
Abstract
Methyl-CpG-binding protein 2 (MeCP2) is a nuclear protein highly expressed in neurons that is involved in transcriptional modulation and chromatin remodeling. Mutations in MECP2 in females are associated with Rett syndrome, a neurological disorder characterized by a normal neonatal period, followed by the arrest of development and regression of acquired skills. Although it was initially thought that MECP2 pathogenic mutations in males were not compatible with life, starting from 1999 about 60 male patients have been identified and their phenotype varies from severe neonatal encephalopathy to mild intellectual disability. Targeted next-generation sequencing of a panel of intellectual disability related genes was performed on two unrelated male patients, and two missense variants in MECP2 were identified (p.Gly185Val and p.Arg167Trp). These variants lie outside the canonical methyl-CpG-binding domain and transcription repression domain domains, where the pathogenicity of missense variants is more difficult to establish. In both families, variants were found in all affected siblings and were inherited from the asymptomatic mother, showing skewed X-chromosome inactivation. We report here the first missense variant located in AT-hook domain 1 and we underline the importance of MECP2 substitutions outside the canonical MeCP2 domains in X-linked intellectual disability.
Similar content being viewed by others


Introduction
MECP2 (methyl-CpG-binding protein 2) mutations in females are associated with Rett Syndrome (RTT, MIM 312750),1 a severe neurodevelopmental disorder characterized by a normal perinatal clinical course (up to the age of 7–18 months), followed by an arrest of development and subsequent deterioration of language skills and purposeful use of the hands, known as regression stage.2 In this stage, patients show for the first time seizures and typical stereotypic hand movements, with a reduction in interpersonal contact.2 As RTT affects almost exclusively girls3 (with an estimated incidence of 1 in 10 000–15 000 female newborns), it was previously thought to result from an X-linked dominant mutation with lethality in hemizygous males.2 In 2001, it was demonstrated that MECP2 mutations are almost exclusively of paternal origin, explaining the rareness of affected males.4 Since 1999 about 60 cases of MECP2 mutated males have been reported5 (http://mecp2.chw.edu.au/). Many of these cases are mosaic or 47,XXY karyotype males showing RTT features.6, 7 Germline MECP2 mutations found in karyotypically normal males were proved to cause a wide spectrum of phenotypes, ranging from severe encephalopathy (children usually die at birth or in the first year of life)8 to X-linked intellectual disability (ID) or autism.5 In these latter cases, MECP2 variants are not found in RTT females and include only substitutions, even outside the canonical domains, or C-terminal deletions.5 This makes it harder to assess the pathogenicity of these variants and some of them turned out to be rare non-pathogenic variants or are still classified as ‘unknown significance’.5, 9
MeCP2 has two well-characterized functional domains: the methyl-CpG-binding domain (MBD) that can bind selectively to DNA at symmetrically methylated CpGs10 and the transcription repression domain (TRD).11 DNA methylation is the major modification of eukaryote genomes and, in vertebrates, this occurs predominantly at position 5′ of CpGs,12 resulting in gene silencing.13 MeCP2 is involved in transcriptional repression in many ways: it interacts by its TRD domain with the corepressor Sin3A and the histone deacetylase complex, recruiting them to the methylated CpG sites;14 it binds through the TRD domain to the co-repressors c-Ski and N-CoR and forms complexes independently from its interaction with Sin3A;15 it can also interact with transcription factor IIB, a key component of the basal transcriptional machinery.16 Nevertheless, MeCP2 was proved not to bind to the majority of promoter regions with the highest methylation levels and, conversely, the majority of the bound promoters lies within transcriptionally active genes.17 It is therefore more correct to consider MeCP2 as a transcriptional regulator, rather than simply a repressor. There is also a conserved region in the C-terminal portion of the protein (C-terminal domain). It is involved in binding both to naked DNA and to the nucleosome core18 and it is considered important since many pathogenic MECP2 mutations occur in this region.19 More recently, two conserved clusters of the protein have been characterized: AT-hook 1 and AT-hook 2, from amino acid 185 to 197 and 265 to 277, respectively20 (www.uniprot.org, entry: P51608). AT-hooks are highly conserved regions among species and have a very important role in DNA binding of non-histone, chromatin-associated proteins of the high-mobility group AT-hook, in which they were first described.21 No pathogenic missense variants have been reported so far in AT-hook 1 cluster.
In the present study, targeted next-generation sequencing of a panel of intellectual disability related genes was performed on two unrelated male patients, and two non-synonymous variants in MECP2 outside the canonical MBD and TRD domains were identified (p.Gly185Val and p.Arg167Trp). Data supporting pathogenicity of these variants and detailed clinical description of the families are provided, helping to better characterize the spectrum of MECP2 mutations in males.
Materials and methods
Targeted next-generation sequencing and data analysis
The technical process to enrich, amplify and sequence the exonic parts of the 565 ID-related candidate or known selected genes, as well as bioinformatic procedures for data analysis, have already been described.22
To assess the pathogenicity of the variants, several bioinformatics tools were consulted. PhyloP (http://compgen.bscb.cornell.edu/phast)23 evaluates the phylogenetic conservation of a specific nucleotide, using multiple sequence alignment. The tool assigns a positive value to conserved positions among species and negative scores to those with high substitution rates, ranging from +6.94 to −3.69. Sorting Intolerant From Tolerant (SIFT, http://sift.jcvi.org/) is a tool that, on the basis of the conservation of the wild-type amino acid, classifies non-synonymous variants as tolerated or deleterious, depending on the effect on the protein function.24 PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) predicts damaging effects of missense substitutions (in terms of structure and function), performing physical and phylogenetic analysis. This tool describes a variant quantitatively (giving a score between 0 and 1) and qualitatively (benign, possibly damaging or probably damaging).25 Mutation Taster (http://www.mutationtaster.org/) can assess the pathogenicity of all kinds of variants, giving as output four kinds of results: ‘predicted disease causing’, ‘predicted polymorphism’, ‘known disease causing’ or ‘known polymorphism’.26 We also checked for the presence of the variants in the National Center for Biotechnology Information Short Genetic Variations database (dbSNP142, http://www.ncbi.nlm.nih.gov/SNP/), Exome Aggregation Consortium data set (http://exac.broadinstitute.org) and an exome in house data set (100 Italian controls).
Variant confirmation and segregation analysis
Sanger sequencing was performed to confirm the presence of the variants in the index cases and segregation analysis was carried out in all the available family members. DNA was amplified by PCR, using appropriate primer pairs (Forward: 5′-TTTGTCAGAGCGTTGTCACC-3′; Reverse: 5′-CTTCCCAGGACTTTTCTCCA-3′) and conditions. PCR products were then analyzed using an ABI PRISM 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA).
X-chromosome inactivation status
To assess whether the non-manifestation of the phenotype in the carrier mothers could be caused by unbalanced X-chromosome inactivation (XCI) pattern, the X-inactivation status was analyzed exploiting a highly polymorphic trinucleotide repeat located in the androgen-receptor gene.27 DNA extracted from lymphocytes was digested overnight with HhaI enzyme that digests the unmethylated, active X chromosome. Digested and undigested DNA was amplified using fluorescent primers for androgen-receptor gene polymorphic region and the products were then separated using an ABI PRISM 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Gene Mapper software (Applied Biosystems, Foster City, CA, USA) was used to perform a comparison of allele areas ratio of undigested and digested DNA.
Computational analysis
The MeCP2 amino-acid sequence (P51608) was retrieved from UniProt database (http://www.uniprot.org/uniprot/). By using Protein Model Portal (PMP) web server,28 we obtained homology model of the region (amino acids 66–200) with the template structure PDB ID 1UB1 (www.rcsb.org/). The last part of this region, the AT-hook 1 motif (185–197), was latter modeled with the template PDB ID 2EZE and then the model was refined by energy minimization with GROMACS v 4.6.7 software.29
The DUET30 and mCSM31 programs were used to predict the possible effect of amino-acids substitutions on the protein structure and function. These latter approaches are novel machine-learning algorithms that use the three-dimensional structure to quantitatively predict the effects of nucleotide changes on protein stability and protein–protein and protein–nucleic acid affinities.
Results
Using targeted next-generation sequencing of 565 known or candidate ID-associated genes, two MECP2 variants (NM_004992.3) located outside the canonical MBD and TRD domains have been identified in two unrelated males showing ID (Figure 1). The clinical and molecular features of these patients are detailed below.

Three-generation pedigree of family 1 (a) and family 2 (b). The genotype at the MECP2 locus is indicated below tested individuals: +/y=hemizygous male; +/−=heterozygous female; −/−=absence of variant in female and −/y=absence of variant in male. Black symbols represent affected individuals, white symbols indicate healthy individuals and white symbols with black dots represent healthy female carriers. In a, the small black circle indicates a spontaneous abortion that occurred at the sixth week of pregnancy.
Pedigrees and clinical features
FAMILY 1 (no. 188)
In the first family (Figure 1a) there are two affected brothers. They were the sole offspring of healthy and unrelated parents, born after full-term and uneventful pregnancies.
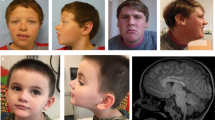
The older patient (Figure 2a) is six-years-old. At birth his weight was 3.650 kg (90th percentile) and the length was 51 cm (75th–90th percentile), occipital frontal circumference (OFC) was not available. Delivery and neonatal period were unremarkable. He started babbling at 14 months and said his first words when he was 16-months-old; independent walking was acquired at 17 months; sphincter control was achieved at 4 years. At 18 months, a global development regression was noticed with loss of the acquired spoken language, impaired social interaction and appearance of motor stereotypies such as hand flapping and body rocking. At the age of 6 he presents learning disability with absent speech and autistic spectrum disorder. Seizures are not reported. Laboratory test revealed occasional microhaematuria. Karyotype, molecular analysis for Fragile X syndrome and comparative genomic hybridization (CGH) array results were normal. Auxological parameters are in the normal range and he does not show peculiar facial features.

Pictures of male patients with MECP2 missense variants. Upper panels show photographs of the two affected siblings of family 1 (no. 188): the older one is 6-years-old (a), the younger one is 4-years-old (b). Note the absence of peculiar facial features. Lower panels show photographs of the three patients of family 2 (no. 177) at the age of 42 years (c), 41 years (d) and 32 years (e), demonstrating similar phenotype with high forehead, downturned nasal tip with wide pinnae, short philtrum, thin lips, truncal obesity and large hands. In c, patient also presents cleft lobe of the left ear. A full color version of this figure is available at the Journal of Human Genetics journal online.
The younger brother (Figure 2b) is 4-years-old. His auxological parameters at birth were: weight 3.560 kg (90th percentile) and length 50 cm (75th–90th percentile), OFC was not available. He started to say his first words when he was 10-months-old; he acquired independent walking at 14 months of age and sphincter control when he was 3-years-old. Similarly to his brother, he experienced a regression of acquired language and social skills at 18 months. Differently from the proband, he does not show stereotypies. Microhaematuria is not present yet.
FAMILY 2 (no. 177)
In the second family there are three affected brothers (Figure 1b). They were all born at term by Cesarean section after uneventful pregnancies. For each of them, a delay in language development and learning disabilities are reported, without regression. Seizures are not observed in any of them.
The eldest brother (Figure 2c) is 42-years-old. He presently shows severe ID (mental age of about 4 years) and impaired social interaction. The language is limited to simple phrases. The behavior is characterized by apathy, anxiety, hypochondria, shyness, obsessive demand for food and hypersomnia. He also suffers from mild-to-moderate pulmonary hypertension. Neurological examination, computed tomography of the brain and electroencephalogram resulted normal. Physical examination showed short stature (<3rd percentile), obesity (body mass index, 30.1), macrocephaly (OFC at 98th percentile), downturned nasal tip with wide pinnae, short philtrum, anteverted ears and large hands.
The second affected brother (Figure 2d) is 41-years-old and shows moderate ID (mental age of about 8 years), poor language and behavioral problems such as aggressiveness, low frustration tolerance, episodes of inappropriate laughter and apathy, obsessive demand for food and hypersomnia. Neurological examination was normal. At physical examination he exhibited short stature (<6th percentile), obesity (body mass index, 31.9), OFC at 60th percentile and craniofacial features with high forehead, downturned nasal tip with wide pinnae, short philtrum, thin lips, posteriorly rotated ears and large hands.
The younger one (Figure 2e) is 32-years-old and shows severe ID (mental age of about 5 years), language limited to simple sentences, social impairments, unmotivated laughter, apathy, obsessive demand for food and hypersomnia. In addition, he suffers from high myopia and was treated for keratoconus. Neurological examination, computed tomography of the brain and electroencephalogram did not show pathological signs. He presents short stature (8th percentile), obesity (body mass index, 32.1), macrocephaly (OFC at 98th percentile) and the same facial appearance with the additional feature of cleft right ear lobe.
The two younger siblings (one female and one male) are healthy and intellectually normal. Parents are fourth degree cousins; the father died of cancer, while the mother is still alive and healthy. She shows normal intelligence without neurological disorders.
Molecular analysis
Targeted next-generation sequencing analysis
The screening for pathogenic variants, performed using a custom panel, led to the identification of the c.554G>T (p.Gly185Val) substitution in exon 4 of MECP2 in the proband of family 1 (no. 188). The variant lies outside the two canonical functional domains of the protein (MBD and TRD) in a conserved cluster that ranges from amino acid 185 to 197, named AT-hook domain 1 (AT-hook 1). The substitution has not been reported neither in dbSNP142, nor in Exome Aggregatium Consortium, nor in internal exome database. The nucleotide is conserved (phyloP score: 4.16) and according to the bioinformatics analysis, the variant is predicted to be deleterious (SIFT, score: 0), disease causing (Mutation Taster, P-value: 1) and probably damaging (PolyPhen-2, score: 0.999).
In the proband of family 2 (no. 177), the c.499C>T (p.Arg167Trp) substitution in exon 4 of MECP2 was identified. The variant is located in the MBD/TRD inter-domain area, outside any known domain. The substitution is reported in dbsnp142 (rs61748420) with unavailable frequency and it is not described in Exome Aggregation Consortium or in internal exome database. The nucleotide is conserved (phyloP score: 3.92) and bioinformatics analysis indicates the variant as deleterious (SIFT, score: 0), disease causing (mutation taster, P-value: 1) and probably damaging (PolyPhen-2, score: 1).
Segregation analysis
The c.554G>T (p.Gly185Val) substitution was found in both affected brothers in hemizygous state and in the healthy mother in heterozygous state (Figure 1a).
All the affected male brothers from family 2 (no. 177) were hemizygous for the c.499C>T (p.Arg167Trp) variant, while the healthy sister and the healthy brother did not carry it. The mother showed the substitution in heterozygous state (Figure 1b).
XCI analysis
To assess if the absence of a clinical phenotype in the two carrier mothers could be due to an unbalanced XCI pattern, the XCI analysis was performed on DNA extracted from lymphocytes. In the mother of the first family (no. 188), carrying the c.554G>T (p.Gly185Val) variant, the XCI status resulted moderately unbalanced (76:24). The mother of family 2 (no. 177), that is healthy carrier of the c.499C>T (p.Arg167Trp) substitution, had a skewed (16:84) XCI pattern.
Computational analysis
The PMP web server28 was used to obtain the molecular model of wild-type (Figures 3a and b) and mutated sequence (Figures 3c and d) of MeCP2 for both ISOFORM_A (NM_004992.3) and ISOFORM_B (NM_001110792.1). Actually the two obtained models were structurally identical since the two isoforms are different only in the N-terminal region that has no effect in remaining protein folding.

Cartoon representation of homology model of the 66E-200K MeCP2 domain obtained by Protein Model Portal. The wild-type structured model of the protein is represented with the amino-acid Arg167 (a) and the amino-acid Gly185 (b) highlighted. (c, d) show the mutated protein for p.Arg167Trp and p.Gly185Val variants, respectively. A full color version of this figure is available at the Journal of Human Genetics journal online.
The prediction programs (DUET30and mCSM31) indicated a pathogenic effect for both variants (Table 1). In particular, the p.Arg167Trp variant was predicted to have a destabilizing effect on protein structure (negative ΔΔG value). For the other variant (p.Gly185Val), in addition to an effect on protein stability, a destabilizing affinity change (negative ΔΔG value) in protein–DNA interaction was predicted, in accordance with the localization inside an AT-hook1 (185–197) motif.
Discussion
Mutations in the MECP2 gene, located on the X chromosome, are mainly associated in females to Rett syndrome (both the classic form and the milder Z-RTT variant).32 Males with MECP2 mutations can show a broader range of clinical presentations, ranging from severe phenotypes (congenital encephalopathy or Rett syndrome) to milder ones (ID or autism)5 (Figure 4). MECP2 mutations in males are mainly located in the two canonical functional domains of the protein: the MBD (amino acids 78–162)33 and the TRD (amino acids 207–310)34 (Figure 4). Rare reported mutations are localized in the connecting peptide between MBD and TRD or in the C-terminal portion of the protein5 (Figure 4). In the present study, we report the identification of MECP2 missense variants (p.Gly185Val and p.Arg167Trp) outside the canonical domains of the protein in two families with ID (Figure 1). Variants are present only in affected siblings and healthy mothers are carriers in both families (Figure 1). Maternal XCI resulted skewed in both cases. Bioinformatic analysis indicates the affected nucleotides as conserved (PhyloP) and all the predictive tools employed (SIFT, PolyPhen-2, Mutation Taster) strongly support the pathogenicity of the two variants. Computational analysis after modeling of wild-type and mutated protein suggested a pathogenic effect for both variants, in terms of structure stability (p.Arg167Trp and p.Gly185Val) and protein–DNA affinity change (p.Gly185Val) (Figure 3 and Table 1). Variants are not present neither in international control reference groups such as the Exome Aggregation Consortium data set (http://exac.broadinstitute.org) nor in an exome in house data set (100 Italian individuals).

MECP2 mutations reported in males and associated phenotypes. In each panel (a, b and c) a schematic representation of the MeCp2 protein is shown. Above protein structure, mutations found in males are indicated (a: neonatal encephalopathy; b: Rett spectrum phenotype; c: ID/autism); below protein structure, the same mutations identified in females are reported. The phenotypes are represented as follows: neonatal encephalopathy in dark gray, Rett phenotype spectrum in light gray, ID in white and autism in striped boxes. Variants described in this paper are highlighted with hatching (c).
The variant p.Gly185Val (family 1) has never been reported in the literature, although a frameshift at this position is described,35 and lies in one of the conserved high-mobility group like AT-hook motifs of MeCP2: AT-hook 1 (amino acids 185–197). These motifs were first identified in high-mobility group chromatin proteins and recognize the minor groove of AT-rich DNA.21 The MeCP2 AT-hook 2 motif (amino acids 265–277) has been recently demonstrated to be crucial for chromatin shaping and DNA binding in whole RTT mouse brain.20 The maintenance of this cluster in MeCP2-G273X mice has been considered the cause of the observed milder phenotype (late onset and longer survival) respect to MeCP2-G270X mice, highlighting these three residues as capable of determining the clinical course of the disease.20 Differently, very little is known about the AT-hook 1 motif and to date it is considered of unknown significance.20 The identification of the first MECP2 missense mutation in the AT-hook 1 motif strongly suggests that this cluster may also have an important role for MeCP2 function.
The p.Arg167Trp variant is located in the MBD/TRD connecting peptide (Figure 4c), outside any known functional or predicted domain. Until now, the variant has been considered of unknown pathogenicity since it was identified in a single family with ID.5, 36 However, the identification of another ID family with the same MECP2 variant segregating with the disease strongly argues in favor of its pathogenicity. The family reported by Couvert et al.36 is a large three-generation family with four males with non-specific ID (age range: 25–50 years). In common with the three affected boys reported in the present study (age range: 32–42), male patients presented with moderate-to-severe ID and obesity.32 However, differently from the family reported by Couvert et al., our patients do not show resting tremors. Since mutation type or XCI cannot be implicated in determining this phenotypic variability, modifier genes elsewhere in the genome probably may have an important role.
To our knowledge, about 60 males mutated in MECP2 have been reported so far5 (http://mecp2.chw.edu.au/). Non-mosaic, karyotypically normal males with MECP2 point mutations can be divided in three groups with decreasing phenotypic severity: (1) Males with neonatal encephalopathy and early death whose mutations are also found in females with RTT (Figure 4a); (2) Males with RTT or RTT-like phenotype whose mutations can be found in females with variable phenotypes, ranging from RTT to non-specific ID (Figure 4b); (3) Males with ID whose mutations are not found in females with RTT but in females with milder phenotypes (Figure 4c). In the first group, mutations are mainly represented by early truncating ones or substitutions in MBD or TRD (Figure 4a). Differently, patients from the second and the third group bear missense changes also located in the C-terminal region or late truncating mutations (Figures 4b and c). The third group is the only one that includes missense changes in the MBD/TRD connecting peptide (Figure 4c).
Besides early truncating mutations whose pathogenicity is not questionable, the clinical interpretation of late truncating or missense changes outside the canonical domains is challenging and some of these changes turned out to be non-pathogenic variants, suggesting to proceed with caution.37 Segregation with disease in the family argues in favor of a pathogenic significance but it cannot exclude a casual association. Given the rareness of these cases, the identification of additional families with the same MECP2 variant and a comparable phenotype is fundamental to assess pathogenicity, as in the case of the variant p.Arg167Trp reported in the present study. As for the second variant, the localization in a predicted functional motif (AT-hook 1) suggests its functional importance and alerts for the identification of additional cases with changes disrupting these specific residues. The absence of both variants in control population of the same ethnic origin is also a crucial factor supporting pathogenicity and it should be always considered in a diagnostic setting.
In conclusion, this paper reports the identification of missense mutations outside the two crucial functional domains of MeCP2 protein in two new families with ID. These mutations lie in the ‘gray area’ connecting the MBD and the TRD domains, where interpretation of missense variant pathogenicity is more difficult and raises important issues in genetic counseling. MECP2 mutations in this region are not present in severe phenotypes (neonatal encephalopathy or RTT) but are found only in patients with ID of different degrees and additional symptoms in some cases (Figure 4). In this paper, we describe in detail the clinical picture of the affected males, expanding the spectrum of phenotypes associated to MECP2 mutations.
Finally, this study reports for the first time a MECP2 missense mutation in the AT-hook 1 motif of the protein, pointing to its possible functional importance as already demonstrated for the AT-hook 2 motif.20
References
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke., U. & Zoghbi, H. Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188 (1999).
Hagberg, B., Aicardi, J., Dias, K. & Ramos, O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett's syndrome: report of 35 cases. Ann. Neurol. 14, 471–479 (1983).
Rett, A. Ueber ein eigenartiges hirnatrophisches syndrom bei hyperammonaemie im kindesalter. Wien. Med. Wochenschr. 116, 723–726 (1966).
Trappe, R., Laccone, F., Colibanschi, J., Meins, M., Huppke, P., Hanefeld, F. et al. MECP2 mutations in sporadic cases of Rett syndrome are almost exclusively of paternal origin. Am. J. Hum. Genet. 68, 1093–1101 (2001).
Villard, L. MECP2 mutations in males. J. Med. Genet. 44, 417–423 (2007).
Clayton-Smith, J., Watson, P., Ramsden, S. & Black, G. C. Somatic mutation in MECP2 as a non-fatal neurodevelopmental disorder in males. Lancet 356, 830–832 (2000).
Topcu, M., Akyerli, C., Sayi, A., Toruner, A., Kocoglu, S. R., Cimbis, M. et al. Somatic mosaicism for a MECP2 mutation associated with classic Rett syndrome in a boy. Eur. J. Hum. Genet. 10, 77–81 (2002).
Villard, L., Cardoso, A. K., Chelly, P. J., Tardieu, P. M. & Fontes, M. Two affected boys in a Rett syndrome family: clinical and molecular findings. Neurology 55, 1188–1193 (2000).
Laccone, F., Zoll, B., Huppke, P., Hanefeld, F., Pepinski, W. & Trappe, R. MECP2 gene nucleotide changes and their pathogenicity in males: proceed with caution. J. Med. Genet. 39, 586–588 (2002).
Meehan, R. R., Lewis, J. D. & Bird, A. P. Characterization of MeCP2, a vertebrate DNA binding protein with affinity for methylated DNA. Nucleic Acids Res. 20, 5085–5092 (1992).
Razin, A. CpG methylation, chromatin structure and gene silencing-a three-way connection. EMBO J. 17, 4905–4908 (1998).
Hendrich, B. & Bird, A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol. Cell. Biol. 18, 6538–6547 (1998).
Bartolomei, M. S. & Tilghman, S. M. Genomic imprinting in mammals. Annu. Rev. Genet. 31, 493–525 (1997).
Nan, X., Ng, H., Johnson, C., Laherty, C., Turner, B., Eisenman, R. et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393, 386–389 (1998).
Kokura, K., Kaul, S., Wadhwa, R., Nomura, T., Khan, M., Shinagawa, T. et al. The Ski protein family is required for MeCP2-mediated transcriptional repression. J. Biol. Chem. 276, 34115–34121 (2001).
Kaludov, N. K. & Wolffe, A. P. MeCP2 driven transcriptional repression in vito: selectivity for methylated DNA, action at a distance and contacts with the basal transcription machinery. Nucleic Acids Res. 28, 1921–1928 (2000).
Yasui, D. H., Peddada, S., Bieda, M. C., Vallero, R. O., Hogart, A., Nagarajan, R. P. et al. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc. Natl Acad. Sci. USA 104, 19416–19421 (2007).
Chandler, S. P., Guschin, D., Landsberger, N. & Wolffe, A. P. The methyl-CpG binding transcriptional repressor MeCP2 stably associates with nucleosomal DNA. Biochemistry 38, 7008–7018 (1999).
Yusufzai, T. M. & Wolffe, A. P. Functional consequences of Rett syndrome mutations on human MeCP2. Nucleic Acids Res. 28, 4172–4179 (2000).
Baker, S. A., Chen, L., Wilkins, A. D., Yu, P., Lichtarge, O. & Zoghbi, H. Y. An AT-hook domain in MeCP2 determines the clinical course of Rett syndrome and related disorders. Cell 152, 984–996 (2013).
Reeves, R. & Nissen, M. S. The A.T-DNA-binding domain of mammalian high mobility group I chromosomal proteins. A novel peptide motif for recognizing DNA structure. J. Biol. Chem. 265, 8573–8582 (1990).
Grozeva, D., Carss, K., Spasic-Boskovic, O., Parker, M. J., Archer, H., Firth, H. V. et al. De novo loss-of-function mutations in SETD5, encoding a methyltransferase in a 3p25 microdeletion syndrome critical region, cause intellectual disability. Am. J. Hum. Genet. 94, 618–624 (2014).
Pollard, K. S., Hubisz, M. J., Rosenbloom, K. R. & Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 20, 110–121 (2010).
Ng, P. C. & Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 11, 863–874 (2001).
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 11, 361–362 (2014).
Allen, R. C., Zoghbi, H. Y., Moseley, A. B., Rosenblatt, H. M. & Belmont, J. W. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human adrogen-recetor gene correlates with X chromosome inactivation. Am. J. Hum. Genet. 51, 1229–1239 (1992).
Haas, J., Roth, S., Arnold, K., Kiefer, F., Schmidt, T., Bordoli, L. et al. The Protein Model Portal—a comprehensive resource for protein structure and model information. Database (Oxford) 2013, bat031 (2013).
Hess, B., Kutzner, C., van der Spoel, D. & Lindahl, E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 (2008).
Pires, D. E., Ascher, D. B. & Blundell, T. L. DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 42, W314–W319 (2014).
Pires, D. E., Ascher, D. B. & Blundell, T. L. mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 30, 335–342 (2014).
De Bona, C., Zappella, M., Hayek, G., Meloni, I., Vitelli, F., Bruttini, M. et al. Preserved speech variant is allelic of classic Rett syndrome. Eur. J. Hum. Genet. 8, 325–330 (2000).
Nan, X., Meehan, R. & Bird, A. Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res. 21, 4886–4892 (1993).
Nan, X., Campoy, F. & Bird, A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 88, 471–481 (1997).
Buyse, I. M., Fang, P., Hoon, K. T., Amir, R. E., Zoghbi, H. Y. & Roa, B. B. Diagnostic testing for Rett syndrome by DHPLC and direct sequencing analysis of the MECP2 gene: identification of several novel mutations and polymorphisms. Am. J. Hum. Genet. 67, 1428–1436 (2000).
Couvert, P., Bienvenu, T., Aquaviva, C., Poirier, K., Moraine, C., Gendrot, C. et al. MECP2 is highly mutated in X-linked mental retardation. Hum. Mol. Genet. 10, 941–946 (2001).
Moncla, A., Kpebe, A., Missirian, C., Mancini, J. & Villard, L. Polymorphisms in the C-terminal domain of MECP2 in mentally handicapped boys: implications for genetic counseling. Eur. J. Hum. Genet. 10, 86–89 (2002).
Acknowledgements
The Cell lines and DNA bank of Rett Syndrome, X-linked mental retardation and other genetic diseases, member of the Telethon Network of Genetic Biobanks (project no. GTB12001), funded by Telethon Italy, and of the EuroBioBank network, provided us with specimens.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Bianciardi, L., Fichera, M., Failla, P. et al. MECP2 missense mutations outside the canonical MBD and TRD domains in males with intellectual disability. J Hum Genet 61, 95–101 (2016). https://doi.org/10.1038/jhg.2015.118
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.118
This article is cited by
-
Treating Rett syndrome: from mouse models to human therapies
Mammalian Genome (2019)
-
Transcriptome analysis of microglia in a mouse model of Rett syndrome: differential expression of genes associated with microglia/macrophage activation and cellular stress
Molecular Autism (2017)
-
Brain phosphorylation of MeCP2 at serine 164 is developmentally regulated and globally alters its chromatin association
Scientific Reports (2016)
