
Abstract
Immunoglobulin A nephropathy (IgAN) is the most common form of primary glomerulonephritis in many parts of the world. Although previous genome-wide association studies (GWAS) identified the major susceptibility loci for IgAN, the causal genes currently remain unknown. We performed a GWAS using 23 465 microsatellite (MS) markers to identify genes related to IgAN in a Japanese population. A pooled sample analysis was conducted in three-stage screenings of three independent case–control populations, and after the final step of individual typing, 11 markers survived. Of these, we focused on two regions on 6p21 and 12q21 because they (i) showed the strongest relationship with IgAN, and (ii) appeared to be highly relevant to IgAN in view of several previous studies. These regions contained the HLA, TSPAN8 and PTPRR genes. This study on GWAS, using >20 000 MS markers, provides a new approach regarding susceptible genes for IgAN for investigators seeking new tools for the prevention and treatment of IgAN.
Similar content being viewed by others
Introduction
Immunoglobulin A nephropathy (IgAN) is the most common form of primary glomerulonephritis worldwide and an important cause of end-stage renal disease in various parts of the world.
Microscopically, IgAN is characterized by predominant IgA deposits in glomerular mesangial areas. Its prevalence is as high as 50% of all cases of glomerulonephritis in Asia, especially in Japan, with significant geographic and ethnic vari'ations being reported in its prevalence.1 Because biopsy is a prerequisite for diagnosis, the true incidence of this disorder remains unknown.
Various mechanisms have been proposed for the characteristic IgA deposits in glomerular mesangial areas. Variations in clinical presentation, progression and therapeutic responses among patients with similar immunohistological features in renal biopsy specimens indicates that a diagnosis may encompass several subtypes that are indistinguishable on the basis of renal biopsy or clinical data alone. Hence, a complicated determination may be required, in which the interplay between multiple different genes and environmental factors possibly underlie different subsets of patients.2 The reported prevalence of IgAN appears to reflect the demographic and ethnic characteristics of the specific populations studied, in which several cases of familial IgAN and identical twins with IgAN have been reported.3, 4, 5 Therefore, IgAN is a complex disorder, the etiology of which involves immunological, environmental and genetic factors.5 Because the combination of various genes and environmental factors critically influences the incidence of IgAN, the interrelationship between the susceptibility locus and disease phenotypes needs to be investigated using appropriate models.6 Although the pathogenesis of IgAN currently remains unknown, evidence for the role of genetic factors in the development and progression of IgAN has accumulated. Previous studies reported the familial aggregations of IgAN, affected sib pairs and offspring pairs from multiple ethnic groups. In a recent family study of patients with biopsy-proven IgAN, a recurrence risk (λ) of approximately 16 was noted in first-degree relatives and of 2.4 in second-degree relatives.7
Several genome-wide linkage studies have been published to date.8 However, the application of a linkage analysis to IgAN has so far only achieved limited success. Furthermore, the examination of known genes in the linkage intervals reported has not revealed any compelling candidates. The advantage of genome-wide linkage studies is that they do not require a priori assumptions about disease pathogenesis. Unfortunately, linkage studies are highly sensitive to the inaccurate specification of clinical phenotypes, and their power is limited to the detection of rare genetic variants with a relatively large effect on the risk of disease.
The most comprehensive candidate gene study reported to date has been on the selectin gene cluster on chromosome 1 by Takei et al.9 Selectins were identified as possible candidate genes in view of their expression patterns and suspected functional role as adhesion molecules in the accumulation of leukocytes within the glomerulus and interstitium of the kidney. However, most candidate gene association studies on IgAN have not been replicated.
Genome-wide association is a powerful method for elucidating the genetic basis of the sporadic forms of diseases such as IgAN. Prior information on the pathogenesis of disease is not required when analyzing the entire genome. The findings of a large-scale genome-wide association study (GWAS) on IgAN were recently reported.10, 11, 12 They identified major histocompatibility complex (MHC) on chromosome 6p21 and CFHR1 and CFHR3 on chromosome 1q32.
Linkage studies, candidate gene studies and GWAS each have unique advantages and disadvantages. In the present study, we used a distinctive new approach using microsatellite (MS) markers instead of single nucleotide polymorphisms (SNPs) as polymorphic markers. MS markers may represent a new and complementary tool for the genetic dissection of common and rare diseases. We herein applied GWAS for IgAN using whole-genome MS markers to identify the susceptibility loci for IgAN.
Materials and methods
Study subject selection
A total of 915 Japanese patients (438 men and 477 women), who were all over 18 years old, were recruited from Yokohama City University, Tokai University, Osaka University and Niigata University. All IgAN patients were diagnosed by renal biopsy. IgAN was defined as glomerulonephritis with predominantly IgA deposits in the mesangium of all glomeruli. Patients with Henoch-Schonlein purpura and secondary IgAN such as hepatic glomerulosclerosis were excluded from the analysis. A total of 481 Japanese healthy volunteers (267 male and 214 female) were analyzed as control subjects. These healthy subjects did not have hematuria, proteinuria or kidney dysfunction. We obtained informed consent from all patients and healthy individuals whose DNA samples were used in the analyses.
Clinical data were also recorded (age, gender, excretion of protein in urine, serum creatinine, serum IgA, systolic blood pressure and diastolic blood pressure). All personal identities associated with medical information and blood samples were carefully eliminated and replaced with anonymous identities at each recruiting institution.
The Ethical Committees of Yokohama City University, Tokai University, Osaka University and Niigata University approved the study protocol, and written informed consent was obtained from all patients.
Marker information
The repeated sequence of MS markers was computationally detected from all of the chromosomes (in four versions of the human genome draft sequence: Golden Path Feb 2009 to the National Center for Biotechnology Information).13 At present, our laboratory has built 23 465 markers. The MS markers were investigated for repeat polymorphisms in 200 healthy Japanese using the DNA pooling method. Our criteria for the selection of MS markers for the IgAN association study were dinucleotide repeats with >10 repeats; tri-, tetra- and pentanucleotide repeats with >5 repeats; and polymorphic MS markers with heterozygosity of >30%, but not those with heterozygosity of >85% to eliminate any unstable and highly mutated MS markers.
Pooled DNA and genotyping
The pooled DNA method for MS typing was performed according to the protocol of Collins et al.13 with a slight modification.14 The key factor in this methodology was the absolute equality of individual DNA quantities; therefore, we used a highly accurate quantitative procedure to construct a pooled DNA template for PCR amplification. DNA was extracted using the QIAamp DNA blood kit (QIAGEN, Valencia, CA, USA) with a standardized protocol to prevent variations in the quality of DNA. This was followed by 0.8% agarose gel electrophoresis to check for DNA degradation and RNA contamination. DNA concentrations were precisely measured using the PicoGreen fluorescence assay (Molecular Probes, Tokyo, Japan) as previously described.15, 16 The DNA concentration was unified by repeating the procedure to reduce concentration variations, and the final concentration variation was ±5%.
The first set for the association study was the DNA pools of 150 patients (81 males and 69 females) and 150 controls (81 males and 69 females). The quality of the pooled DNA was confirmed by comparing allelic distributions between individual and pooled typing results using 23 MS markers, unless there was the absence of any significant difference (P⩽0.05) in allele frequencies between pooled and individual DNA typing. In the first round screening, 23 465 MS markers were used. All MS markers and the methods for MS genotyping used in this study have been described by Tamiya et al.15 PCR was performed on pooled DNAs in a 20-μl reaction mixture containing 100 ng pooled DNA, 0.5 U AmpliTaq DNA polymerase, 1 × reaction buffer with 1.5 mM MgCl2 provided by the manufacturer (Applied Biosystems, Tokyo, Japan), 5 μM of each primer and 0.25 mM of each deoxyribonucleotide triphosphate in 96-well plates. PCR amplification was performed on a GeneAmp PCR System 9700 (Applied Biosystems) with the following conditions: 96 °C for 9 min (hot start), 57 °C for 1 min and 72 °C for 1 min followed by 30 cycles of 96 °C for 45 s, 57 °C for 45 s and 72 °C for 1 min. Pooled MS genotyping procedures were carried out after PCR amplification according to standard protocols using an ABI3730 DNA analyzer (Applied Biosystems). Various kinds of information, such as peak positions and heights, were manually extracted by the PickPeak and MultiPeaks programs, developed by Applied Biosystems Japan, from the multipeak pattern in chromatogram ABI fsa files. Multiple peak patterns in the pooled DNA showed the distribution of allele frequencies in the subjects. Among them, markers showing significance of P⩽0.05 were subjected to the second stage.
A second set was also grouped from the samples of 150 independent patients (82 males and 68 females) and 150 controls (82 males and 68 females). The markers showing significance of P⩽0.05 in the second screening were subjected to a third stage.
A third set was also grouped from the samples of 150 independent patients (81 males and 69 females) and 150 controls (81 males and 69 females). We confirmed that the positive allele was the same at each screening stage.
All of the positive markers that remained significant (P⩽0.05) in the third screening were confirmed by individual genotyping using the same set of 450 patients (244 males and 206 females) and 450 controls (244 males and 206 females) as the final step. PCR was performed on individual DNAs in a 10-μl reaction mixture containing 1 ng individual DNA, 0.25 U AmpliTaq DNA polymerase, 1 × reaction buffer with 1.5 mM MgCl2 provided by the manufacturer (Applied Biosystems), 1.25 μM of each primer and 0.25 mM of each deoxyribonucleotide triphosphate in 96-well plates. PCR amplification was performed on GeneAmp PCR System 9700 (Applied Biosystems) with the following conditions: 95 °C for 9 min (hot start), adapted x°C for 1 min and 72 °C for 1 min followed by 40 cycles of 96 °C for 45 s, adapted x°C for 45 sec and 72 °C for 1 min. Individual MS genotyping procedures were carried out after PCR amplification according to standard protocols using an ABI3730 DNA analyzer (Applied Biosystems). Peak positions and heights were manually extracted by the MultiTyper programs, developed by Applied Biosystems Japan. We also confirmed that the positive allele in individual typing was the same as that at each screening stage.
Statistical analysis
Measurements of the heights of multiple peaks in the pooled DNA were applied to an association analysis. Allele frequencies in pooled DNA typing were estimated from the height of the peaks (fluorescence intensity): each allele frequency was determined by dividing the height of each allele by the summed height of all alleles.16 To calculate P values, we used two types of Fisher’s exact test for 2 × 2 contingency tables for each individual allele and 2 × m contingency tables for each locus, in which m referred to the number of marker alleles observed in a population. The Markov chain/Monte Carlo simulation method was used to execute Fisher’s exact test for the 2 × m contingency table. The simple 'allelic', but not 'genotype' association was presented for the 2 × 2 contingency tables for MS markers. These analyses were executed using the software package, AStat. The method of Pritchard and Rosenberg was used for the detection of stratification in case and control populations using 23 MS markers.17 The Hardy–Weinberg test for allele frequency distributions at the MS loci was performed by the P test for differentiation, as determined by GenePop 3.4 (the Laboratiore de Genetique et Environment, Montpellier, France). Other basic analyses were carried out using Microsoft Excel. The authors had full access to the data and take responsibility for its integrity.
Results
Three-stage screening: pooled DNA typing
We initially identified 14 markers as IgAN susceptibility loci by three-stage screening of the three independent case–control populations (stage 1: 150, stage 2: 150, stage 3: 150 patients with IgAN and the same number of healthy individuals). Three-stage screenings were intended to sequentially replicate the results in the three independent sample populations and eliminate pseudopositive markers resulting from type I errors.18 The number of markers decreased from 23 465 to 2612 markers in the first screening, then to 521 markers in the second screening and to 210 markers in the third screening. We obtained 210 positive markers in the third screening, but the concordance of the allele associated with the disease in the same marker through all pooled DNA screenings was ignored. The narrowed-down markers indicated completely identical markers in the associated allele and the direction of differences in allele frequency between cases and controls through all pooled DNA screenings. We eventually obtained 14 positive markers after the three-stage screening (Table 1). We also performed a comparison of expected positives with observed positives. The number of biologically relevant positive markers with P-values being below 0.05 in each screening is shown (Supplementary Table 4).
Individual typing
As the results of pooled typing were presumptive, we genotyped a total of 900 individuals (450 case subjects versus 450 control subjects) and reanalyzed the 14 markers used in the three-stage screening procedure. These individuals were the same individuals as those used in pooled typing and were not from a new cohort. We reduced the number of positive markers from 14 to 11 loci by using individual genotyping in GWAS for IgAN (Table 2). All 11 of the markers were significant (P<0.05) by a 2 × 2 analysis. Eleven loci were observed on chromosomes 2, 4, 5, 6, 10, 11, 12, 17 and 18. The observed and expected frequencies of each genotype in the case and control subjects followed the Hardy–Weinberg equilibrium (data not shown).
HLA and TSPAN8-PTPRR gene regions
Among the 11 positive markers that remained significant after individual typing, two loci on 6p21 and 12q21, representing the human leukocyte antigen (HLA) and TSPAN8 genes, respectively, were selected for further analyses because they were functionally interesting and significantly different between IgAN patients and the controls.
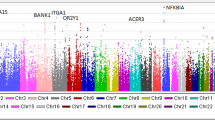
We set the 12 new MS markers on 6p21 and 12q21 (Tables 3 and 4 and Supplementary Table 1), and performed individual typing for these 12 MS markers in 900 individuals. Six out of the eight markers on 6p21 (Table 3 and Figure 1) and three out of the four markers on 12q21 showed significant differences in allele frequency (Table 4 and Figure 2).

Results of individual typing of nine MS markers on 6p21. D6S0483i was a positive marker in the three-stage screening of GWAS and individual typing (Table 2). We set eight new MS markers on 6p21.32-33 around D6S0483i and performed individual typing of these eight MS markers in 900 individuals. As a result, six out of eight markers on 6p21.32 showed significant differences in allele frequency. The locations of the known genes (NOTCH4, C6orf10, BTNL2, HLA-DRA, HLA-DRB5, HLA-DRB1, HLA-DQA1 and HLA-DQB1) are shown by lines at the bottom of this figure.

Results of individual typing of five MS markers on 12q21.1. D12S0933i was a positive marker in the three-stage screening of GWAS and individual typing (Table 2). We designed PCR primers for four new MS markers on 12q21.1 around D12S0933i and performed individual typing of these four MS markers in 900 individuals. As a result, three out of four markers on 12q21.1 showed significant differences in allele frequency. The locations of the known genes (PTPRR and TSPAN8) are shown by lines at the bottom of this figure.
A conditional analysis of the associations with these new MS markers across the HLA on chromosome 6p21 in our study suggested the presence of a susceptibility locus within this region. The most strongly associated MS marker, D6S2913, was mapped within 81 kb downstream of HLA-DRA, 159 kb downstream of HLA-DRB1, 283 kb downstream of HLA-DQA1 and 301 kb downstream of HLA-DQB1. The second most strongly associated MS marker, D6S0588, was mapped within 192 kb upstream of HLA-DRA, 114 kb upstream of HLA-DRB1, 11 kb downstream of HLA-DQA1 and 28 kb downstream of HLA-DQB1 (Figure 1). Therefore, the 165 kb region around the C6orf10 gene, which was defined by D6S2913, D6S0483i and D6S2892 on chromosome 6p21.32 and showed the strongest association with IgAN, may be a critical region for disease-susceptibility loci. Furthermore, the HLA –DR and –DQ subregions around the D6S0588 marker may be other critical regions in the HLA region.
In the locus 12q21, the results from three new MS markers, AC001, AGGF1 and G4A2F1, next to D12S0933i also showed significant relationships, suggesting that the susceptible region was located around the TSPAN8 and PTPRR gene region (Figure 2). The most strongly associated MS marker, AC001, was located in the region 42 kb upstream of PTPRR and 162 kb downstream of TSPAN8.
We showed the linkage disequilibrium (LD) across each marker on chromosome 6 (Supplementary Table 2) and chromosome 12 (Supplementary Table 3). LD was calculated using GENEPOP. This analysis demonstrated LD between additional markers and genome-wide markers in each locus (chromosomes 6 and 12) that were associated with IgAN. Moreover, we also estimated haplotypes between AC001 and D12S0933i using PHASE v2.1, which demonstrated that a haplotype harboring alleles associated with both loci was strongly associated with IgAN (case: 0.0244, control: 0.0033, odds ratio=7.43 (95% confidence interval 2.22–39.0), P-value: 7.91E-5). Therefore, D12S0933i in the genome-wide set may capture AC001, which is strongly associated with IgAN.
Discussion
Genome-wide association is a powerful method for elucidating the genetic basis of the sporadic forms of diseases such as IgAN. Prior information on the pathogenesis of disease is not required when analyzing the entire genome. A large-scale GWAS on IgAN recently reported its findings.10, 11, 12 The first IgAN GWAS used identified association signals at the MHC locus.10 The second IgAN GWAS identified three independent loci in the MHC on chromosome 6p21, as well as the common deletion of CFHR1 and CFHR3 (on chromosome 1q32, respectively).11 The third IgAN GWAS identified replicated four loci in the MHC region, and new loci on chromosome 17p13 and 8q23.12 These findings delineated the genetic architecture of sporadic IgAN, uncovering new pathogenic pathways and connections to other immune-mediated disorders.
Linkage studies, candidate gene studies and GWAS each have unique advantages and disadvantages. In the present study, we used a distinctive new approach using MS markers instead of SNPs as polymorphic markers. MS markers are highly polymorphic, showing a high degree of heterozygosity and LD lengths in the range of 100–200 kb.15 GWAS using MS can, thus, be used as a complementary tool for the genetic dissection of common and rare diseases. We herein applied GWAS for IgAN using whole-genome MS markers to identify the susceptibility loci for IgAN.
We performed a GWAS with 23 465 MS markers to detect candidate loci for IgAN. The pooled DNA method was applied to reduce the cost and technical burden of genome-wide genotyping. The pooled DNA method was established by Collins et al.,13 and has since been used by other studies.19, 20 We performed three-phase screening, which does not allow us to avoid false negative. But the method and the individual genotyping in the final step can solve the problem of false positives. Association results with the pooled DNA method and subsequent re-genotyping of individual DNAs using the same set of 900 screened individuals revealed that 11 markers showed significant differences by Fisher’s exact test (Table 1). Of the 11 positive markers of individual typing, we selected two loci that showed stronger associations with the disease on 6p21 and 12q21 for a more detailed analysis. We set 11 new MS markers in addition to 2 MS markers around the two loci, and then selected and performed individual typing. Among these 13 MS markers, 10 markers showed significant differences in allele frequency (Tables 2 and 3,Figures 1 and 2).
Human leukocyte antigen (HLA)
One of the two loci we selected from the 11 markers that were positive throughout the first to third screenings and individual typing steps was mapped to 6p21 on the short arm of chromosome 6, representing the HLA region, especially around the HLA class II region. The HLA, the human MHC, contains a large number of genes related to immune system function. This HLA region carries a large number of immunologically important genes including HLA genes encoding cell-surface antigen-presenting proteins. Therefore, HLA genes or other genes in the HLA region may be involved in the pathogenesis of IgAN.21
Classical HLA class II antigens consist of HLA-DR, -DQ, and -DP, and are known to have a crucial role in the presentation of processed antigens to helper T cells. As the IgA antibody-mediated response is helper T-cell-dependent, the HLA class II allelic polymorphism of the HLA gene may affect the pathogenesis of IgAN. Seven out of nine MS markers in the HLA region showed significant differences in allele frequency (Table 3). These markers showed bimodal peaks in the P-value, as shown in Figure 1, with one peak on the C6orf10 gene just outside the HLA class II gene region and the other between the HLA-DRB1 and HLA-DQA1 genes in the HLA class II gene region.
In the past few decades, many clinical studies have provided further evidence to support the strong association between HLA gene polymorphisms and IgAN.2, 5, 22 Akiyama et al.23 reported a relationship between the HLA-DRA gene and IgAN. They analyzed the extent of LD in the region of chromosome 6p21.3 in a Japanese test population and found extended LD blocks within the class II locus.23 They designed a case–control association study on SNPs in each of these LD blocks, and determined that SNPs located in the HLA-DRA gene correlated with an increased risk of IgAN.23 A previous study identified a strong relationship between HLA-DQB1*0301 and IgAN patients.24 Other studies have examined the relationship between HLA alleles and IgAN in various ethnic groups, but the lack of concordance across many of these studies may be due to racial differences in the genetic background of different ethnic groups and the differential interactions of IgAN susceptibility genes with environmental factors.
As GWAS has recently been identified as a feasible tool for the detection of genetic factors for multi-gene diseases due to large-scale SNP discovery efforts from the definition of LD blocks by the HapMap Project and technological advances in DNA typing, the successful findings of a large-scale GWAS on HLA and IgAN were recently reported.10, 11, 12 We adopted MS markers for GWAS, instead of SNPs. To the best of our knowledge, GWAS has not yet been conducted on IgAN using whole-genome MS markers. The complex genetic pattern of IgAN is reflected by the multiple pathways involved in its immunopathogenesis. From this point of view, several genetic loci significantly contribute to the disease susceptibility that underlies the primary immunological defects observed in IgAN. Variations in these major genetic loci may not be sufficient for the development and progression of IgAN, and the contribution of a potentially large number of modifying genes with modest genetic effects, but high prevalence may also be needed. The various allelic combinations of these loci may underlie the different disease phenotypes observed in IgAN. To identify new candidate genes for diseases with a complex genetic pattern, we adopted an original and unique analysis approach using MS markers. MS markers also show longer LD than SNPs, necessitating much fewer markers for a genome-wide association analysis than SNP. Gulcher et al.25 demonstrated that MSs had higher mutation rates than SNPs, which allowed them to break up apparently immutable common SNP haplotypes into lower frequencies that may better match the functional variants of the intermediate frequency or rare functional variants they are meant to detect. Therefore, MS marker-based GWAS provides a complementary tool for the genetic dissection of common and rare diseases, and may be one of the reasons why we successfully detected new loci susceptible to IgAN, whereas previous studies did not.
Overall, our results provide clear evidence for the association of IgAN with genes around the HLA region (Figure 1). We replicated the previously identified IgAN susceptibility loci on chromosome 6p21, specifically close to HLA-DRB, DQA and DQB.10, 11, 12 The physical position at the loci of the positive MS markers with the strongest association in our study was different from that of those defined by previous GWAS studies using SNPs. High-resolution mapping, the genotyping of reported polymorphic SNPs and detailed HLA-DNA typing are needed to further dissect this interval and pinpoint disease-susceptible variants.
TSPAN8–PTPRR
The other susceptible loci identified from the 11 markers that were positive throughout the first to third screenings and individual typing were in the TSPAN8–PTPRR gene region on 12q21, suggesting that the susceptible locus for IgAN is the TSPAN8 or PTPRR gene. The protein encoded by the TSPAN8 gene is a member of the transmembrane 4 superfamily, also known as the tetraspanin family. Tetraspanins are type III proteins that contain four membrane-spanning segments with conserved CCG motifs, transmembrane polar residues and multiple acylation sites. The transmembrane proteins of the tetraspanin superfamily directly regulate all cellular events, such as coordination between tumor cell-cell adhesion, cell-matrix adhesion, matrix degradation and cell migration. These proteins mediate the signal transduction events that have a role in the regulation of cell development, activation, growth and motility. This cell surface glycoprotein is known to complex with integrins. They are widely expressed in all eukaryotes.26 Tetraspanins regulate not only the motility-related behavior of tumor cells, but also the interactions between tumor cells and their microenvironment. Tspan8 was originally identified as a tumor-associated antigen. Its expression has been correlated with the promotion of tumor progression.27, 28 Through the tumor-secreted exosomes that are enriched with integrin alpha 4 and VCAM-1, Tspan8-expressing tumor cells upregulate the expression of vascular endothelial growth factor, its receptor, chemokines and their receptors, von Willebrand factor, and Tspan8 itself in endothelial cells, vascular endothelial growth factor, matrix metalloproteinases, and urokinase plasminogen activator in tumor-adjacent fibroblasts.27, 29, 30 However, the mechanisms of action of tetraspanins currently remain unknown. Although the molecular mechanism by which TSPAN8 is involved in the pathogenesis of IgAN has not yet been elucidated in detail, TSPAN8 may contribute to renal dysfunction through the upregulation of vascular endothelial growth factor by TSPAN8 in well-vascularized tissue. We performed immunohistochemistry for tetraspanin-8 on human renal biopsy specimens associated with IgAN.31 In this study, we found that tetraspanin-8 was often expressed in vascular smooth muscle cells and occasionally in tubule cells in normal kidneys, and the degree of tubular staining negatively correlated with the estimated glomerular filtration rate, independently of the type of nephropathy.31 Further studies are needed to clarify the molecular mechanism underlying the tetraspanin-mediated development of IgAN. Furthermore, studies are required to elucidate their function in the pathogenesis or progression of the disease due to interactions between Tspan8 and CD151 as a risk for IgAN.
The protein encoded by the other candidate gene, PTPRR (protein tyrosine phosphatase receptor-type R), is a member of the protein tyrosine phosphatase (PTP) family. Information regarding the putative roles of PTPRR isoforms in health and disease is limited. PTPs have been identified as signaling molecules that regulate various cellular processes including cell growth, differentiation, the mitotic cycle and oncogenic transformation. Tyrosine phosphorylation is a powerful mechanism that modulates the proliferation, differentiation and functioning of neurons. Orthologues of the PTPRR gene are expressed in many vertebrates. The human PTPRR gene has been excluded as a candidate for a type 2 diabetes locus on chromosome 12q15, but has been implicated in the pathogenesis of acute myelogenous leukemia. The silencing of this gene has been associated with colorectal cancer.
There were several limitations to the present study. One potential limitation is the small sample size for GWAS. The sample sizes required are further increased by the larger number of markers that are tested in GWAS because P-values must be corrected for multiple-hypothesis testing. To overcome these issues, we used a three-stage screening process in which a more modest threshold for passing markers as positive was used in screening of the entire genome for associations. We also qualified all samples of patients with biopsy-proven IgAN. In the future, studies with a larger sample size will be needed for more strict GWAS, and replication studies in different races are also necessary. Furthermore, we set the significance level of 0.05, and regarded alleles associated with P<0.05 as 'positive' and those with P>0.05 as 'negative'. If we performed a Bonferroni correction, the significance level would be 3.0E-07 in all the screening steps. This significance level is based on not the number of markers but the total number of alleles in all multi-allelic markers. Each test in multi-alleles of the same locus (the same MS) was not completely independent. Therefore, Bonferroni correction may not be more suitable for this study; however, we cannot find the best method for MS study. Moreover, although false negatives were inevitable when a significance level of 0.05 was used, the screening was repeated several times to reduce false positives and obtain true positive markers. This study found one locus yielding high odds ratio (7.65) against general GWASs, although the P-value (8.7E-7) is not significant after Bonferroni correction. Therefore, this association with IgAN should be confirmed by additional population including other ethnic group in future studies. Thus, we could not completely elucidate whether associations with IgAN shown in this study were false positive or not, taking into consideration that the number of observed positive markers was similar to expected positive markers (Supplementary Table 4). The only way to confirm this association between TSPAN8/PTPRR and IgAN is to study other independent cohorts. Another limitation is that clinical follow-up data were not included. The progression and outcomes of patients with IgAN vary significantly. Therefore, by following up the clinical data, we plan to test the possibility of utilizing genes for the differential diagnosis and treatment of IgAN.
Our next step is to narrow down the candidate regions with other MS markers between these genes to discriminate which of the TSPAN8 and PTPRR genes is responsible for the development of IgAN. Furthermore, to clarify the pathogenesis of IgAN in relation to rare alleles or variants in the HLA, TSPAN8 or PTPRR gene, these regions need to be resequenced and the gene expression of both genes needs to be analyzed in the tissues of renal biopsy specimens. Further studies are warranted to explore the immunological mechanisms underlying this genotype-disease phenotype association.
This study on GWAS, using >20 000 MS markers, provides a new approach regarding the susceptible genes for IgAN for investigators seeking new tools for the prevention and treatment of IgAN. We identified HLA, TSPAN8 and PTPRR as new candidate genes for diseases with a complex genetic pattern. Although SNP and MS were previously used in a disease-related gene search method of IgAN, this is the first GWAS to use pooled DNA markers. GWAS using MS could be a very powerful approach especially for the case that MS is directly associated with a disease. Because a number of triplet repeat expansion disorders have been identified, the risk of common disease may be also affected by MS. GWAS using MS would be a suitable tool for identifying susceptible genes of common disease.
References
Barratt, J. & Feehally, J. IgA nephropathy. J. Am. Soc. Nephrol. 16, 2088–2097 (2005).
Beerman, I., Novak, J., Wyatt, R. J., Julian, B. A. & Gharavi, A. G. The genetics of IgA nephropathy. Nat. Clin. Pract. Nephrol. 3, 325–338 (2007).
Tolkoff-Rubin, N. E., Cosimi, A. B., Fuller, T., Rublin, R. H. & Colvin, R. B. IGA nephropathy in HLA-identical siblings. Transplantation 26, 430–433 (1978).
Julian, B. A., Quiggins, P. A., Thompson, J. S., Woodford, S. Y., Gleason, K. & Wyatt, R. J. Familial IgA nephropathy. Evidence of an inherited mechanism of disease. N. Engl. J. Med. 312, 202–208 (1985).
Hsu, S. I., Ramirez, S. B., Winn, M. P., Bonventre, J. V. & Owen, W. F. Evidence for genetic factors in the development and progression of IgA nephropathy. Kidney Int. 57, 1818–1835 (2000).
Suzuki, H., Suzuki, Y., Yamanaka, T., Hirose, S., Nishimura, H., Toei, J. et al. Genome-wide scan in a novel IgA nephropathy model identifies a susceptibility locus on murine chromosome 10, in a region syntenic to human IGAN1 on chromosome 6q22-23. J. Am. Soc. Nephrol. 16, 1289–1299 (2005).
Schena, F. P., Cerullo, G., Rossini, M., Lanzilotta, S. G., D'Altri, C. & Manno, C. Increased risk of end-stage renal disease in familial IgA nephropathy. J. Am. Soc. Nephrol. 13, 453–460 (2002).
Gharavi, A. G., Yan, Y., Scolari, F., Schena, F. P., Frasca, G. M., Ghiggeri, G. M. et al. IgA nephropathy, the most common cause of glomerulonephritis, is linked to 6q22-23. Nat. Genet. 26, 354–357 (2000).
Takei, T., Iida, A., Nitta, K., Tanaka, T., Ohnishi, Y., Yamada, R. et al. Association between single-nucleotide polymorphisms in selectin genes and immunoglobulin A nephropathy. Am. J. Hum. Genet. 70, 781–786 (2002).
Feehally, J., Farrall, M., Boland, A., Gale, D. P., Gut, I., Heath, S. et al. HLA has strongest association with IgA nephropathy in genome-wide analysis. J. Am. Soc. Nephrol. 21, 1791–1797 (2010).
Gharavi, A. G., Kiryluk, K., Choi, M., Li, Y., Hou, P., Xie, J. et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat. Genet. 43, 321–327 (2011).
Yu, X. Q., Li, M., Zhang, H., Low, H. Q., Wei, X., Wang, J. Q. et al. A genome-wide association study in Han Chinese identifies multiple susceptibility loci for IgA nephropathy. Nat. Genet. 44, 178–182 (2012).
Collins, H. E., Li, H., Inda, S. E., Anderson, J., Laiho, K., Tuomilehto, J. et al. A simple and accurate method for determination of microsatellite total allele content differences between DNA pools. Hum. Genet. 106, 218–226 (2000).
Oka, A., Hayashi, H., Tomizawa, M., Okamoto, K., Suyun, L., Hui, J. et al. Localization of a non-melanoma skin cancer susceptibility region within the major histocompatibility complex by association analysis using microsatellite markers. Tissue Antigens 61, 203–210 (2003).
Tamiya, G., Shinya, M., Imanishi, T., Ikuta, T., Makino, S., Okamoto, K. et al. Whole genome association study of rheumatoid arthritis using 27 039 microsatellites. Hum. Mol. Genet. 14, 2305–2321 (2005).
Kawashima, M., Tamiya, G., Oka, A., Hohjoh, H., Juji, T., Ebisawa, T. et al. Genomewide association analysis of human narcolepsy and a new resistance gene. Am. J. Hum. Genet. 79, 252–263 (2006).
Pritchard, J. K. & Rosenberg, N. A. Use of unlinked genetic markers to detect population stratification in association studies. Am. J. Hum. Genet. 65, 220–228 (1999).
Saito, A. & Kamatani, N. Strategies for genome-wide association studies: optimization of study designs by the stepwise focusing method. J. Hum. Genet. 47, 360–365 (2002).
Goertsches, R., Villoslada, P., Comabella, M., Montalban, X., Navarro, A., de la Concha, E. G. et al. A genomic screen of Spanish multiple sclerosis patients reveals multiple loci associated with the disease. J. Neuroimmunol. 143, 124–128 (2003).
Godde, R., Brune, S., Jagiello, P., Sindern, E., Haupts, M., Schimrigk, S. et al. An extended association screen in multiple sclerosis using 202 microsatellite markers targeting apoptosis-related genes does not reveal new predisposing factors. J. Negat. Results Biomed. 4, 7 (2005).
Horton, R., Wilming, L., Rand, V., Lovering, R. C., Bruford, E. A., Khodiyar, V. K. et al. Gene map of the extended human MHC. Nat. Rev. Genet. 5, 889–899 (2004).
Maxwell, P. H. & Wang, Y. Genetic studies of IgA nephropathy. Nephron Exp. Nephrol. 102, e76–e80 (2006).
Akiyama, F., Tanaka, T., Yamada, R., Ohnishi, Y., Tsunoda, T., Maeda, S. et al. Single-nucleotide polymorphisms in the class II region of the major histocompatibility complex in Japanese patients with immunoglobulin A nephropathy. J. Hum. Genet. 47, 532–538 (2002).
Raguenes, O., Mercier, B., Cledes, J., Whebe, B. & Ferec, C. HLA class II typing and idiopathic IgA nephropathy (IgAN): DQB1*0301, a possible marker of unfavorable outcome. Tissue Antigens 45, 246–249 (1995).
Gulcher, J. Microsatellite markers for linkage and association studies. Cold Spring Harb. Protoc. 2012, 425–432 (2012).
Zoller, M. Tetraspanins: push and pull in suppressing and promoting metastasis. Nat. Rev. Cancer. 9, 40–55 (2009).
Gesierich, S., Berezovskiy, I., Ryschich, E. & Zoller, M. Systemic induction of the angiogenesis switch by the tetraspanin D6.1A/CO-029. Cancer Res. 66, 7083–7094 (2006).
Le Naour, F., Andre, M., Greco, C., Billard, M., Sordat, B., Emile, J. F. et al. Profiling of the tetraspanin web of human colon cancer cells. Mol. Cell. Proteomics 5, 845–857 (2006).
Nazarenko, I., Rana, S., Baumann, A., McAlear, J., Hellwig, A., Trendelenburg, M. et al. Cell surface tetraspanin Tspan8 contributes to molecular pathways of exosome-induced endothelial cell activation. Cancer Res. 70, 1668–1678 (2010).
Champy, M. F., Le Voci, L., Selloum, M., Peterson, L. B., Cumiskey, A. M. & Blom, D. Reduced body weight in male Tspan8-deficient mice. Int. J. Obes. 35, 605–617 (2011).
Hirukawa, T., Wu, Q., Sawada, K., Matsusaka, T., Saka, S., Oka, A. et al. Kidney diseases enhance expression of tetraspanin-8: a possible protective effect against tubular injury. Nephron Extra 4, 70–81 (2014).
Acknowledgements
We are grateful to the patients and relatives for their cooperation in this study, as well as all of the control subjects who donated blood samples. We thank all members of the laboratory for technical support and discussion. We also thank all staff and doctors who contributed to blood sample collection from the subjects. Our work was supported in part by a grant for Research from Yokohama City University, Japan and a grant from the study group on IgA nephropathy in Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
SU discloses conflict of interest with research grants from Novartis Pharmaceuticals Japan Co. and Pfizer Japan Inc., and TM discloses conflict of interest with a research grant from Chugai Pharmaceutical Co., Ltd. The other authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Saka, S., Hirawa, N., Oka, A. et al. Genome-wide association study of IgA nephropathy using 23 465 microsatellite markers in a Japanese population. J Hum Genet 60, 573–580 (2015). https://doi.org/10.1038/jhg.2015.88
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.88
