amyloid enhancing factor (AEF) accelerates experimentally induced mouse AA amyloidosis (Elliott-Bryant and Cathcart, 1998). Although SDS-PAGE analysis of AApoAII fibrils used in our experiments showed that apoA-II was the major and sole band (Higuchi et al, 1997; Naiki et al, 1989), the possibility that contaminant molecules in AApoAII amyloid fibrils might induce amyloidosis cannot be ruled out. Because denaturation of amyloid fibrils inactivates their template ability (Higuchi et al, 1998), a method for further purification is not known at this time. The use of recombinant-derived apoA-II would confirm the amyloidosis-enhancing ability of AApoAII fibrils, but we have not succeeded yet in making fibrils from recombinant proteins.
Surprisingly, young R1.P1-Apoa2c mice had amyloidosis after being reared for 3 months with old, amyloid-laden R1.P1-Apoa2c mice. This transmissibility of amyloidosis from old mice to young mice was investigated carefully: (a) we performed five independent experiments, and (b) we used female mice to avoid biting and scratching in fights because AA amyloid fibrils or AEF induced by inflammation through injury may promote AApoAII deposition. Amyloid deposition in the young mice was revealed in all five experiments. Fights were not observed during the experiments, and AA amyloid deposition was not detected in any of the mice. The mechanism for amyloidosis transmission from old to young mice was not readily apparent. However, we considered that one possible route for amyloidosis transmission might be the eating of feces, which is a characteristic behavior of mice. Severe amyloid deposition in the lamina propria of the small intestine of old mice may be excreted to the feces (Fig. 2D). AApoAII was detected in the feces of old R1.P1-Apoa2c mice, but not in old SAMR1 mice. The similar AApoAII-isoforms in the liver and feces suggest that there was no modification or degradation of AApoAII in the feces. To evaluate this trans-fecal hypothesis, we isolated amyloid fibril fractions from the feces and found that injection of them into young mice induced AApoAII deposition.
Materials and Methods
Animals
R1.P1-Apoa2c is a congenic strain of mice that has the amyloidogenic allele of the apoA-II gene (Apoa2c) in the SAMP1 strain on the genetic background of the amyloid-resistant SAMR1 strain (Higuchi et al, 1993, 1995). R1.P1-Apoa2c and SAMR1 were maintained by sister-brother mating under conventional conditions at 24° C ±2° C with a light-controlled regimen (12-hour light/dark cycle). A commercial diet (CE-2; Nihon CLEA, Tokyo, Japan) and tap water were available ad libitum.
Animal studies were conducted in accordance with the guidelines for the use of laboratory animals of both Shinshu University School of Medicine and Kyoto University. Mice were killed by cardiac puncture under diethyl ether anesthesia.
Isolation of Amyloid Fibrils (AApoAII)
The AApoAII amyloid fibril fraction was isolated as a water suspension from the livers of 18- to 21-month-old R1.P1-Apoa2c mice with severe amyloid deposition. Isolated amyloid fibril fractions were further purified by sucrose gradient ultracentrifugation as described elsewhere (Higuchi et al, 1997). Purified AApoAII fibrils were resuspended in DW at a concentration of 1.0 mg/ml by mixing thoroughly with an ultradispenser (Ultra-Turrax T25; Janke & Kunkel Gmbh Company, KG, Staufen, Germany). One milliliter of this solution was placed into an Eppendorf tube (size, 1.5 ml) and sonicated on ice for 30 seconds with a Microtip-equipped Astrason Ultrasonic Processor W-380 (Heat System-Ultrasonics Inc., Farmingdale, New York) at maximum power. This procedure was repeated five times at 30-second intervals. Sonicated AApoAII samples were used immediately.
Oral Injection of AApoAII
Male R1.P1-Apoa2c mice were weaned at 4 weeks of age. AApoAII amyloid fibrils were sonicated and diluted with DW to a concentration of 0.1 mg/1.0 ml. Using disposable feeding needles with silicon tips and Teflon tubes (Fuchigami Ltd., Kyoto, Japan), 0.2 ml of diluted AApoAII was introduced into the stomachs of 2-month-old male R1.P1-Apoa2c mice. Once-daily intragastric injections were given for 5 consecutive days. Mice were killed by cardiac puncture under diethyl ether anesthesia at 2, 3, and 4 months after the first injection of AApoAII. Five 2-month-old male mice in each cage were deprived of drinking water for 24 hours before they were given 2.5-mg AApoAII fibrils suspended in 50 ml DW. After we checked that mice had consumed completely the contents of the bottles (usually within 2 days), DW was given without limit. At 1, 2, and 4 months after the injections, mice were killed and amyloid depositions were determined.
Transmission of Amyloidosis from Old Mice to Young Mice
Four to seven 2-month-old female R1.P1-Apoa2c mice were kept in the same aluminum cage (20 cm wide × 30 cm high × 10 cm deep) spread with thin wood chips with two old (12- to 19-month-old) female SAMR1 or R1.P1-Apoa2c mice for 3 months and then killed. The transmutability of amyloidosis was examined in five independent experiments.
Detection of Amyloid Deposition
Tissues of the whole body were fixed in 10% neutral-buffered formalin, embedded in paraffin, and cut into 4-μm sections. Deposition of amyloid fibrils was identified by green birefringence in Congo red-stained sections under polarizing microscopy. Amyloid fibril proteins, AApoAII, were immunohistochemically identified by the avidin-biotinylated horseradish peroxidase complex (ABC) method with specific antiserum against AApoAII (Higuchi et al, 1983). The intensity of the AApoAII amyloid deposition was determined using the amyloid index (AI) as a parameter. AI was determined to represent the average of the AApoAII deposition graded 0 to 4 in sections of seven organs (liver, spleen, skin, heart, stomach, small intestine, and tongue) stained with Congo red after immunohistochemical confirmation of the AApoAII deposition (Higuchi et al, 1998). Two observers who had no information about the examined tissue graded AI independently and averaged AI for each mouse.
AApoAII in the Feces
Fresh feces were collected from the cages in which three to five old (15- to 20-month-old) R1.P1-Apoa2c and SAMR1 mice had been kept for 24 hours. Amyloid fibril fractions were isolated as water suspensions from the feces by the same procedure as for isolation of AApoAII from mouse liver, but sucrose gradient ultracentrifugation was omitted. AApoAII was detected by Western blot using anti-apoA-II rabbit serum after 16.5% SDS-PAGE of 5 μg protein with a Tris-Tricine buffer system using 8 M urea instead of glycerol (Schagger and von Jagow, 1987). Rehydrated Immobiline Drystrip (pH range 3–10 NL, 13cm; Amersham Pharmacia Biotech, Tokyo, Japan) was applied to the Multiphor II 2-D electrophoresis system (Amersham Pharmacia Biotech), and isoelectric focusing with 180 μg protein was conducted according to the manual. After equilibrating the strip with dithiothreitol (DTT) and iodoacetamide (IAA) equilibrating buffer, two-dimensional 16.5% Tris-Tricine-Urea/SDS-PAGE was performed. Then the separated spots were electrotransferred to polyvinylidene difluoride (PVDF) membrane (Bio-Rad, Hercules, California) and immuno-detected by anti-apoA-II antiserum. Amyloid fractions (0.1 mg protein in 0.1 ml DW) isolated from the feces of old R1.P1-Apoa2c mice were sonicated and injected into 2-month-old male R1.P1-Apoa2c mice intraperitoneally. Mice were killed after 2 months and amyloid deposition was examined.
References
Booth DR, Sunde M, Bellotti V, Robinson CV, Hutchinson WL, Fraser PE, Hawkins PN, Dobson CM, Radford SE, Blake CC, and Pepys MB (1997). Instability, unfolding and aggregation of human lysoyme variants underlying amyloid fibrillogenesis. Nature 385: 787–793.
Caughey B (2000). Transmissible spongiform encephalopathies, amyloidosis and yeast prions: Common threads? Nat Med 6: 751–754.
Chesebro B (1998). BSE and prions: Uncertainties about the agent. Science 279: 42–43.
DePace AH, Santoso A, Hillner P, and Weissman JS (1998). A critical role for amino-terminal glutamine/asparagine repeats in the formation and propagation of a yeast prion. Cell 93: 1241–1252.
Diringer H, Roechmel J, and Beekes M (1998). Effect of repeated oral infection of hamsters with scrapie. J Gen Virol 79: 609–612.
Elliott-Bryant R, and Cathcart ES (1998). Amyloid enhancing factor and dietary transmission in accelerated amyloid A amyloidosis. Clin Immunol Immunopathol 88: 65–69.
Glenner GG (1980). Amyloid deposits and amyloidosis: The β-fibrillosis. N Engl J Med 302: 1283–1292.
Hashimoto N, Naiki H, and Gejyo F (1999). Modification of beta 2-microglobulin with D-glucose or 3-deoxyglucosone inhibits A beta 2M amyloid fibril extension in vitro. Amyloid 6: 256–264.
Higuchi K, Hosokawa M, and Takeda T (1999). Senescence-accelerated mouse. Methods Enzymol 309: 674–686.
Higuchi K, Kitado H, Kitagawa K, Kogishi K, Naiki H, and Takeda T (1993). Development of congenic strains of mice carrying amyloidogenic apolipoprotein A-II (Apoa2c). Apoa2c reduces the plasma level and the size of high-density lipoprotein. FEBS Lett 317: 207–210.
Higuchi K, Kitagawa K, Naiki H, Hanada K, Hosokawa M, and Takeda T (1991a). Polymorphism of apolipoprotein A-II (apoA-II) among inbred strains of mice. Relationship between the molecular type of apoA-II and mouse senile amyloidosis. Biochem J 279: 427–433.
Higuchi K, Kogishi K, Wang J, Chen X, Chiba T, Matsushita T, Hoshii Y, Kawano H, Ishihara T, and Yokota T (1998). Fibrilization in mouse senile amyloidosis is fibril conformation-dependent. Lab Invest 78: 1535–1542.
Higuchi K, Kogishi K, Wang J, Xia C, Chiba T, Matsushita T, and Hosokawa M (1997). Accumulation of pro-apolipoprotein A-II in mouse senile amyloid fibrils. Biochem J 325: 653–659.
Higuchi K, Matsumura A, Honma A, Takeshita S, Hashimoto K, Hosokawa M, Yasuhira K, and Takeda T (1983). Systemic senile amyloid in senescence-accelerated mice. A unique fibril protein demonstrated in tissues from various organs by the unlabeled immunoperoxidase method. Lab Invest 48: 231–240.
Higuchi K, Naiki H, Kitagawa K, Hosokawa M, and Takeda T (1991b). Mouse senile amyloidosis. ASSAM amyloidosis in mice presents universally as a systemic age-associated amyloidosis. Virchows Arch B Cell Pathol Incl Mol Pathol 60: 231–238.
Higuchi K, Naiki H, Kitagawa K, Kitado H, Kogishi K, Matsushita T, and Takeda T (1995). Apolipoprotein A-II gene and development of amyloidosis and senescence in a congenic strain of mice carrying amyloidogenic ApoA-II. Lab Invest 72: 75–82.
Higuchi K, Yonezu T, Kogishi K, Matsumura A, Takeshita S, Higuchi K, Kohno A, Matsushita M, Hosokawa M, and Takeda T (1986). Purification and characterization of a senile amyloid-related antigenic substance (apoSASSAM) from mouse serum: apoSASSAM is an apoA-II apolipoprotein of mouse high-density lipoproteins. J Biol Chem 261: 12834–12840.
Higuchi K, Wang J, Kitagawa K, Matsushita T, Kogishi K, Naiki H, Kitado H, and Hosokawa M (1996). Accelerated senile amyloidosis induced by amyloidogenic apoA-II gene shortens the life span of mice but does not accelerate the rate of senescence. J Gerontol 51A: B295–B302.
Horwich AL and Weissman JS (1997). Deadly conformations-protein misfolding in prion disease. Cell 89: 499–510.
Ionescu-Zanetti C, Khurana R, Gillespie JR, Petrick JS, Trabachino LC, Minert LJ, Carter SA, and Fink AL (1999). Monitoring the assembly of Ig light-chain amyloid fibrils by atomic force microscopy. Proc Natl Acad Sci USA 96: 3175–3179.
Jarrett JT and Lansbury PJ (1993). Seeding one-dimensional crystallization of amyloid: A pathogenic mechanism in Alzheimer's disease and scrapie. Cell 73: 1055–1058.
Johan K, Westermark G, Engstrom U, Gustavsson A, Hultman P, and Westermark P (1998). Acceleration of amyloid protein A amyloidosis by amyloid-like synthetic fibrils. Proc Natl Acad Sci USA 95: 2558–2563.
Kelly JW and Lansbury PTJ (1994). A chemical approach to elucidate the mechanism of transthyretin and β-protein amyloid fibril formation. Amyloid 3: 186–205.
Kisilevsky R, Lemieux L, Boudreau L, Yang DS, and Fraser P (1999). New clothes for amyloid enhancing factor (AEF): Silk as AEF. Amyloid 6: 98–106.
Naiki H, Higuchi K, Hosokawa M, and Takeda T (1989). Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavin T. Anal Biochem 177: 244–249.
Naiki H, Higuchi K, Nakakuki K, and Takeda T (1991). Kinetic analysis of amyloid fibril polymerization in vitro. Lab Invest 65: 104–110.
Naiki H, Higuchi K, Shimada A, Takeda T, and Nakakuki K (1993). Genetic analysis of murine senile amyloidosis. Lab Invest 68: 332–337.
Naiki H and Nakakuki K (1996). First-order kinetic model of Alzheimer's beta-amyloid fibril extension in vitro. Lab Invest 74: 374–383.
Prusiner SB (1997). Prion disease and the BSE crisis. Science 278: 245–251.
Prusiner SB, Scott MR, DeArmond SJ, and Cohen FE (1998). Prion protein biology. Cell 93: 37–48.
Rochet JR, and Lansbury PT Jr (2000). Amyloid fibrillogenesis: Themes and variation. Curr Opin Struct Biol 10: 60–68.
Santoso A, Chien P, Osherovich LZ, and Weissman JS (2000). Molecular basis of a yeast prion species barrier. Cell 100: 277–288.
Schagger H and von Jagow G (1987). Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem 166: 368–379.
Scherzinger E, Sittler A, Schweiger K, Heiser V, Lurz R, Hasenbank R, Bates GP, Lehrach H, and Wanker EE (1999). Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: Implications for Huntington's disease pathology. Proc Natl Acad Sci USA 96: 4604–4609.
Supattapone S, Bosque P, Muramoto T, Wille H, Aagaard C, Peretz D, Nguyen HO, Heinrich C, Torchia M, Safar J, Cohen FE, DeArmond SJ, Prusiner SB, and Scott M (1999). Prion protein of 106 residues creates an artificial transmission barrier for prion replication in transgenic mice. Cell 96: 869–878.
Weissmann C (1999). Moleculargenetics of transmissible spongiform encephalopathies. J Biol Chem 274: 3–6.
Westermark P (1998). The pathogenesis of amyloidosis: Understanding general general principles. Am J Pathol 152: 1125–1127.
Acknowledgements
This work was supported by Grants-in-Aid for Priority Areas (09276209) and Scientific Research (B) (114700596) from the Ministry of Education of Japan and by a grant from the Ministry of Health and Welfare of Japan.
We are grateful to Dr. T. Takeda of Senescence-Accelerated Mouse (SAM) research council for helpful comments and to Mr. E. Deguchi and Mr. S. Yasuoka of Kyoto University for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Xing, Y., Nakamura, A., Chiba, T. et al. Transmission of Mouse Senile Amyloidosis. Lab Invest 81, 493–499 (2001). https://doi.org/10.1038/labinvest.3780257
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3780257
This article is cited by
-
Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases
Nature Neuroscience (2018)
-
Extracellular deposition of mouse senile AApoAII amyloid fibrils induced different unfolded protein responses in the liver, kidney, and heart
Laboratory Investigation (2015)
-
Peripheral administration of tau aggregates triggers intracerebral tauopathy in transgenic mice
Acta Neuropathologica (2014)
-
Self-propagation of pathogenic protein aggregates in neurodegenerative diseases
Nature (2013)
-
Renal amyloidosis caused by apolipoprotein A-II without a genetic mutation in the coding sequence
Clinical and Experimental Nephrology (2011)
