
Abstract
The Middle East (ME) is an important crossroad where modern humans migrated ‘out of Africa’ and spread into Europe and Asia. After the initial peopling and long-term isolation leading to well-differentiated populations, the ME also had a crucial role in subsequent human migrations among Africa, Europe and Asia; thus, recent population admixture has been common in the ME. On the other hand, consanguinity, a well-known practice in the ME, often reduces genetic diversity and works in opposition to admixture. Here, we explored the degree to which admixture and consanguinity jointly affected genetic diversity in ME populations. Genome-wide single-nucleotide polymorphism data were generated in two representative ME populations (Arabian and Iranian), with comparisons made with populations worldwide. Our results revealed an overall higher genetic diversity in both ME populations relative to other non-African populations. We identified a much larger number of long runs of homozygosity in ME populations than in any other populations, which was most likely attributed to high levels of consanguineous marriages that significantly decreased both individual and population heterozygosity. Additionally, we were able to distinguish African, European and Asian ancestries in ME populations and quantify the impact of admixture and consanguinity with statistical approaches. Interestingly, genomic regions with significantly excessive ancestry from individual source populations are functionally enriched in olfactory pathways, which were suspected to be under natural selection. Our findings suggest that genetic admixture, consanguinity and natural selection have collectively shaped the genetic diversity of ME populations, which has important implications in both evolutionary studies and medical practices.
Similar content being viewed by others

Introduction
Studies of both mitochondrial DNA (mtDNA) and Y-chromosome lineages indicate that after modern human migrating out of Africa, tens of thousands of years ago, they arrived in the Middle East (ME), and then dispersed into Europe and Asia.1,2 Over thousands of years, most of human populations have been relatively isolated, evolved independently and generated the distinct genomic characteristics as can be noted today. However, Africa, Asia and Europe are geographically connected by the ME, which provides opportunities for population contact and thus population admixture, with this effect being more pronounced following trade and the establishment of the Silk Road. Previous studies have identified admixture events in ME populations when examining both uniparental markers and genome-wide single-nucleotide polymorphisms (SNPs).3, 4, 5, 6 A previous study of Uyghurs and African Americans reported that admixture could increase the genetic diversity of the admixed populations.7 Moreover, ME populations generally have large family sizes, with marriages between relatives very common;8 thus, consanguinity is highly prevalent in this region. A similar situation is encountered in Central Asia, South Asia and the Americas,9,10 especially in Islamic-influenced areas. It is deemed that consanguinity usually decreases the genetic diversity and results in many recessive diseases such as neuromuscular disorders, metabolic disorders, osteopetrosis syndromes and chondrodystrophia.8,11 However, to our knowledge, few studies have focused on the influences of both admixture and consanguinity on population genetic diversity simultaneously. In the present study, we attempt to qualify and quantify the influence of these two forces in two representative ME populations residing in Kuwait, with evidence showing that both populations experienced admixture and consanguinity.3, 4, 5,11 Ultimately, mathematical modeling was used to elucidate the degree to which admixture and consanguinity shaped the genetic diversity and structure of the two ME human populations.
Materials and methods
Samples and quality controls
DNA samples were collected from 42 Kuwaitis whose ancestry had been traced back at least four generations to the Arabian Peninsula (ARB) and 22 Kuwaitis whose ancestry had been traced back at least four generations to Persia (IRN) via pedigree analysis. These samples were genotyped by Affymetrix Genome-Wide Human SNP Array 6.0 (Santa Clara, CA, USA) according to standard protocols. Additionally, the raw data of 200 unrelated samples from four populations in the International HapMap Project phase III12 were downloaded to include 50 CHB (Han Chinese in Beijing, China), 50 JPT (Japanese in Tokyo, Japan), 50 YRI (Yoruba in Ibadan, Nigeria) and 50 CEU (Utah residents with Northern and Western European ancestries), which were also genotyped by Affymetrix Genome-Wide Human SNP Array 6.0. All the raw array data were called by Affymetrix Power Tools (APT, Version 1.15.0) with the Affymetrix platform annotation file (Genome-Wide SNP 6 annotation na32, with genome references to UCSC hg19 or NCBI GRCh37). One sample from the ARB sample group was removed from subsequent analyses owing to a calling rate <85%. Data filtering was performed within each population, samples with a missing rate >5% per individual, SNPs with missing rate >5% and SNPs failing the Hardy–Weinberg equilibrium test (P-value <1.0E−6) were excluded from the analysis. Data from the six populations was merged according to the intercepted SNPs (721 989 autosomal SNPs), SNPs whose minor allele frequency (MAF) <0.01 was excluded (11 362 autosomal SNPs). Finally, 40 ARB samples, 22 IRN samples, all 200 selected HapMap Phase III samples and 710 627 autosomal SNPs were used for subsequent analyses.
Population structure analysis
Markers with r2>0.5 calculated by a 50-SNP sliding window shifted at a 5-SNP interval were removed to reduce strong linkage disequilibrium (LD). Principal component analysis (PCA) was performed on the six populations with the thinned autosomal SNPs (334 705 markers) using smartpca in the EIGENSOFT package (version 4.2),13,14 The population structure was also inferred with ADMIXTURE (version 1.2.3),15 which implemented a maximum-likelihood method to estimate individual ancestries. The analysis was performed with LD-pruned SNPs, with ten fold cross validation error (–cv=10), a K from 2 to 6 and other parameters set to default.
Assessing genetic diversity
To eliminate the effects that the ascertainment bias of genotyping data might bring to the measurement of genetic diversity, we merged the SNPs of the four reference populations (CEU, CHB, JPT and YRI) called from the next-generation sequencing database.16,17 Next, we calculated the MAF and divided the MAF ⩾0.05 into small intervals: increase each interval by 0.01; that is, [0.05, 0.06), [0.06, 0.07), …, and [0.49, 0.50]. Next, we randomly sampled SNPs from the genotyping data of the same merged four populations according to the proportion in each interval decided by sequencing called SNPs, with the sampling processes repeated 10 times.
The expected heterozygosity for each SNP (HSe) was used to measure the SNP-based diversity of each population by the formula:  , where n denotes the number of SNPs in a sliding window and pj denotes the allele frequency of jth allele. We chose a 100 kb sliding window, with at least five SNPs per 100 kb. Individual-based diversity was measured by dividing the number of heterozygous SNPs by the total number of non-missing SNPs per individual. The data were then phased using Beagle (version 3.3.2)18,19 with default parameters and the expected haplotype heterozygosity (HHe) calculated for the windows of 10, 20, 30, 40, 50, 100, 200 and 500 kb as previously described.7 All measurements were performed independently on the 10 sampling repeats.
, where n denotes the number of SNPs in a sliding window and pj denotes the allele frequency of jth allele. We chose a 100 kb sliding window, with at least five SNPs per 100 kb. Individual-based diversity was measured by dividing the number of heterozygous SNPs by the total number of non-missing SNPs per individual. The data were then phased using Beagle (version 3.3.2)18,19 with default parameters and the expected haplotype heterozygosity (HHe) calculated for the windows of 10, 20, 30, 40, 50, 100, 200 and 500 kb as previously described.7 All measurements were performed independently on the 10 sampling repeats.
Assessing consanguinity
If a child received two copies of the same segment from father and mother, it would create a run of homozygosity (ROH).20 The probability for a child from consanguineous marriage receiving the same segment was significantly elevated; thus, ROH could be used to measure the level of consanguinity. Here ROH was calculated by PLINK (version 1.07)21 with a sliding window of 500 kb and at least 50 SNPs, one heterozygote and no more than five missing SNPs allowed per window. Satisfactory ROHs contained a span of at least 500 kb, with a minimum density of one SNP per 50 kb and a maximum distance between two adjacent SNPs of 100 kb. Two adjacent ROH segments were merged if the proportion of overlap was >0.05. ROH fragments were then clustered into three classes with Mclust (an R package) using the methods described by Pemberton et al.9
Testing population admixture
To test whether ARB and IRN were admixed populations, the three-population test (F3 test), a test that can provide strong evidence of population admixture by modeling genetic drift paths, was used.22 The F3 test has a general form F3(C; A, B), in which C denotes the target population and A and B denote two reference populations; thus, ARB and IRN were deemed target populations. In this study, we selected CEU, CHB and YRI as the reference populations (surrogate ancestral populations) because they are less admixed and high-density SNP data are available. Significantly negative F3 scores support population admixture, with gene flow occurring between the two reference populations.
Inferring local ancestry
To determine local ancestry for each SNP from each individual, ELAI, a two-layer hidden Markov model, was used by modeling the LD within and among groups.23 YRI, CEU and CHB were set as surrogate ancestral reference populations, with 50 EM steps, and 3 upper and 30 lower clusters. Previous studies on the admixture events of other ME populations reported the events to have occurred about 100 generations ago;24,25 thus, this time estimate was used as a priori in our local ancestry inference.
Linear regression analysis
To determine how admixture and consanguinity jointly influenced genetic diversity, linear regression analysis was performed at both the SNP and individual levels as follows:
(1) For SNP diversity, ROH scores were defined as

and the admixture effect was defined as

where αi is the ancestral contribution to that SNP, fi is the MAF of that SNP and i is the ancestral population. Then, the diversity of an SNP (Ys) was modeled according to

(2) For the individual diversity, ROH scores were defined as

and the admixture effect was defined as

where αi is the ancestral contribution to that individual, Hi is the mean individual diversity and i is the ancestral population. Similarly, the diversity of an individual (Yi) was modeled according to

The proposed null hypothesis (H0) for these models assumes the ROH score and admixture effect to have no impact on the observed SNP and individual diversities. Based on this hypothesis, linear regression analysis was performed separately on the two ME populations, ARB and IRN, for the 10 sampling repeats.
Results
Population structure of ME populations
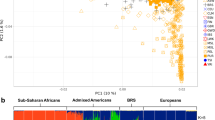
PCA was performed at the individual level to investigate the population structure. A plot displaying the two most significant principal components (PCs) (Figure 1a) showed individuals from Africa, Europe and Asia to tightly cluster in their groups. PC1 clearly separated Africans and non-Africans, whereas PC2 separated Asians and Europeans. However, individuals from the two ME populations (ARB and IRN) clustered loosely, with ARB samples located along the edge between YRI and CEU, while the IRN samples shifted slightly towards the Eastern Asian populations (CHB and JPT) (Figure 1a). The long tails exhibited by the two ME populations in the PCA plot imply possible admixture events, or the occurrence of gene flow from other populations. When performing the ADMIXTURE analysis, the lowest cross-validation error could be found when K=3 (Supplementary Figure S1). These results clearly show that the genetic ancestries of the two ME populations share mainly European (blue) and African (gray) ancestries, as well as a slight Eastern Asian (red) ancestry (Figure 1b), which was consistent with the observed PCA results and suggested admixture events supported by the ADMIXTURE analysis.

Population structure analysis. (a) Principal component analysis (PCA) with samples from the two Middle East (ME) populations: Arabian (ARB), Iranian (IRN) and 200 samples of four reference populations (CEU, CHB, JPT and YRI) from the International HapMap Project III. (b) ADMIXTURE analysis with data pruned based on linkage disequilibrium (LD); the lowest cross-validation error was observed at K=3.
ME populations show higher genetic diversity than the other non-African populations
To compare the genetic diversity of ME populations relative to others, SNP-based, haplotype-based and individual-based heterozygosity assessments were measured. All diversities were calculated from ascertainment bias-corrected SNP subsets, with independent sampling repeated 10 times. Furthermore, the mean SNP-based diversity (HSe) in the two ME populations were higher than those in CEU, CHB and JPT populations, but slightly lower than that in the YRI population (Figure 2a), with the same pattern for individual-based heterozygosity noted (Figure 2b). Remarkably, when SNPs in ROH regions were excluded for each individual to control potential consanguinity, the two ME populations exhibited even higher individual heterozygosity than the other non-African populations and showed comparable levels to the African population (Figure 3b). When examining haplotype-based heterozygosity (HHe), similar patterns were observed despite window size (Figure 2c), with increasing values approaching 1 correlated with an increased window size and a value of 1 almost reached with a window size exceeding 500 kb. One possible interpretation of these results is that the two ME populations are admixed populations with ancestral contributions from African, European and Asian populations, with the increased genetic diversity due to admixture counteracted by the substantial increase of consanguineous marriage practices, which is consistent with previous findings.3,5

Single-nucleotide polymorphism (SNP) level, individual level and haplotype level of genetic diversity obtained from 10 independent random samplings. (a) Mean SNP heterozygosity of a 100 kb sliding window. (b) Mean individual heterozygosity calculated from non-missing SNPs with and without runs of homozygosity (ROH) regions considered; and (c) mean haplotype heterozygosity. Haplotype heterozygosity was calculated by sliding windows of 10, 20, 30, 40, 50, 100, 200 and 500 kb.

Runs of homozygous fragments (ROHs). (a–c) Total length (top) and total number (bottom) of short (a), intermediate (b) and long (c) ROHs per individual, respectively. (d–f) Scatterplot of total length against the total number of short (d), intermediate (e) and long (f) ROHs per individual, respectively. Legends in (e) and (f) are the same as those in (d).
ME populations show higher consanguinity
To compare consanguinity, we measured consanguinity using ROH and clustered the ROH fragments into three classes. It was clearly observed that both the total number and total length of ROHs per individual gradually increased with an increase in geographical distance from Africa for both short (Figure 3a) and intermediate (Figure 3b) ROH classes. These observed patterns were consistent with a previous study based on the HGDP data set.9 However, the two ME populations presented large variations in both the total number and total length of ROHs per individual (Figure 3c). When the total ROH length was plotted against the total ROH number per individual, a strong correlation was noted for both the short and intermediate classes, with the distance along the fitted line proportional to the geographical distance from Africa (Figures 3d and e). For long ROH class, the two ME populations showed a greater total ROH number and longer total ROH length per individual than the other four populations (Figure 3f), with the long ROH fragments most likely arising from a recent background relatedness;that is, consanguinity.
Thus, the possibility of consanguinity having reduced the genetic diversity of the two originally admixed ME populations is plausible, with these populations exhibiting a lower genetic diversity than their surrogate ancestral YRI population. However, the two ME populations still showed higher genetic diversity than the other non-African populations. This may be explained by both European and Asian populations having possibly experienced a bottleneck event since their divergence from the ME populations, in addition to the time and strength of consanguinity being unable to counteract completely the diversity introduced by admixture in the two ME populations.
Evidence of admixture in the ME populations
ADMIXTURE analysis revealed that the two ME populations had the highest genetic similarities to Europeans, followed by Africans and Asians. To formally test for admixture in these populations, we first calculated F3 (ARB or IRN; YRI, CEU) and observed significant negative values for both ARB and IRN; then we calculated F3 (ARB or IRN; YRI, CHB or JPT), but none of them were negative; and finally we calculated F3 (ARB or IRN; CEU, CHB or JPT), and only the value for IRN was significantly negative no matter whether the Asian reference population was CHB or JPT (Table 1), thus indicating both of these populations admixed. In summary, the ARB population received ancestral contributions from European and African populations, whereas the IRN population received ancestral contributions from European, African and Asian populations. These results were in accordance with the PCA and ADMIXTURE analyses. Moreover, some individuals showed excessive African ancestry (Figure 1b), suggesting recent gene flow from African population, which was consistent with previous mtDNA studies.3 For the ARB population, negative values were not obtained during the F3 testing using Asian reference populations, possibly because of low levels of gene flow that could not be detected. Furthermore, negative F3 values were not obtained for either of the ME populations with the YRI, CHB or JPT reference populations, which could be attributed to the fact that the admixture events were mainly between European and African populations, with only low-level gene flow occurring with Asian populations.
The direction and magnitude of influences of admixture and consanguinity on genetic diversity
To investigate the direction and magnitude of influences that admixture and consanguinity had on genetic diversity, a linear model was proposed, with the ROH score and admixture effect fitted to the observed diversity. Regression analysis was performed on the two ME populations separately with the 10 independent samplings to investigate relationships at both the SNP and individual levels. At the SNP level, the results for both ME populations were highly concordant among 10 independent samplings, with the intercept (βs0), ROH score (Xs1) coefficient (βs1) and admixture effect (Xs2) coefficient (βs2) all statistically significant (Supplementary Table S1). Owing to the level of consistency among the 10 independent samplings, the regression model parameters βs0, βs1 and βs2 were averaged to generate the final regression model for the SNP diversity as: Ys(ARB)=0.06722−0.05680*Xs1+0.80069*Xs2 (mean adjusted R2=0.66885) and Ys(IRN)=0.05560−0.03963*Xs1+0.82681*Xs2 (mean adjusted R2=0.70289).
Similar results were obtained for the individual diversity. Both the regression models for ARB and IRN showed statistical significance and the 10 independent samplings were highly concordant (Supplementary Table S2). Again, the regression model parameters βi0, βi1 and βi2 were averaged to obtain the final individual diversity models: Yi(ARB)=−1.05647−0.36337*Xi1+4.71255*Xi2 (mean adjusted R2=0.90692) and Yi(IRN)=−1.76090−0.28837*Xi1+7.16242*Xi2 (mean adjusted R2=0.97046).
The positive coefficients for the admixture effect confirmed an increased genetic diversity owing to admixture, which was consistent with the previous study,7 whereas the negative ROH score coefficients confirmed a decrease in genetic diversity owing to consanguinity. Overall, linear modeling enabled the quantification of the influences of admixture and consanguinity on the genetic diversity in the two ME populations.
Genome-wide distribution of local ancestry in ME populations
The local ancestry at each SNP for each individual was estimated by ELAI. The local ancestry contributions from different ancestries were not uniformly distributed across the genome, with some genomic regions showing excessive ancestry contribution from a given parental population (Figure 4). For the ARB and IRN populations, the loci showing excessive or scarce ancestry contribution beyond the 0.5% quantile were collected and the collected ARB and IRN SNPs were found to be highly overlapped (Supplementary Figure S2). These overlaps could contribute to the populations adapting to the same local environment. Functional annotation of these overlapping SNPs were identified using the DAVID database26,27 and showed all of the top 10 categories to relate to olfactory perception pathway (Benjamini corrected P-value <6.50 × 10−15; false discovery rate P-value <6.60 × 10−14) (Table 2). The genes enriched among the top 10 functional categories were mostly from olfactory families (2, 4, 5, 8, 9, 10, 11 and 12), with the exception of GABBR1 and MAS1L (Supplementary Table S3). GABBR1 is a γ-aminobutyric acid B receptor, which is the main inhibitory neurotransmitter in the mammalian central nervous system, whereas MAS1L is a G-protein-coupled receptor and is associated with the G-protein coupled receptor protein signaling pathway. It is likely that these two genes are also associated with the olfactory perception pathways indirectly.

Mean ancestry contributions. (a) Mean ancestry contributions for each single-nucleotide polymorphism (SNP) in Arabian (ARB). Top: Mean European ancestry contribution to ARB; bottom: mean Asian ancestry contribution to ARB; (b) mean ancestry contribution for each SNP in Iranian (IRN). Top: Mean European ancestry contribution to IRN; bottom: mean Asian ancestry contribution to IRN. Black solid line denotes average mean ancestry contribution across genome; blue solid line denotes 99.5% quantile; and red solid line denotes 0.005% quantile.
Discussion
In this study, we attempted to explore the combined effect of genetic admixture and consanguinity on human genetic diversity. We analyzed genome-wide SNP data of two ME populations, ARB and IRN, and our results showed that the genetic diversity of the two ME populations was higher than that of the other non-African populations, which was consistent with an admixture scenario. At the same time, long ROH fragments were also identified in a vast number of genomic regions in the two ME populations, which was also consistent with the expected consequence of consanguineous marriage. These results suggest that the demographic history of the two ME populations is very complex.
Considering the geographical location of the ME, the observed higher genetic diversity in the two ME populations could simply be explained by a possible scenario that these populations were surrogate ancestral populations of the European and Asian populations. Moreover, signatures of population admixture were also very pronounced based on PCA, ADMIXTURE analysis and F3 testing. Therefore, a more likely yet complex scenario was that the two ME populations were admixed populations with gene flow contributions from European, Asian and African populations. These admixture events increased the genetic diversity of the ME populations to levels comparable to or higher than those of African populations, with this diversity gradually decreased because of the prevalent cultural practices of consanguinity. Our results support the second scenario and are consistent with previous findings.3, 4, 5 However, the genetic architectures of modern ME populations could result from ancient migration, subsequent gene flow (or admixture) between well-differentiated populations and entangle with recent consanguineous marriages. Social and historical documentation in conjunction with other genetic findings all support this interpretation.8,28
A challenge when analyzing genetic diversity based on genotyping data is potential ascertainment bias. The availability of public sequencing data in worldwide populations (e.g., CEU, CHB, JPT and YRI) made it possible to correct for this bias by referencing the MAF spectrum of sequence data (Supplementary Figure S3). This bias was corrected by randomly sampling a subset of SNPs from the genotyping data according to the distribution of MAFs from the sequencing data, with this approach repeated 10 times. Interestingly, even after correction, the CEU diversity was still slightly higher than that of CHB and JPT. One possible cause of these differences between European and Asian populations may be attributed to differences in strength and lasting time of a population bottleneck. To address this problem, the individual diversities of the four reference populations were calculated from sequencing data, with the same pattern observed as when randomly sampling genotyping data (Supplementary Figure S4). Therefore, it seemed that the higher genetic diversity in CEU populations was intrinsic, suggesting that recent gene flow in Europeans could be an important factor, with genetic contributions from other sources such as African or even possibly some archaic humans29,30 having significantly influenced the European gene pool.
Admixture has been a common phenomenon throughout the history of modern humans, with previously isolated populations often come into contact through colonization and migration. It is especially common in the ME since it has been a melting pot of cultures, languages and people. Both prehistoric and recent genetic admixture have greatly influenced the genetic makeup of regional ME populations. On the other hand, consanguineous marriage is prevalent in many ME countries, which is expected to decrease ME genetic diversity. As a retrospective study based on modern human genomic data, it is difficult to fully distinguish the influences of admixture and consanguinity on genetic diversity, as each has a confounding effect on the other. However, we were able to confirm the generated theoretical predictions and roughly estimate the magnitudes of the influence of admixture and consanguinity based on the statistical approaches used in this study. Our analyses revealed that the current genetic architectures of the two ME populations were shaped by a joint effect of the two forces that resulted from historical, cultural and potentially also from religious reasons.
Additionally, we further explored the possibility of the influence of a third type of force on regional genetic diversity, natural selection. Our approach to search for footprints of natural selection in both ME populations was based on admixture analysis seeking to identify genomic regions with local ancestry significantly deviated from the mean genome-wide distribution. While this approach could only detect natural selection signatures after population admixture, it is extremely interesting that the top candidate genes underlying natural selection in the two ME populations were associated with olfactory pathways. While we could not provide a convincing interpretation for these selection signatures, the noted statistical signals could not be explained by stochastic process. This suggests the presence of environmental pressures on these genes in the history of the two ME populations. Taken together, genetic admixture, consanguinity and natural selection have jointly shaped the genetic diversity of the two ME populations, with admixture and consanguinity having opposing effects on diversity, while natural selection exhibits a more regional effect relative to the genome-wide influences seen from the former two factors.
References
Oppenheimer S . Out-of-Africa, the peopling of continents and islands: tracing uniparental gene trees across the map. Philos. Trans. R. Soc. Lond. Ser. B 367, 770–784 (2012).
Wilder J. A., Kingan S. B., Mobasher Z., Pilkington M. M. & Hammer M. F. Global patterns of human mitochondrial DNA and Y-chromosome structure are not influenced by higher migration rates of females versus males. Nat. Genet. 36, 1122–1125 (2004).
Theyab J. B., Al-Bustan S. & Crawford M.H . The genetic structure of the Kuwaiti population: mtDNA inter- and intra-population variation. Hum. Biol. 84, 379–403 (2012).
Triki-Fendri S., Alfadhli S., Ayadi I., Kharrat N., Ayadi H. & Rebai A . Genetic structure of Kuwaiti population revealed by Y-STR diversity. Ann. Hum. Biol. 37, 827–835 (2010).
Alsmadi O., Thareja G., Alkayal F., Rajagopalan R., John S.E., Hebbar P. et al. Genetic substructure of Kuwaiti population reveals migration history. PLoS ONE 8, e74913 (2013).
Hellenthal G., Busby G.B., Band G., Wilson J.F., Capelli C., Falush D. et al. A genetic atlas of human admixture history. Science 343, 747–751 (2014).
Xu S., Jin W. & Jin L . Haplotype-sharing analysis showing Uyghurs are unlikely genetic donors. Mol. Biol. Evol. 26, 2197–2206 (2009).
Teebi A. S. & Teebi S.A . Genetic diversity among the Arabs. Community Genet. 8, 21–26 (2005).
Pemberton T. J., Absher D., Feldman M.W., Myers R.M., Rosenberg N.A. & Li J.Z . Genomic patterns of homozygosity in worldwide human populations. Am. J. Hum. Genet. 91, 275–292 (2012).
Leutenegger A.L., Sahbatou M., Gazal S., Cann H. & Genin E . Consanguinity around the world: what do the genomic data of the HGDP–CEPH diversity panel tell us? Eur. J. Hum. Genet. 19, 583–587 (2011).
Al-Kandari Y. Y. & Crews D.E . The effect of consanguinity on congenital disabilities in the Kuwaiti population. J. Biosoc. Sci. 43, 65–73 (2011).
International HapMap, C., Altshuler D. M., Gibbs R.A., Peltonen L., Altshuler D.M., Gibbs R.A. et al. Integrating common and rare genetic variation in diverse human populations. Nature 467, 52–58 (2010).
Patterson N., Price A.L. & Reich D . Population structure and eigenanalysis. PLoS Genet. 2, e190 (2006).
Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A. & Reich D . Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006).
Alexander D.H., Novembre J. & Lange K . Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Genomes Project, C., Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M. et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012).
Genomes Project, C., Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M. et al. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010).
Browning B.L. & Browning S.R . A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am. J. Hum. Genet. 84, 210–223 (2009).
Browning S.R. & Browning B.L . Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 81, 1084–1097 (2007).
Kirin M., McQuillan R., Franklin C.S., Campbell H., McKeigue P.M. & Wilson J.F . Genomic runs of homozygosity record population history and consanguinity. PLoS ONE 5, e13996 (2010).
Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Patterson N., Moorjani P., Luo Y., Mallick S., Rohland N., Zhan Y. et al. Ancient admixture in human history. Genetics 192, 1065–1093 (2012).
Guan Y . Detecting structure of haplotypes and local ancestry. Genetics 196, 625–642 (2014).
Jin W., Wang S., Wang H., Jin L. & Xu S . Exploring population admixture dynamics via empirical and simulated genome-wide distribution of ancestral chromosomal segments. Am. J. Hum. Genet. 91, 849–862 (2012).
Price A.L., Tandon A., Patterson N., Barnes K.C., Rafaels N., Ruczinski I. et al. Sensitive detection of chromosomal segments of distinct ancestry in admixed populations. PLoS Genet. 5, e1000519 (2009).
Huang D.W., Sherman B.T. & Lempicki R.A . Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Huang D.W., Sherman B.T. & Lempicki R.A . Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 (2009).
Al-Bustan M., Majeed S., Bitar M.S. & Al-Asousi A . Socio-demographic features and knowledge of diabetes mellitus among diabetic patients in kuwait. Int. Q. Community Health Educ. 17, 65–76 (1997).
Der Sarkissian C., Balanovsky O., Brandt G., Khartanovich V., Buzhilova A., Koshel S. et al. Ancient DNA reveals prehistoric gene-flow from siberia in the complex human population history of North East Europe. PLoS Genet. 9, e1003296 (2013).
Botigue L.R., Henn B.M., Gravel S., Maples B.K., Gignoux C.R., Corona E. et al. Gene flow from North Africa contributes to differential human genetic diversity in southern Europe. Proc. Natl Acad. Sci. USA 110, 11791–11796 (2013).
Acknowledgements
These studies were supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (CAS) (XDB13040100) and by the National Science Foundation of China (NSFC) grants (91331204; 31171218). This research was supported in part by the Ministry of Science and Technology (MoST) International Cooperation Base of China and by National Center for Mathematics and Interdisciplinary Sciences (NCMIS), Academy of Mathematics and Systems Science, CAS. SX is Max-Planck Independent Research Group Leader and member of CAS Youth Innovation Promotion Association. SX also gratefully acknowledges the support of the National Program for Top-notch Young Innovative Talents of The ‘Ten-Thousand-Talents’ Project and the support of KC Wong Education Foundation, Hong Kong. Fahd Al-Mulla was supported by the Kuwait Foundation for Advancement of Sciences (No. 2011-1302-06). WG was supported by the Fundamental Research Funds for the Central Universities (2011JBZ019). We thank LetPub (http: //www.letpub.com) for its linguistic assistance during the preparation of this manuscript. All funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author contributions:
SX conceived and designed the study; FA-M, SA-B and MM collected and genotyped the samples; XY, QF and WG analyzed the data; XY and SX wrote the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interests.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Yang, X., Al-Bustan, S., Feng, Q. et al. The influence of admixture and consanguinity on population genetic diversity in Middle East. J Hum Genet 59, 615–622 (2014). https://doi.org/10.1038/jhg.2014.81
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2014.81
This article is cited by
-
al mena: a comprehensive resource of human genetic variants integrating genomes and exomes from Arab, Middle Eastern and North African populations
Journal of Human Genetics (2017)
-
Impact of Genetic Polymorphisms on Phenytoin Pharmacokinetics and Clinical Outcomes in the Middle East and North Africa Region
Drugs in R&D (2017)
-
Genome-wide insights into the genetic history of human populations
Investigative Genetics (2015)

{kind=link}
{kind=link}