
Abstract
The complete mitochondrial genome of one 700-year-old individual found in Tashkurgan, Xinjiang was target enriched and sequenced in order to shed light on the population history of Tashkurgan and determine the phylogenetic relationship of haplogroup U5a. The ancient sample was assigned to a subclade of haplogroup U5a2a1, which is defined by two rare and stable transversions at 16114A and 13928C. Phylogenetic analysis shows a distribution pattern for U5a2a that is indicative of an origin in the Volga–Ural region and exhibits a clear eastward geographical expansion that correlates with the pastoral culture also entering the Eurasian steppe. The haplogroup U5a2a present in the ancient Tashkurgan individual reveals prehistoric migration in the East Pamir by pastoralists. This study shows that studying an ancient mitochondrial genome is a useful approach for studying the evolutionary process and population history of Eastern Pamir.
Similar content being viewed by others
Introduction
The Tashkurgan Tajik Autonomous County is located at the western edge of the oasis zone of the Tarim Basin and the eastern edge of the Pamir Plateau, where the Kunlun, Karakoram, Hindukush and Tian Shan mountains meet. This region’s unique geographic location and long history of human occupation make it a junction for a series of trade and cultural transmission routes such as the ancient Silk Road, which was not only important for cultural interaction but also the main migration route connecting the West and East.1 Therefore, whether the origin and development of the Tashkurgan population was affected by admixture from ethnically different people has been a complex question of heated discussion.
In our genetic study on 700-year-old cemetery from Tashkurgan, an ancient individual was assigned to the haplogroup U5a2a based on the sequence of mitochondrial hypervariable region I (mtDNA HVRI). Haplogroup U5 is the oldest European-specific haplogroup, and it is estimated to be ~50 000 years old. In early hunter-gatherer populations of Europe, the dominant haplogroup is U5, with its subclades U5a and U5b.2 U5a and U5b frequencies are highest in northern Europe, but it is also found in lower frequencies throughout Europe, Near East and Central Asia. Until now, the subclade of U5a that was most widespread in Central Asia is U5a1, which is also found in both the ancient and extant populations of the Tarim Basin, Xinjiang. The other subclade of U5a, U5a2, is rare in ancient and extant populations of Central Asia2 and has not been found in Xinjiang.3 Therefore, finding this ancient U5a2a in Tashkurgan offers an important clue as to the source and flow of the Tashkurgan population.
In this study, we use target enriched and sequenced the complete mitochondrial genome of the Tashkurgan individual assigned to U5a2a, in order to shed further light on the population history of Tashkurgan and to provide fine resolution of phylogenetic relationship of haplogroup U5a2a.
Materials and Methods
Archaeological samples
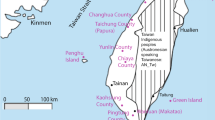
The Xiabandi cemetery is located in Tashkurgan, Xinjiang (37°55′12″N, 75°10′48″E), and is separated into A and B parts by the Tashkurgan River (Figure 1). Xiabandi A was occupied during Bronze Age (3500–2600 bp), carrying characteristics of typical Andronovo culture. The tooth sample analyzed in this study was collected from the Xiabandi B cemetery, radiocarbon dated to 720–610 Cal bp (95.4% confidence interval).4 The archaeological site was located at the eastern edge of Pamir with an average altitude above 3000 m and an annual average temperature of 3.3 °C. The cold and dry climate found here is favorable for DNA conservation. The sample was excavated by the Xinjiang Cultural Relics and Archaeology Institute in 2004.

Geographic location of the archaeological sites studied. The map on the top right indicates the position of East Pamir in the Eurasia. A full color version of this figure is available at the Journal of Human Genetics journal online.
Ancient DNA extraction
Tooth samples were treated before DNA extraction as described by Li et al.5 Tooth powder (0.1 g) was incubated for 24 h in 3 ml of a solution containing 0.45 m EDTA, 0.5% SDS and 0.7 mg ml−1 proteinase K in a shaker (220 r.p.m. min−1) at 50 °C. Afterwards, DNA was extracted using the QIAquickPCR Purification Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol.
Preparation of the genomic library, ancient mtDNA enrichment and illumina sequencing
Two separate libraries were prepared from the same 50 μl of ancient DNA extract following the NEBNext Ultra DNA Library preparation protocol (New England Biolabs, Beijing, China), except that a 1:20 diluted adapter was applied to the ends of DNA fragments during ligation. Multiple Ampure Bead XP cleanups (Beckman Coulter, Brea, CA, USA) were conducted to remove any adapter dimer that may have developed. PCR amplification of the genomic library was prepared in the Ancient DNA laboratory (25 μl reaction with 10 mm primers, 5 × PCR buffer, 10 mm DNTPs, AmpliTaq Gold DNA Polymerase, genomic library) and then transported to thermocyclers in the contemporary laboratory, where libraries were amplified for 15 cycles6 and then cleaned with the MinElute PCR Purification Kit (Qiagen, Chatsworth, CA, USA).
The quality and concentration of the two libraries were determined on an Agilent Bioanalyzer 2100 (Agilent Technologies, Palo Alto, CA, USA). Subsequently, target enrichment of the mitochondrial genome was performed on the amplified library using MyGenostics Human Mitochondria Capture Kit (MyGenostics Inc., Beijing, China). A final post-enrichment amplification was performed for 15 cycles. The post-enrichment amplified product was then quantified using quantitative PCR. Sequencing was carried out using an Illumina HiSeq 2000 platform at MyGenostics Inc. The 100-bp (base pair) paired-end reads and a single index read were generated according to the manufacturer’s instructions.
Bioinformatics and sequence analysis
Raw data from the Illumina HiSeq 2000 platform were analyzed with CASAVA 1.8.2. Adapter sequences were trimmed using Adapter-Removal7 with a minimum length of 257. Sequence reads were mapped to the revised Cambridge Reference Sequence using the Burrows-Wheeler Aligner 0.6.2 (Li H. Wellcome Trust Sanger Institute, Cambridge, UK) with default parameters except for seed length, which was set to 1000.8 All duplicates were removed using SAMtools v0.1.18 (Li H. Wellcome Trust Sanger Institute). Coverage depth was directly estimated from the read-mapping file by counting the number of reads covering each mtDNA position. Single-nucleotide polymorphisms (SNP) and insertions and deletions (INDELs) were called using the SNVer package 0.4.1 (Zhi Wei. New Jersey Institute of Technology, Newark, NJ, USA).9 Single-nucleotide polymorphism quality thresholds were set with a haploid model, a read depth of 20 and a base quality of 20. This sequence was submitted to GenBank (http://www.ncbi.nlm.nih.gov/nuccore), and the assigned accession number is KT023499. A specific pattern of DNA damage has been showed a pattern of increased DNA damage at the ends of the degraded DNA fragments, which is also found in other ancient DNA studies.10
Phylogenetic analysis
The network of U5a2a was constructed by reduced median-joining method11 using NETWORK v. 4.5.1.6 (Fluxus Technology Ltd., Kiel, Germany). A nucleotide diversity map based on massive available hypervariable segment-I data (Supplementary Table S1) was generated by using the Kriging algorithm of the Surfer 8.0 package (Golden Software, Golden, CO, USA). In addition, our sample together with 88 whole mitochondrial sequences from MitoTool database12 (http://www.mitotool.org/) were used as implemented in BEAST 1.7.13, 14 The General Time Reversible sequence evolution model with a fixed fraction of invariable sites was determined by the best-fit model approach of jModeltest-2.1.415, 16 and ran 40 000 000 generations of the Markov Chain Monte Carlo with the first 4 000 000 generations discarded as burn-in. The alignment was analyzed using a strict molecular clock with a substitution rate of the 13 coding regions of 1.691 × 10−8 substitutions per site per year with the ND6 gene readjusted to present the same reading direction as the other genes.17, 18, 19 The nucleotide and sequence diversity of mtDNA was determined using Arlequin software (Excoffier L. Zoological Institute, University of Berne, Switzerland).20 The most parsimonious trees were reconstructed using mtPhyl l 3.0 software (http://eltsov.org). Eighty-eight U5a2a genome sequences was used to build the phylogenetic tree for haplogroup U5a2a and its subclades.21 The coalescence time of each lineage was estimated using the ρ statistic-based method. The s.d. was calculated following Saillard et al.22 Then the time for most recent common ancestor of each lineage was estimated using the Soares rate for complete mitochondrial genomes.23
Contamination control and independent replication
Appropriate precautions were taken to ensure the authenticity of the ancient DNA results.24 All pre-PCR steps were performed in a positive pressure laboratory dedicated to ancient DNA located in the Research Center for Chinese Frontier Archaeology of Jilin University. Different rooms were used for sample preparation, DNA extraction and PCR setup. Post-PCR procedures were carried out in a different building. Surfaces were cleaned regularly with a 10% sodium hypochlorite solution and ultraviolet light (254 nm), and full-body protective clothing, facemasks and gloves were worn. Gloves were changed frequently. All consumables were purchased as DNA free.
Contamination controls were used with every DNA extraction and PCR setup in order to detect any contamination. To check for reproducibility, the experiments were performed in parallel using duplicate teeth of each individual in the Molecular Forensic Lab at the School of Life Sciences in Jilin University. DNA from samples was extracted twice and the results were confirmed by sequencing the hypervariable segment-I portion using the Sanger method.
Results
Authentication of the ancient mtDNA sequence
Strict procedures were used to prevent contamination from modern DNA. We assess the authenticity of our results using a number of different observations: (a) the negative extraction and amplification control were free of contamination; (b) the same results for the hypervariable segment-I region of the ancient sample were achieved when sequencing by the same protocol as in Li et al.5; and (c) the sequences of the ancient individual were confirmed to be different from those of the laboratory researchers.
The nucleotide misincorporation pattern at the 3′- and 5′-ends of the ancient DNA fragments was evaluated in order to assess the preservation conditions and check for possible contamination in the ancient sample. We observed a C-to-T substitution frequency of 14.029% at the 5′-end (Supplementary Figure S1), which correlates well with the expected age of the sample relative to the substitution frequency, as suggested by Sawyer et al.25 The results of the DNA damage analyses support that the sample preservation conditions were good and that the aDNA data presented here are authentic.
Mitochondrial genome of the ancient Tashkurgan sample
Target enrichment of the Tashkurgan ancient DNA, followed by sequencing on Illumina Hiseq 2000 platform, allowed the unambiguous determination of 100% of the ancient mtDNA genome with 363.06 Mb of unique reads assembled to revised Cambridge Reference Sequence26 at an average coverage of 180 × and an average read length of more than 100 bp. Adequate depth and coverage of the mtDNA genome sequence data prevented false-positive base calls. The ancient mtDNA haplotype of our ancient sample differed from the revised Cambridge Reference Sequence at 22 nucleotide positions (Table 1) and was further characterized as U5a2a1 by MitoTool 1.1.2.21
Phylogenetic analysis of the ancient Tashkurgan sample
The ancient Tashkurgan sample can be assigned to a subclade of haplogroup U5a2a1 in accordance with the current phylogeny (www.PhyloTree.org), and this subclade is mainly defined by two rare and stable transversions at 16114A and 13928C. In the current study, we reconstructed the phylogeny of subclade U5a2a (Figure 2). Information from complete mtDNA sequencing reveals that the ancient Tashkurgan sample matches six extant samples in the NCBI database. Of these six, two individuals (KF161325 and GU296615) have confirmed locations of Denmark and Russia (European region). The coalescence time of U5a2a1, estimated using the average sequence divergence of mitochondrial genomes, is ~6377.15±1149.2 years before present, corresponding to the end of the Late Neolithic and the beginning of the Early Bronze Age in the Eurasian continent (Figure 2).

Phylogeny of complete mitochondrial genomes with U5a2a. Mutations are transitions unless specified. Transversions are indicated by an A, G, C or T after the nucleotide position. Insertions are indicated by an ‘i’. Each circle represents a different haplotype, and the size of each circle is proportional to the number of individuals sharing the corresponding haplotype, with the smallest size corresponding to one individual. The haplotype labeled ‘aXBD’ is from the ancient sample in this study.
The network analysis of haplogroup U5a2a revealed a star-like pattern and showed a signal of population expansion of this haplogroup. The Volga–Ural population harbors the most kinds of haplotypes (Supplementary Figure S2a), which is consistent with the highest nucleotide and sequence diversity of this population. Owing to low frequency of U5a2a and its sub-haplogroups, the counter map here was drawn based on internal diversity distribution of subclade U5a2a (Supplementary Figure S2b). The counter map showed similar expansion pattern as the network.
Discussion
The dispersion of subclade U5a2a1 accompanies the Pastoralist Indo–European expansion in the Eurasian steppe
As one subclade of U5a2 (U5a2a–U5a2e), U5a2a is estimated to be about 12 000 years old,2 but its main subclade U5a2a1 (nearly 97%) is the youngest and is only about 7000 years old based on the complete mitochondrial genomes, which mostly belongs to the central and eastern Europeans. The sample set is not evenly distributed across Eurasia, so an expansion survey was performed using the U5a2a hypervariable segment-I region motifs reported in different populations (Supplementary Table S1) in order to evaluate more comprehensively the distribution of subclade U5a2a.27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39 The highest nucleotide and sequence diversity of this subclade was found in the Volga–Ural region, although the frequency of U5a2a was higher in eastern Europe (Table 2). U5a2a is also present in low frequency in other Eurasian populations such as central Siberia and central Asia, such as Kazakhstan, Kyrgyzstan and Uzbekistan. No U5a2a is detected in any other samples from both ancient and modern Eurasian populations. The geographic distribution of U5a2a, supported with the network analysis, may indicate a Volga–Ural origin for subclade U5a2a and exhibits a clear eastward geographic expansion pattern.
The Pontic–Caspian steppe, where the Volga–Ural region is located, is known as the origin of Kurgan culture, which was a stockbreeding steppe culture in the Late Neolithic and Early Bronze Age. Beginning from the fifth millennium bc, domesticated horses and metallurgical technology gave rise to a new mobile lifestyle that would eventually lead to the great Indo–European migrations eastward along the Eurasian steppe.40 During the Bronze Age, the Andronovo Culture, a sedentary pastoralist society succeeding the Kurgan culture, flourished on the Eurasian steppe. This culture became the dominant archaeological culture of central Asia and Pamir from around 2000 bc.41 From the Bayesian skyline analysis (Figure 3), the expansion time of U5a2a was around 5000–3000 bc, which corresponds to the period of pastoral culture found across the Eurasian steppe. Furthermore, Keyser et al.39 found the U5a2a in a 3000-year-old pastoral population in the Krasnoyarsk region of southern Siberia. It is likely that the dispersion of U5a2a accompanied the migration of the steppe population.

Estimated effective population size (Ne) for the complete Eurasian mtDNA data set containing U5a2a. The x axis shows time in years before present, the y axis shows the effective population size, Ne. The center line represents the mean Ne estimate, and the upper and lower lines are the 95% posterior density intervals. We assumed a mutation rate in the coding regions of 1.691 × 10−8 substitutions per site and year with a generation time of 20 years.46 A full color version of this figure is available at the Journal of Human Genetics journal online.
The ancient haplotype U5a2a present in the Tashkurgan region reveals prehistoric migration in the East Pamir by pastoralists
Archaeological studies show that most Bronze-Age cemeteries (second millennium bc) found around the Pamir region, such as the Vakhsh site in Tajikistan, the Sapalli site in Uzbekistan and the Xiangbaobao site in Tashkurgan, exhibit the Andronovo cultural style.42 Thus, the Andronovo culture reached south Central Asia and the Pamir plateau. The dispersion and distribution of U5a2a may mostly be a consequence of migrations southward by Andronovo pastoralists during the Bronze Age. Another possibility for U5a2a entering the Pamir region is a massive nomadic movement that happened in the second century bc.43 After the Yuezhi tribe was defeated by the Xiongnu, and created a domino effect where the Yuezhi were forced to move south, displacing the Scythians, who were later driven down to south Central Asia. Scythians are believed to be ethnically Indo–European nomads exhibiting Europoid traits. Genetic analyses have shown that the Scythians are genetically more closely related to modern populations of Eastern Europe than those of central and southern Asia.44
In the Iron Age, farmers came from the Bactria–Margiana Archaeological Complex (also known as the Oxus civilization) and gradually started to influence this region. Anthropological studies show that the physical features observed from the Iron Age in Tashkurgan45 and other sites43 on the Pamir plateau are a mix of Eastern Mediterranean and Mongol features, which were characterized by dolichocrany combines with narrow face, which is different from the proto-Europoid features observed in the main populations belonging to the Andronovo culture. Genetic studies on Iron-Age samples and extant populations show that the main groups belonging to the western European mitochondrial lineages originated in the Near East and Iran regions.3 Since then, this area had been occasionally conquered by northern nomads until the sixteenth century. These nomads such as the Turkic and Mongols mainly came from the east Eurasian steppe and are unlikely to have brought U5a2a into the Pamir region.
As there are no extant individuals in the Tashkurgan and adjacent regions that possess the haplogroup U5a2a, except for one extant eastern Pamir individual from Tajikistan, we conclude that our ancient 700-year-old specimen containing the haplogroup U5a2a could be a residual evidence for prehistoric immigration by pastoralists, yet there is also a possibility that this ancient U5a2a is the descendant of central Asian who had Andronovo ancestry. Because of U5a2a’s low frequency, the earliest presence of the haplogroup is likely to have been affected by demographic processes, such as genetic drift or population replacement that may have occurred in the later agricultural population. Eventually, U5a2a may have reached extremely low frequencies or have gone extinct, thus preventing it from being detected in the extant Tarim Basin populations. The existence of U5a2a in our ancient sample and the extant southeastern Tajikistan population indicates this ancient nomad lineage continued in the eastern Pamir region.
In general, we provide a clear case study of an ancient mitochondrial genome that reveals a trace of prehistoric migration by pastoralists in the eastern Pamir region and shows that combining phylogenetic and archaeological data is a useful approach for ascertaining the phylogeny of mitochondrial subclades. Additional genomic information from extant and ancient individuals will be needed to provide greater insight into the evolutionary process and the population history of the Eastern Pamir region.
Accession codes
References
Museum, X. U. A. R. The investigation of Paleolithic Age ruins in Jirzanka of Tashkurgan Tajik autonomous county. Xinjiang Relics 1, 6 (1985).
Malyarchuk, B., Derenko, M., Grzybowski, T., Perkova, M., Rogalla, U., Vanecek, T. et al. The peopling of Europe from the mitochondrial haplogroup U5 perspective. PloS ONE 5, e10285 (2010).
Cui, Y., Li, C., Gao, S., Xie, C. & Zhou, H. Early Eurasian migration traces in the Tarim Basin revealed by mtDNA polymorphisms. Am. J. Phys. Anthropol. 142, 558–564 (2010).
Wu, Y. On the Bronze Culture of Xiabandi Cemetery in Kashi, Xinjiang. Western Reg. Stud. 04, 36–44 (2012).
Li, H., Zhao, X., Zhao, Y., Li, C., Si, D., Zhou, H. et al. Genetic characteristics and migration history of a bronze culture population in the West Liao-River valley revealed by ancient DNA. J. Hum. Genet. 56, 815–822 (2011).
Cui, Y., Lindo, J., Hughes, C. E., Johnson, J. W., Hernandez, A. G., Kemp, B. M. et al. Ancient DNA analysis of mid-holocene individuals from the Northwest Coast of North America reveals different evolutionary paths for mitogenomes. PloS ONE 8, e66948 (2013).
Lindgreen, S. AdapterRemoval: easy cleaning of next-generation sequencing reads. BMC Res. Notes 5, 337 (2012).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Wei, Z., Wang, W., Hu, P., Lyon, G. J. & Hakonarson, H. SNVer: a statistical tool for variant calling in analysis of pooled or individual next-generation sequencing data. Nucleic Acids Res. 39, e132 (2011).
Jonsson, H., Ginolhac, A., Schubert, M., Johnson, P. L. & Orlando, L. mapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29, 1682–1684 (2013).
Bandelt, H. J., Forster, P. & Rohl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16, 37–48 (1999).
Fan, L & Yao, Y. G. MitoTool: a web server for the analysis and retrieval of human mitochondrial DNA sequence variations. Mitochondrion 11, 351–356 (2011).
Drummond, A. J. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214 (2007).
Drummond, A. J., Suchard, M. A., Xie, D. & Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973 (2012).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods. 9, 772 (2012).
Guindon, S. & Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704 (2003).
Atkinson, Q. D., Gray, R. D. & Drummond, A. J. mtDNA variation predicts population size in humans and reveals a major southern Asian chapter in human prehistory. Mol. Biol. Evol. 25, 468–474 (2008).
Atkinson, Q. D., Gray, R. D. & Drummond, A. J. Bayesian coalescent inference of major human mitochondrial DNA haplogroup expansions in Africa. Proc. Biol. Sci. 276, 367–373 (2009).
Peng, M.-S. & Zhang, Y.-P. Inferring the population expansions in peopling of Japan. PloS ONE 6, e21509 (2011).
Excoffier, L., Estoup, A. & Cornuet, J. M. Bayesian analysis of an admixture model with mutations and arbitrarily linked markers. Genetics 169, 1727–1738 (2005).
Fan, L. & Yao, Y. G. An update to MitoTool: using a new scoring system for faster mtDNA haplogroup determination. Mitochondrion 13, 360–363 (2013).
Saillard, J., Forster, P., Lynnerup, N., Bandelt, H. J. & Norby, S. mtDNA variation among Greenland Eskimos: the edge of the Beringian expansion. Am. J. Hum. Genet. 67, 718–726 (2000).
Soares, P., Ermini, L., Thomson, N., Mormina, M., Rito, T., Rohl, A. et al. Correcting for purifying selection: an improved human mitochondrial molecular clock. Am. J. Hum. Genet. 84, 740–759 (2009).
Cooper, A. & Poinar, H. N. Ancient DNA: do it right or not at all. Science 289, 1139 (2000).
Sawyer, S., Krause, J., Guschanski, K., Savolainen, V. & Paabo, S. Temporal patterns of nucleotide misincorporations and DNA fragmentation in ancient DNA. PloS ONE 7, e34131 (2012).
Behar, D. M., van Oven, M., Rosset, S., Metspalu, M., Loogvali, E. L., Silva, N. M. et al. A "Copernican" reassessment of the human mitochondrial DNA tree from its root. Am. J. Hum. Genet. 90, 675–684 (2012).
Quintana-Murci, L., Chaix, R., Wells, R. S., Behar, D. M., Sayar, H., Scozzari, R. et al. Where west meets east: the complex mtDNA landscape of the southwest and Central Asian corridor. Am. J. Hum. Genet. 74, 827–845 (2004).
Comas, D., Calafell, F., Mateu, E., Perez-Lezaun, A., Bosch, E., Martinez-Arias, R. et al. Trading genes along the silk road: mtDNA sequences and the origin of central Asian populations. Am. J. Hum. Genet. 63, 1824–1838 (1998).
Ingman, M. & Gyllensten, U. Rate variation between mitochondrial domains and adaptive evolution in humans. Hum. Mol. Genet. 16, 2281–2287 (2007).
Irwin, J. A., Ikramov, A., Saunier, J., Bodner, M., Amory, S., Rock, A. et al. The mtDNA composition of Uzbekistan: a microcosm of Central Asian patterns. Int. J. Legal Med. 124, 195–204 (2010).
Malyarchuk, B. A. & Derenko, M. V. Mitochondrial DNA variability in Russians and Ukrainians: implication to the origin of the Eastern Slavs. Ann. Hum. Genet. 65, 63–78 (2001).
Malyarchuk, B. A., Grzybowski, T., Derenko, M. V., Czarny, J., Wozniak, M. & Miscicka-Sliwka, D. Mitochondrial DNA variability in Poles and Russians. Ann. Hum. Genet. 66, 261–283 (2002).
Pakendorf, B., Wiebe, V., Tarskaia, L. A., Spitsyn, V. A., Soodyall, H., Rodewald, A. et al. Mitochondrial DNA evidence for admixed origins of central Siberian populations. Am. J. Phys. Anthropol. 120, 211–224 (2003).
Melchior, L., Lynnerup, N., Siegismund, H. R., Kivisild, T. & Dissing, J. Genetic diversity among ancient Nordic populations. PloS ONE 5, e11898 (2010).
Bramanti, B., Thomas, M. G., Haak, W., Unterlaender, M., Jores, P., Tambets, K. et al. Genetic discontinuity between local hunter-gatherers and central Europe's first farmers. Science 326, 137–140 (2009).
Bermisheva, M., Tambets, K., Villems, R. & Khusnutdinova, E. [Diversity of mitochondrial DNA haplotypes in ethnic populations of the Volga-Ural region of Russia]. Mol. Biol. (Mosk.) 36, 990–1001 (2002).
Malyarchuk, B., Derenko, M., Grzybowski, T., Lunkina, A., Czarny, J., Rychkov, S. et al. Differentiation of mitochondrial DNA and Y chromosomes in Russian populations. Hum. Biol. 76, 877–900 (2004).
Grzybowski, T., Malyarchuk, B. A., Derenko, M. V., Perkova, M. A., Bednarek, J. & Wozniak, M. Complex interactions of the Eastern and Western Slavic populations with other European groups as revealed by mitochondrial DNA analysis. Forensic Sci. Int. Genet. 1, 141–147 (2007).
Keyser, C., Bouakaze, C., Crubezy, E., Nikolaev, V. G., Montagnon, D., Reis, T. et al. Ancient DNA provides new insights into the history of south Siberian Kurgan people. Hum. Genet. 126, 395–410 (2009).
Chernykh, E. N. Formation of the Eurasian "Steppe Belt" of stockbreeding cultures: viewed through the prism of archaeometallurgy and radiocarbon dating. Archaeol Ethnol Anthropol Eurasia 35, 36–53 (2008).
Mei, J. & Colin, S. The existence of Andronovo cultural influence in Xinjiang during the 2nd millennium BC. Antiquity 73, 570–578 (1999).
Guo, W. Archaeological Research on the Societies of the Late Prehistoric Xinjiang, (Shanghai Shiji Press, Shanghai, China, 2012).
Johnson, J. W. The Scythian: his rise and fall. J. Hist. Ideas 20, 250–257 (1959).
Ricaut, F. X., Keyser-Tracqui, C., Cammaert, L., Crubezy, E. & Ludes, B. Genetic analysis and ethnic affinities from two Scytho-Siberian skeletons. Am. J. Phys. Anthropol. 123, 351–360 (2004).
Han, K. Anceint Race Study on the Silk Road, (Xijiang People's Publishing House, Xinjiang, China, 1993).
Hey, J. On the number of New World founders: a population genetic portrait of the peopling of the Americas. PLoS Biol. 3, e193 (2005).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (Grant Nos. 41472024 and 31301025).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Ning, C., Gao, S., Deng, B. et al. Ancient mitochondrial genome reveals trace of prehistoric migration in the east Pamir by pastoralists. J Hum Genet 61, 103–108 (2016). https://doi.org/10.1038/jhg.2015.128
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.128
This article is cited by
-
Genetic kinship and admixture in Iron Age Scytho-Siberians
Human Genetics (2019)
-
Mitochondrial genomes uncover the maternal history of the Pamir populations
European Journal of Human Genetics (2018)
-
Cannabis in Eurasia: origin of human use and Bronze Age trans-continental connections
Vegetation History and Archaeobotany (2017)

{kind=link}
{kind=link}