
Abstract
T follicular helper cells (Tfh cells), which are a prototypic subset of effector CD4+ T cells, regulate the production of high-affinity antibodies by controlling B cells at initial and recall phases. Since the discovery of Tfh cells in human tonsils, many notable studies focusing on Tfh cells have clarified mechanisms underlying Tfh-cell-related physiological and pathological settings. Results of these studies revealed a chief regulatory function of BCL6 in Tfh cells and the involvement of Tfh cells in the pathogenesis of various disorders including autoimmune diseases, allergies and cancers. Further, accumulating evidence has revealed microRNAs (miRNAs) of functional noncoding RNAs (ncRNAs) to be cardinal regulators of Tfh cells during the processes of development, differentiation and plasticity. In this review article, we summarize and discuss the results of recent studies about miRNAs operating Tfh-cell function and their relationships in diseases. Through the window of such functional ncRNAs, the functional significance of Tfh cells in CD4+ T-cell biology is becoming apparent. Studies to determine the complex background of the genetic program of Tfh cells operated by functional RNAs should lead to an understanding of the manifestations of Tfh cells with unidentified pathophysiological relevance.
Similar content being viewed by others

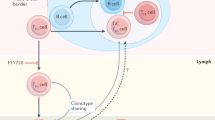

Introduction
T lymphocytes take a distinctive arm in the immune system through molecule-specific recognition mediated by highly divergent T-cell receptors. Multifunctional helper CD4+ T cells have a critical role in orchestrating protective immunoregulation, and their functional deficits cause various types of pathologic perturbations.1 Since the discovery of type 1 and type 2 helper T cells (Th1 and Th2 cells, respectively) in the 1980s, a physical condition characterized by a Th1/Th2 balance has been widely accepted as the underlying immune setting of various disorders.2 Subsequent studies have identified other types of effector CD4+ T cells including regulatory T cells (Treg cells), T follicular helper cells (Tfh cells) and IL-17-producing helper T cells (Th17 cells).3, 4, 5 It then became recognized that functional alteration and/or polarization of such effector CD4+ T cells causes a variety of diseases including autoimmune diseases and allergies. Effector CD4+ T cells are primarily differentiated from naïve CD4+ T cells after encounter with antigen-presenting cells such as dendritic cells and macrophages. In addition to the activation signals generated by engagement of the T-cell receptors and major histocompatibility complex (MHC) class II molecules, costimulatory molecules and cytokines derived from antigen-presenting cells and surrounding cells in the inflammatory milieu profoundly influence the exclusive processes of CD4+ T-cell differentiation into distinct subsets. Besides regulatory mechanisms of the differentiation pathway, mechanisms of the maintenance as well as plasticity of distinct effector CD4+ T cells have been studied to know physiological and pathological conditions.1, 6, 7
The landscape of effector CD4+ T-cell regulation has not been fully clarified. However, it is noteworthy that studies on CD4+ T-cell biology identified a bimodal fate decision in a range of effector CD4+ T-cell subsets, thereby shedding light on the functional significance of Tfh cells.8 This is supported by evidence that Tfh cells preferentially express B-cell lymphoma 6 (BCL6) of a transcription repressor and that other effector CD4+ T-cell subsets (non-Tfh cells) including Th1, Th2, Treg and Th17 cells are regulated by B lymphocyte-induced maturation protein 1 (BLIMP1), which has a characteristic property to antagonize BCL6.9, 10 In this context, Tfh cells, which confer T-cell-dependent humoral immunity at both initial and recall phases, are postulated to be an independent subset among effector CD4+ T cells. With respect to host clearance of many unfavorable pathogens, antigen-specific humoral immunity constitutes a backbone of defense, depending on Tfh cells. The results of a recent study have shown that pathognomonic antigen-specific Th2 cells residing in affected areas such as the lung originate from Tfh cells, that have already been activated in regional lymphoid tissues.11 Such a transitional property indicates functional importance of Tfh cells, probably relating to their plasticity. Although it is still controversial whether the Tfh cell is a separate cell or a cell in a stage of effector CD4+ T-cell differentiation, the concept of Th-cell lineages has recently been challenged by data suggesting plasticity between different effector cell populations based on Tfh cells. In addition to the fact that no long-lived plasma cells and memory B cells specific to antigens are generated without Tfh cells, this is another reason why Tfh cells have been focused on in terms of health and disease.
Tfh cells and their subsets
Tfh cells were first reported as activated T cells (CD4+CD45RO+) expressing C-X-C motif chemokine receptor 5 (CXCR5) that are preferentially localized in germinal centers of lymphoid follicles.12, 13, 14 Because CXCR5 is commonly shared by B cells, this feature of Tfh cells and B cells ensures their cognate interaction in lymphoid tissues, and thereby their close contact around the boundary between T zones and B-cell follicles (T-B border) is instructed.15, 16 C-X-C motif chemokine ligand 13 (CXCL13) of a CXCR5 ligand, which is highly produced by follicular dendritic cells and also Tfh cells, efficiently facilitates this process and can indeed be utilized as a biomarker for monitoring germinal center reactions.17, 18 To facilitate interaction of Tfh cells and B cells, sphingosine-1-phosphate receptor-2 (S1PR2) on Tfh cells further allows their migration to B-cell follicles.19 Owing to an awesome capacity of Tfh cells to secrete interleukin 21 (IL-21), Tfh cells shape germinal centers. In germinal centers, B cells promote isotype switching and somatic hypermutation of immunoglobulins and further clue memory B cells as well as long-lived plasma cells. Tfh cells also present CD40 ligand (CD40L, CD156), B- and T-lymphocyte attenuator (BTLA, CD272), inducible costimulator (ICOS, CD278), and programmed cell death-1 (PD-1, CD279) on the cell surface, and they are directly linked to Tfh cell biology.19, 20 According to these molecules, Tfh cells can migrate to B-cell follicles and provide signals for initiation and maintenance of germinal center B cells to efficiently produce antigen-specific antibodies in lymphoid tissues. In addition to a chief regulator of BCL6, other transcription factors such as interferon regulatory factor 4 (IRF4), v-Maf avian musculoaponeurotic fibrosarcoma oncogene homolog (MAF), basic leucine zipper transcription factor (BATF), and signal transducer and activator of transcription 3 and 5 (STAT3/5) are also involved after the activation events leading to Tfh cell development.19 In addition, we recently found B-cell Oct-binding protein 1 (BOB1, OBF1) as an intrinsic regulator of Tfh cell numbers in vivo, as suggested by experiments using human and mouse lymphocytes.21 In this way, regulatory molecules operating Tfh cells have been discovered, and their functional roles as well as intermolecular relationships have been investigated in terms of Tfh cell biology and pathology.
Of note, within peripheral blood, there are CD4+CXCR5+ T cells termed blood memory Tfh or circulating Tfh-like cells (2–3% and 0.8–1.5% of peripheral blood mononuclear cells in humans and mice, respectively), some of which indeed possess ICOS and/or PD-1 in humans.22 In an analogy of conventional CD4+ T-cell subsets, blood memory Tfh cells in humans have phenotypes similar to those of CD4+ T-cell subsets based on the expression profiles of C-C motif chemokine receptor type 6 (CCR6) and C-X-C motif chemokine receptor 3 (CXCR3). These are type 1 Tfh cells (Tfh1 cells, CCR6−CXCR3+), type 2 Tfh cells (Tfh2 cells, CCR6−CXCR3−) and IL-17-producing Tfh cells (Tfh17 cells, CCR6+CXCR3−), which have the capacity to secrete IFN-γ, IL-4 and IL-17, respectively.23 Tfh1, Tfh2 and Tfh17 cells that commonly express CXCR5 reside in peripheral blood, though most of them are not detectable in lymphoid tissues such as tonsils. To know their functional relevance in life, we examined such blood Tfh cells (CD4+CXCR5+) and their subsets (Tfh1, Tfh2 and Tfh17 cells) in healthy volunteers (n=175) as shown in Figure 1. As assessed by analysis of variance (ANOVA) in each age group, the data showed that the percentage of total Tfh cells during childhood (<10 years old, average of 12.8% in total CD3+CD4+ cells) was increased in the next age group (11–20 years old, average of 17.3%) and that the levels were thereafter maintained until over 71 years of age (Figure 1a). More interestingly, the percentage of Tfh2 cells gradually decreased from childhood (<10 years old, average of 47.2% in total CD4+CXCR5+ cells) to adulthood (31–40 years old, average of 34.8%) and then Tfh2 cell levels were preserved through life (Figure 1b). In contrast, the percentage of Tfh17 cells seemed to reciprocally increase in the corresponding age groups, from childhood (average of 20.6%) to adulthood (average of 34.3%). In each age group from childhood to elderly, however, the percentage of Tfh1 cells was steadily maintained within a certain range (average of 19.2–24.3% for all age groups). Such a tendency was also observed in CCR6+ Tfh1 cells (CCR6+CXCR3+) as an additional subset of Tfh cells; the percentage was maintained within a range (average of 7.9–11.1%) for all age groups. These results suggest that the natural history of blood Tfh cells and their subsets does not uniformly proceed with age. Characteristically, dominant skewing of Tfh2 cells would mitigate in the term from childhood to early adulthood, when an increase of Tfh17 cells occurs (‘Tfh2/Tfh17 reciprocity’). Since both Tfh2 cells and Tfh17 cells work more efficiently than Tfh1 cells as B-cell helpers, the manner of dependency on efficient B-cell helpers seems to be dynamically changed during this period. The immunological significance and biological background of this phenomenon remain unknown, though these subsets may support healthy settings at different ages. There is accumulating evidence indicating functional polarization of such blood Tfh cell subsets, which is associated with the pathogenesis of various immune diseases including autoimmune diseases and allergies.23, 24, 25, 26, 27 We recently found that the percentage of CCR6+ Tfh1 cells was significantly increased in peripheral blood from patients with Sjögren's syndrome (unpublished observation). However, little is known about the genetic program in the control of blood Tfh cell subsets.

Percentages of Tfh cells and Tfh cell subsets in blood from healthy volunteers of different ages. (a) Total Tfh cells (CD4+CXCR5+), (b) Tfh cell subsets including Tfh1 cells (CCR6−CXCR3+), Tfh2 cells (CCR6−CXCR3−), Tfh17 cells (CCR6+CXCR3−) and CCR6+ Tfh1 cells (CCR6+CXCR3+). Blood specimens were obtained from healthy volunteers (n=175). None of them had abnormal physical or chest X-ray findings, and results of allergy testing (serum-specific IgEs to aeroallergens, animal dander and foods) were negative. Preparation, staining and flow cytometry analysis of cells were performed as described previously.24 All blood samples were obtained after receiving informed consent and with the approval of the institutional review boards of Sapporo Medical University in Japan. Graphs are depicted with means±s.d. in each age group, which consisted of 40 (age<10 years old), 8 (11–20), 26 (21–30), 35 (31–40), 15 (41–50), 24 (51–60), 17 (61–70) and 10 (71<) persons. Statistical analysis was performed for each age group (one-way analysis of variance; *P<0.05, **P<0.01, ***P<0.001).
Studies on Tfh cells provided a starting point for the salient discovery of CD4+CXCR5+FoxP3+ T cells, which have been shown to be T follicular regulatory cells (Tfr cells) in lymphoid tissues and blood.28, 29, 30 Tfr cells control Tfh cell activities during the formation of germinal centers in a cytotoxic T-lymphocyte-associated protein 4 (CTLA-4)-dependent manner, indicating that Tfh cells and Tfr cells collaborate in tuning to establish specific humoral immune responses and control self-tolerance simultaneously.31, 32 Such Tfr cells express both BCL6 and BLIMP1, and Tfr cells are thus postulated to be near-of-kin Tfh cells in addition to natural killer T (NKT)-Tfh cells that have been recently reported.33, 34 The fundamental question of whether these cells originate from archetypal Tfh cells has not been fully addressed; however, Tfh cells by themselves may have a capacity to take part in a plastic program for other effector CD4+ T cells and eventually allow self-transformation by covering a part of the functional spectrum of CD4+ T-cell subsets.35 Thus, elucidation of the genetic instructions of Tfh cells should provide fundamentals for overviewing CD4+ T-cell biology as a whole.
Control of Tfh cells mediated by miRNAs
Recent studies on functional noncoding RNAs have shown their unequivocal involvement in the operation programs of immune cells. The importance of endogenous microRNAs (miRNAs), which are noncoding single-stranded RNAs (~22 nucleotides in length), as a layer of regulatory elements for controlling genetic programs in T cells is becoming apparent.36 Generally, miRNAs have a role in the regulation of many aspects of development, differentiation, survival and proliferation. More than 2500 miRNAs in humans have been identified, and each miRNA can influence a transcriptome and robustly change genetic programs. Regarding CD4+ T cells, it has been demonstrated that deficiencies of enzymes such as double-stranded RNA-specific endoribonuclease (Dorsha), Dicer (DCR-1) and DiGeorge syndrome critical region gene 8 (DGCR8), which process nascent transcripts to yield mature miRNAs, cause significant instability of CD4+ T cells.37, 38 Namely, such enzyme deficits directly give rise to functional loss of Tfh cells, accelerated differentiation into Th1 cells and unstable expression of FoxP3 in Treg cells, clearly indicating active involvement of miRNAs in the maintenance of Tfh cells and other effector CD4+ T-cell subsets. Indeed Tfh cells exhibit unique signatures of miRNA and messenger RNA (mRNA) expression with a specific effort of the transcriptional repressor BCL6.39, 40 Therefore, functional miRNAs have been focused on and their role in the regulation of Tfh cells has been extensively studied. It is now evident that there is a complex network of transcription factors, epigenetic changes and post-transcriptional modulations to fulfill characteristic programs of gene expression in Tfh cells of CD4+ T cells.41 Hereafter, we summarize recent findings about miRNAs that regulate the differentiation and function of Tfh cells. Certain miRNAs are involved in Tfh cell biology at the point of plasticity, development and maintenance by tight control of cell numbers for keeping peripheral tolerance.
miR-10a
It has become clear that an miRNA(s) interacting with a gene transcript encoding BCL6 of a lineage-defining regulator of Tfh cells plays a key role in the molding of CD4+ T-cell species. During the initial process of naïve CD4+ T-cell differentiation prone to Tfh cells, achaete-scute homologue 2 (ASCL2 in humans, Mash2 in mice) of an E-box protein starts to upregulate CXCR5 and downregulate C-C chemokine receptor type 7 (CCR7) and P-selectin glycoprotein ligand-1 (PSGL1) prior to the achievement of induction of BCL6.42 BCL6 administers a robust program for differentiation and function of Tfh cells by virtue of vigorous control of AP1 activity over the genome.43 Repeated encounters with antigen-presenting cells and the principal feature of BCL6 in upregulating its own production further reinforce BCL6 expression to establish Tfh cell identity in an exclusive manner.44 It has been reported that miR-10a can directly bind to the 3′-UTR sequence of the gene transcript encoding BCL6 and inhibit the expression of BCL6, indicating that miR-10a is involved in dynamic regulation of Tfh cells.45 Functional loss of miR-10a in Treg cells, which express miR-10a at high levels, preferentially leads to their phenotypic transition to Tfh cells. Indeed, the microenvironment of the intestine deeply influences the plasticity of Treg cells converting to Tfh cells, and miR-10a would secure a balance of Treg cells and Tfh cells in the intestine, which is a site permitting constant contact of various foreign antigens protected by IgA.46, 47 Although the role of miR-10a in Treg cells has been unclear, recent studies suggest that phosphatase and tensin homolog (PTEN), which is regulated by miR-10a, might preserve the integrity of Treg cells.48, 49 Genetic ablation of miR-10a does not seem to directly induce Treg defects or autoimmunity, because miR-10a likely shares redundant function with miR-10b. It should be noted that miR-10 is one of the most highly conserved miRNAs across mammalian species with respect to its chromosomal localization within Hox clusters.50 A sequence immediately downstream from a 5′-terminal oligopyrimidine (5′-TOP) element is also a functional target of miR-10a.51 Thus, miR-10a indirectly regulates translation of a number of 5′-TOP mRNAs such as mRNAs encoding ribosomal proteins to govern general translation in cells. However, the functional significance of 5′-TOP mRNAs in the regulatory mechanism of Tfh cell biology remains unknown.
miR-17~92 cluster
The miR-17~92 polycistronic cluster, which encodes six miRNAs (miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1 and miR-92-1), is well conserved among vertebrates.52 Increased expression of the miR-17~92 cluster results in lymphoproliferative disease and a systemic lupus-like autoimmune condition characterized by functional anomaly of CD4+ T cells and high serum levels of anti-DNA antibodies.53 This indicates direct involvement of the miR-17~92 cluster in the regulation of CD4+ T-cell subsets. Mice lacking the miR-17~92 cluster (Mir17~92−/− mice) in CD4+ T cells show selective reduction in Tfh cells and defective long-lived antibody responses against infection with lymphocytic choriomeningitis virus and also immunization of foreign protein antigens.40, 54 The expression of miR-17~92 is induced early in T cell activation and is thereafter repressed before the end of Tfh cell differentiation. During CD4+ T-cell differentiation, the miR-17~92 cluster has a role in promoting Tfh cell differentiation directly by inhibiting gene expression programs for non-Tfh cells. This is suggested by the results of studies with Mir17~92−/− mice showing that the miR-17~92 cluster represses the genes encoding CCR6, IL-1R1, IL-1R2, IL-22 and RAR-related orphan receptor α (RORα) of a transcription factor, which are normally expressed in non-Tfh cells, indicating a limiting factor of Tfh cells.40, 54 RORα has the capacity to regulate the expression of genes encoding CCR6 and other molecules associated with Th17 cell function. It is also recognized that miR-17~92 indirectly upregulates essential molecules related to Tfh cell lineage: BCL6, CXCR5 and IL-21. Furthermore, miR-17~92 directly represses phosphatase and tensin homolog (PTEN) and PHLPP2, both of which are phosphatases that inactivate PI3 kinase.54 Given that the PI3 kinase pathway is downstream of ICOS to activate Akt during Tfh cell differentiation, miR-17~92 would enhance the PI3 kinase pathway.55, 56 PI3 kinase and Akt pathways subsequently induce the phosphorylation and degradation of FoxO1, resulting in enhanced BCL6 expression and exaggerated differentiation of Tfh cells.57
miR-146a
Like BCL6, there are molecules that Tfh cells and germinal center B cells mutually share in abundance, and thereby antigen-specific immune responses are centrally operated. One such molecule is miR-146a, which was found by a comprehensive approach.58 The absence of miR-146a in mice (Mir-146a−/− mice) causes spontaneous accumulation of Tfh cells and germinal center B cells. Further experiments using mixed bone marrow chimera mice showed autonomous expansion of Mir146a−/− Tfh cells in vivo.58 This mechanism is attributable to direct control of the amounts of gene transcripts encoding ICOS by miR-146a, eventually enhancing signaling for proliferation of Mir146a−/− Tfh cells.58 Such an intrinsic mechanism to control Tfh cell numbers has been focused on to obtain a better understanding of immunological tolerance in peripheral tissues. Excess accumulation of Tfh cells is thought to lower the threshold for B-cell tolerance, causing unfavorable leakage of self- and/or cross-reactive B cells. Thus, the numerical limitation of Tfh cells is considered to be a fundamental checkpoint to prevent tissues from autoinjury.59 Since abnormalities of miR-146a are observed in multiple sclerosis, miR-146a is involved in the pathogenesis of autoimmunity as a cardinal post-transcriptional regulator.25, 60 Among multiple bona fide genes targeted by miR-146a, activation of NF-κB through T-cell receptor signaling leads to upregulation of miR-146a, which in turn restricts NF-κB activity.61, 62 This type of negative feedback loop regulating the NF-κB pathway might also be a part of the regulatory mechanism of Tfh cells. It has been reported that miR-146a targets a 3′-UTR region of the gene encoding Fas-associated death domain to modulate FAS-induced cell death in Jurkat T-cell leukemia cells.63 Since Tfh cells are postulated to be the origin of angioimmunoblastic T-cell lymphoma, this mechanism might underlie tumor cell growth to escape FAS-induced cell death.
miR-155
Along with other miRNAs such as miR-146a, miR-155 also act as an active mediator of inflammation and immunity by the control of T and B cells as well as macrophages.64, 65 Whereas the thymus exhibits high levels of miR-155 expression in comparison to levels in other tissues, activated lymphocytes in peripheral tissues, especially helper CD4+ T cells and B cells, preferentially express miR-155. Experiments using mice lacking miR-155 (Mir155−/− mice) have unraveled its canonical role in the formation of germinal centers to establish antigen-specific humoral immunity, suggesting the possible involvement of miR-155 in the regulation of Tfh cells.66 By using miR-155 conditional knockout T cells in mice (CD4-Cre Mir155fl/fl mice), it has been convincingly revealed that CD4+ T cells require intrinsic miR-155 for Tfh cell differentiation, accompanying significant reduction of germinal center formation even when immunized with a foreign antigen.67 Initially, miR-155 was derived from a long intergenic noncoding RNA (lncRNA) named bic (B-cell integration cluster), which abundantly accumulates in B-cell lymphomas.68 Since then, mRNA targets of miR-155 have been extensively investigated and many of them have been shown to be involved in multiple processes of inflammation, immunity and also regulatory mechanisms of numerous diseases.69 There are two specific targets of miR-155 within Tfh cells, Fos-like antigen 2 (FOSL2) and pellino E3 ubiquitin protein ligase 1 (PELI1), both of which have an inhibitory effect on the expression of BATF and IRF4.67 Therefore, suppression of Tfh cell differentiation in Mir155−/− mice is attributable to the weak function of BATF and IRF4. Interestingly, inflammatory and autoimmune pathologies in Mir146a−/− mice are prevented by the lack of miR-155.67 The opposing roles of miR-155 and miR-146a in the function and development of Tfh cells have been clarified. These miRNAs that are concurrently upregulated in response to proinflammatory cues may counterbalance each other during immune responses established by high-affinity antibodies. A concept of low-grade inflammation (inflamm-aging phenomenon) is applied to illustrate systemic roles of such miRNAs in the regulation of Tfh cells.70
miR-346
The investigation of noncoding RNAs such as miRNAs, which are responsible for regulating the settings of circulating Tfh cells in peripheral blood, has just started. Actually, little is known about miRNAs regulating blood CD4+CXCR5+ T cells in healthy and disease conditions, though it has been reported that miR-346 possibly targets BCL6 in blood CD4+CXCR5+ T cells of Graves’ disease.71 In that study, a unique correlation of disease activities with expression levels of miR-346 in blood CD4+ T cells and plasma was also found. Given that genetic polymorphisms related to miRNAs including miR-346 and miR-146a are involved in the pathogenesis of rheumatoid arthritis, functional disturbances of such miRNAs may broadly underlie autoimmune diseases.72 As mentioned above, Tfh cell polarization characteristic to ages might be related to healthy settings through life (Figure 1). Further, there is accumulating evidence that suggests functional skewing of blood Tfh-cell subsets in a variety of disorders, such as autoimmune diseases and allergies.22, 23, 24, 25, 26, 27 With miR-346 as the first opportunity, it is expected that investigation of the genetic background of blood Tfh-cell subsets will advance to elucidate the mechanism by which noncoding RNAs contribute to the regulation of polarization of blood Tfh cells and their biomedical significance. Since Tfh-cell subsets are characterized by the expression profiles of CCR6 and CXCR3 of CD4+CXCR5+ Tfh cells, miRNAs regulating the expression of these molecules might be associated with the specification of peripheral blood Tfh cells.
Concluding remarks
Tfh cells are attractive targets for the treatment of refractory immune-related disorders and even cardinal to durable immune responses by successful vaccinations. Therefore, regulatory mechanisms of Tfh cells and their circulating subsets need considerable attention. While the functional execution of Tfh cells depends on complex machinery governed by both intracellular and extracellular factors, our current knowledge of how functional RNAs are enrolled in the phenotypic control of Tfh cells is expanding. Generally, each miRNA has a potential capacity to target tens or hundreds of mRNAs, and collaborative regulation of numerous direct targets in gene networks must occur in cells.73 Besides miRNAs, studies on other functional RNAs of CD4+ T cells should provide critical information for the understanding of Tfh cell regulation. Evidence that lnc-MAF-4 operates the differentiation of CD4+ T cells may uncover the expression mechanism of a MAF transcription factor in Tfh cells.19, 20, 74 As studies on functional RNAs advance, another factor(s) of regulators might be elucidated and enable a specific approach for control of Tfh cells and their subsets.
References
Vahedi, G., C. Poholek, A., Hand, T. W., Laurence, A., Kanno, Y., O'Shea, J. J. et al. Helper T-cell identity and evolution of differential transcriptomes and epigenomes. Immunol. Rev. 252, 24–40 (2013).
Mosmann, T. R., Cherwinski, H., Bond, M. W., Giedlin, M. A. & Coffman, R. L. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 136, 2348–2357 (1986).
Sakaguchi, S., Sakaguchi, N., Asano, M., Itoh, M. & Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 155, 1151–1564 (1995).
Breitfeld, D., Ohl, L., Kremmer, E., Ellwart, J., Sallusto, F., Lipp, M. et al. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J. Exp. Med. 192, 1545–1552 (2000).
Harrington, L. E., Hatton, R. D., Mangan, P. R., Turner, H., Murphy, T. L., Murphy, K. M. et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 6, 1123–1132 (2005).
Tuomela, S. & Lahesmaa, R. Early T helper cell programming of gene expression in human. Semin. Immunol. 25, 282–290 (2013).
Witte, S., O'Shea, J. J. & Vahedi, G. Super-enhancers: asset management in immune cell genomes. Trends Immunol. 36, 519–526 (2015).
Crotty, S., Johnston, R. J. & Schoenberger, S. P. Effectors and memories: Bcl-6 and Blimp-1 in T and B lymphocyte differentiation. Nat. Immunol. 11, 114–120 (2010).
Nurieva, R. I., Chung, Y., Martinez., G. J., Yang, X. O., Tanaka, S., Matskevitch, T. D. et al. Bcl6 mediates the development of T follicular helper cells. Science 325, 1001–1005 (2009).
Johnston, R. J., Poholek, A. C., DiToro, D., Yusuf, I., Eto, D., Barnett, B. et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 325, 1006–1010 (2009).
Ballesteros-Tato, A., Randall, T. D., Lund, F. E., Spolski, R., Leonard, W. J. & León, B. T follicular helper cell plasticity shapes pathogenic T helper 2 cell-mediated immunity to inhaled house dust mite. Immunity 44, 259–273 (2016).
Förster, R., Mattis, A. E., Kremmer, E., Wolf, E., Brem, G. & Lipp, M. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell 87, 1037–1047 (1996).
Ansel, K. M., McHeyzer-Williams, L. J., Ngo, V. N., McHeyzer-Williams, M. G. & Cyster, J. G. In vivo-activated CD4 T cells upregulate CXC chemokine receptor 5 and reprogram their response to lymphoid chemokines. J. Exp. Med. 190, 1123–1134 (1999).
Schaerli, P., Willimann, K., Lang, A. B., Lipp, M., Loetscher, P. & Moser, B. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J. Exp. Med. 192, 1553–1562 (2000).
Rasheed, A. U., Rahn, H. P., Sallusto, F., Lipp, M. & Müller, G. Follicular B helper T cell activity is confined to CXCR5hiICOShi CD4 T cells and is independent of CD57 expression. Eur. J. Immunol. 36, 1892–1903 (2006).
Kerfoot, S. M., Yaari, G., Patel, J. R., Johnson, K. L., Gonzalez, D. G., Kleinstein, S. H. et al. Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity. 34, 947–960 (2011).
Kim, C. H., Lim, H. W., Kim, J. R., Rott, L., Hillsamer, P. & Butcher, E. C. Unique gene expression program of human germinal center T helper cells. Blood 104, 1952–1960 (2004).
Havenar-Daughton, C., Lindqvist, M., Heit, A., Wu, J. E., Reiss, S. M., Kendric, K. et al. CXCL13 is a plasma biomarker of germinal center activity. Proc. Natl Acad. Sci. USA 113, 2702–2707 (2016).
Vinuesa, C. G., Linterman, M. A., Yu, D. & MacLennan, I. C. Follicular helper T cells. Annu. Rev. Immunol. 34, 335–368 (2016).
Ueno, H., Banchereau, J. & Vinuesa, C. G. Pathophysiology of T follicular helper cells in humans and mice. Nat. Immunol. 16, 142–152 (2015).
Yamashita, K., Kawata, K., Matsumiya, H., Kamekura, R., Jitsukawa, S., Nagaya, T. et al. Bob1 limits cellular frequency of T follicular helper cells. Eur. J. Immunol. 46, 1361–1370 (2016).
Ueno, H. Human circulating T follicular helper cell subsets in health and disease. J. Clin. Immunol. 36 (Suppl 1), 34–39 (2016).
Morita, R., Schmitt, N., Bentebibel, S. E., Ranganathan, R., Bourdery, L., Zurawski, G. et al. Human blood CXCR5+CD4+ T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity 34, 108–121 (2011).
Kamekura, R., Shigehara, K., Miyajima, S., Jitsukawa, S., Kawata, K., Yamashita, K. et al. Alteration of circulating type 2 follicular helper T cells and regulatory B cells underlies the comorbid association of allergic rhinitis with bronchial asthma. Clin. Immunol. 158, 204–211 (2015).
Fan, X., Jin, T., Zhao, S., Liu, C., Han, J., Jiang, X. et al. Circulating CCR7+ICOS+ Memory T follicular helper cells in patients with multiple sclerosis. PLoS ONE 10, e0134523 (2015).
Szabó, K., Papp, G., Szántó, A., Tarr, T. & Zeher, M. A comprehensive investigation on the distribution of circulating follicular T helper cells and B cell subsets in primary Sjögren's syndrome and systemic lupus erythematosus. Clin. Exp. Immunol. 183, 76–89 (2016).
Che, Y., Qiu, J., Jin, T., Yin, F., Li, M. & Jiang, Y. Circulating memory T follicular helper subsets, Tfh2 and Tfh17, participate in the pathogenesis of Guillain-Barré syndrome. Sci. Rep. 6, 20963 (2016).
Wollenberg, I., Agua-Doce, A., Hernández, A., Almeida, C., Oliveira, V. G., Faro, J. et al. Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J. Immunol. 187, 4553–4560 (2011).
Linterman, M. A., Pierson, W., Lee, S. K., Kallies, A., Kawamoto, S., Rayner, T. F. et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat. Med. 17, 975–982 (2011).
Chung, Y., Tanaka, S., Chu, F., Nurieva, R. I., Martinez, G. J., Rawal, S. et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat. Med. 17, 983–988 (2011).
Wing, J. B., Ise, W., Kurosaki, T. & Sakaguchi, S. Regulatory T cells control antigen-specific expansion of Tfh cell number and humoral immune responses via the coreceptor CTLA-4. Immunity 41, 1013–1025 (2014).
Sage, P. T., Paterson, A. M., Lovitch, S. B. & Sharpe, A. H. The coinhibitory receptor CTLA-4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity 41, 1026–1039 (2014).
Sage, P. T. & Sharpe, A. H. T follicular regulatory cells in the regulation of B cell responses. Trends Immunol. 36, 410–418 (2015).
Nair, S., Boddupalli, C. S., Verma, R., Liu, J., Yang, R., Pastores, G. M. et al. Type II NKT-TFH cells against Gaucher lipids regulate B-cell immunity and inflammation. Blood 125, 1256–1271 (2015).
Cannons, J. L., Lu, K. T. & Schwartzberg, P. L. T follicular helper cell diversity and plasticity. Trends Immunol. 34, 200–207 (2013).
O'Connell, R. M., Rao, D. S. & Baltimore, D. microRNA regulation of inflammatory responses. Annu. Rev. Immunol. 30, 295–312 (2012).
Muljo, S. A., Ansel, K. M., Kanellopoulou, C., Livingston, D. M., Rao, A. & Rajewsky, K. Aberrant T cell differentiation in the absence of Dicer. J. Exp. Med. 202, 261–269 (2005).
Chong, M. M., Rasmussen, J. P., Rudensky, A. Y. & Littman, D. R. The RNAseIII enzyme Drosha is critical in T cells for preventing lethal inflammatory disease. J. Exp. Med. 205, 2005–2017 (2008).
Yu, D., Rao, S., Tsai, L. M., Lee, S. K., He, Y., Sutcliffe, E. L. et al. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity 31, 457–468 (2009).
Baumjohann, D., Kageyama, R., Clingan, J. M., Morar, M. M., Patel, S., de Kouchkovsky, D. et al. The microRNA cluster miR-17-92 promotes TFH cell differentiation and represses subset-inappropriate gene expression. Nat. Immunol. 14, 840–848 (2013).
Baumjohann, D. & Ansel, K. M. microRNA-mediated regulation of T helper cell differentiation and plasticity. Nat. Rev. Immunol. 13, 666–678 (2013).
Liu, X., Chen, X., Zhong, B., Wang, A., Wang, X., Chu, F. et al. Transcription factor achaete-scute homologue 2 initiates follicular T-helper-cell development. Nature 507, 513–518 (2014).
Hatzi, K., Nance, J. P., Kroenke, M. A., Bothwell, M., Haddad, E. K., Melnick, A. et al. BCL6 orchestrates Tfh cell differentiation via multiple distinct mechanisms. J. Exp. Med. 212, 539–553 (2015).
Baumjohann, D., Okada, T. & Ansel, K. M. Distinct waves of BCL6 expression during T follicular helper cell development. J. Immunol. 187, 2089–2092 (2011).
Takahashi, H., Kanno, T., Nakayamada, S., Hirahara, K., Sciumè, G., Muljo, S. A. et al. TGF-β and retinoic acid induce the microRNA miR-10a, which targets Bcl-6 and constrains the plasticity of helper T cells. Nat. Immunol. 13, 587–595 (2012).
Tsuji, M., Komatsu, N., Kawamoto, S., Suzuki, K., Kanagawa, O., Honjo, T. et al. Preferential generation of follicular B helper T cells from Foxp3+ T cells in gut Peyer’s patches. Science 323, 1488–1492 (2009).
Kawamoto, S., Maruya, M., Kato, L. M., Suda, W., Atarashi, K., Doi, Y. et al. Foxp3+ T cells regulate immunoglobulin a selection and facilitate diversification of bacterial species responsible for immune homeostasis. Immunity 41, 152–165 (2014).
Yu, T., Liu, L., Li, J., Yan, M., Lin, H., Liu, Y. et al. MiRNA-10a is upregulated in NSCLC and may promote cancer by targeting PTEN. Oncotarget 6, 30239–30250 (2015).
Shrestha, S., Yang, K., Guy, C., Vogel, P., Neale, G. & Chi, H. Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat. Immunol. 16, 178–187 (2015).
Tanzer, A., Amemiya, C. T., Kim, C. B. & Stadler, P. F. Evolution of microRNAs located within Hox gene clusters. J. Exp. Zool. B Mol. Dev. Evol. 304, 75–85 (2005).
Ørom, U. A., Nielsen, F. C. & Lund, A. H. MicroRNA-10a binds the 5'UTR of ribosomal protein mRNAs and enhances their translation. Mol. Cell 30, 460–471 (2008).
He, L., Thomson, J. M., Hemann, M. T., Hernando-Monge, E., Mu, D., Goodson, S. et al. A microRNA polycistron as a potential human oncogene. Nature 435, 828–833 (2005).
Xiao, C., Srinivasan, L., Calado, D. P., Patterson, H. C., Zhang, B., Wang, J. et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat. Immunol. 9, 405–414 (2008).
Kang, S. G., Liu, W. H., Lu, P., Jin, H. Y., Lim, H. W., Shepherd, J. et al. MicroRNAs of the miR-17~92 family are critical regulators of Tfh differentiation. Nat. Immunol. 14, 849–857 (2013).
Bauquet, A. T., Jin, H., Paterson, A. M., Mitsdoerffer, M., Ho, I. C., Sharpe, A. H. et al. The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH-17 cells. Nat. Immunol. 10, 167–175 (2009).
Gigoux, M., Shang, J., Pak, Y., Xu, M., Choe, J., Mak, T. W. et al. Inducible costimulator promotes helper T-cell differentiation through phosphoinositide 3-kinase. Proc. Natl Acad. Sci. USA 106, 20371–20376 (2009).
Stone, E. L., Pepper, M., Katayama, C. D., Kerdiles, Y. M., Lai, C. Y., Emslie, E. et al. ICOS coreceptor signaling inactivates the transcription factor FOXO1 to promote Tfh cell differentiation. Immunity 42, 239–251 (2015).
Pratama, A., Srivastava, M., Williams, N. J., Papa, I., Lee, S. K., Dinh, X. T. et al. MicroRNA-146a regulates ICOS-ICOSL signaling to limit accumulation of T follicular helper cells and germinal centers. Nat. Commun. 6, 6436 (2015).
Pratama, A. & Vinuesa, C. G. Control of TFH cell numbers: why and how? Immunol. Cell Biol. 92, 40–48 (2014).
Fenoglio, C., Cantoni, C., De Riz, M., Ridolfi, E., Cortini, F., Serpente, M. et al. Expression and genetic analysis of miRNAs involved in CD4+ cell activation in patients with multiple sclerosis. Neurosci. Lett. 504, 9–12 (2011).
Yang, L., Boldin, M. P., Yu, Y., Liu, C. S., Ea, C. K., Ramakrishnan, P. et al. miR-146a controls the resolution of T cell responses in mice. J. Exp. Med. 209, 1655–1670 (2012).
Taganov, K. D., Boldin, M. P., Chang, K. J. & Baltimore, D. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl Acad. Sci. USA 103, 12481–12486 (2006).
Curtale, G., Citarella, F., Carissimi, C., Goldoni, M., Carucci, N., Fulci, V. et al. An emerging player in the adaptive immune response: microRNA-146a is a modulator of IL-2 expression and activation-induced cell death in T lymphocytes. Blood 115, 265–273 (2010).
Rodriguez, A., Vigorito, E., Clare, S., Warren, M. V., Couttet, P., Soond, D. R. et al. Requirement of bic/microRNA-155 for normal immune function. Science 316, 608–611 (2007).
He, S., Yang, L., Li, D. & Li, M. Kruppel-like factor 2-mediated suppression of microRNA-155 reduces the proinflammatory activation of macrophages. PLoS One 10, e0139060 (2015).
Thai, T. H., Calado, D. P., Casola, S., Ansel, K. M., Xiao, C., Xue, Y. et al. Regulation of the germinal center response by microRNA-155. Science 316, 604–608 (2007).
Hu, R., Kagele, D. A., Huffaker, T. B., Runtsch, M. C., Alexander, M., Liu, J. et al. miR-155 promotes T follicular helper cell accumulation during chronic, low-grade inflammation. Immunity 41, 605–619 (2014).
Eis, P. S., Tam, W., Sun, L., Chadburn, A., Li, Z., Gomez, M. F. et al. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc. Natl Acad. Sci. USA 102, 3627–3632 (2005).
Mashima, R. Physiological roles of miR-155. Immunology 145, 323–333 (2015).
Yu, D. MicroRNAs in Tfh cells: micromanaging inflammaging. Immunity 41, 509–511 (2014).
Chen, J., Tian, J., Tang, X., Rui, K., Ma, J., Mao, C. et al. MiR-346 regulates CD4+CXCR5+ T cells in the pathogenesis of Graves' disease. Endocrine 49, 752–760 (2015).
Chatzikyriakidou, A., Voulgari, P. V., Georgiou, I. & Drosos, A. A. miRNAs and related polymorphisms in rheumatoid arthritis susceptibility. Autoimmun. Rev. 11, 636–641 (2012).
Ebert, M. S. & Sharp, P. A. Roles for microRNAs in conferring robustness to biological processes. Cell 149, 515–524 (2012).
Ranzani, V., Rossetti, G., Panzeri, I., Arrigoni, A., Bonnal, R. J., Curti, S. et al. The long intergenic noncoding RNA landscape of human lymphocytes highlights the regulation of T cell differentiation by linc-MAF-4. Nat. Immunol. 16, 318–325 (2015).
Acknowledgements
This work was supported by the Japan Society for the Promotion of Science (JSPS) Grants #26670178 (S.I.), #15K10787 (R.K.), #15K20214 (K.K.) and #16K15723 (T.H.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Ichimiya, S., Kamekura, R., Kawata, K. et al. Functional RNAs control T follicular helper cells. J Hum Genet 62, 81–86 (2017). https://doi.org/10.1038/jhg.2016.88
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2016.88
