
Abstract
Genome-wide screening for copy number variations (CNVs) in ten Indian dyslexic families revealed the presence of five de novo CNVs in regions harboring GABARAP, NEGR1, ACCN1, DCDC5, and one in already known candidate gene CNTNAP2. These genes are located on regions of chromosomes 17p13.1, 1p31.1, 17q11.21, 11p14.1 and 7q35, respectively, and are implicated in learning, cognition and memory processes through dendritic spinal plasticity, though not formally associated with dyslexia. Molecular network analysis of these and other dyslexia-related module genes suggests them to be associated with synaptic transmission, axon guidance and cell adhesion. Thus, we suggest that dyslexia may also be caused by neuronal disconnection in addition to the earlier view that it is due to neuronal migrational disorder.
Similar content being viewed by others


Introduction
Developmental dyslexia (DD) (MIM#127700) is a common neurogenetic disorder that affects 5–12% of school-aged children.1, 2 The major deficit is impaired phonological processing, which interferes with the mapping between orthography and phonology. Linkage studies have identified nine following regions on different chromosomes for DD (DYX1–DYX9): DYX1 on 15q21 contains DYX1C1; DYX2 on 6p22.2–p21.3 contains DCDC2 and KIAA0319; DYX3 on 2p16–p15 contains MRPL19 and C2ORF3; DYX4 on 6q11.2–q12; DYX5 on 3p12–q13 contains ROBO1; DYX6 on 18p11.2; DYX7 on 11p15.5; DYX8 on 1p36–p34 contains KIAA0319L and DYX9 on Xq27.2–q28.3 These loci were identified in samples covering the American, Canadian, British, German and the Finnish families. A growing body of evidence suggests that structural variations across the genome, including copy number variations (CNVs) likely contributes to human disorders.4, 5 CNVs refer to the presence of DNA segments longer than 1 kb with >90% sequence identity that differ in the number of copies between the genomes of different individuals.6 It affects more nucleotides per genome than single-nucleotide polymorphism (SNP), and contributes significantly to variations in levels of gene expression and phenotypes of medical relevance among individuals.7 Even in studies that have shown negative linkage, the possible contribution of undetected CNVs cannot be dismissed.3 Genome-wide screenings for CNVs have demonstrated that deletions and duplications that disrupt gene functioning are found more in patients than in healthy subjects.8, 9, 10, 11, 12, 13 The pathogenicity of CNVs is correlated with their presence, location and size.14, 15 Our search for de novo CNVs in persons with dyslexia and healthy controls from India identified five novel genes, GABARAP, ACCN1, NEGR1 and DCDC5, and also one already identified candidate gene CNTNAP2.16 Protein interaction network and pathway analyses of these genes suggest that dyslexia is not only a neuronal migration disorder but also a disorder due to defects in neurotransmission and cell adhesion.
Materials and methods
Thirty-eight members consisting of 14 DD subjects and 24 related/unrelated non-dyslexic controls from 10 families were initially recruited for the study. DD subjects were certified as having dyslexia by authorized National Institutes viz., All Indian Institute of Speech and Hearing (AIISH), Mysore and National Institute of Mental Health and Neuro Sciences (NIMHANS), Bangalore. DD subjects were between the ages of 11–23 years. Their school records, class note books as well as test papers, wherever available were examined and informal assessment of reading and writing using their text books was performed as a preliminary step to judge their present status. We also tested these individuals on standard tests of reading and reading-related tests in Kannada (the mother tongue of the participants), such as phonological awareness and rapid automatized naming measures. All the children with dyslexia (except case 5) were normal on nonverbal intelligence measure and reading at least two s.d. below the mean of reference group consisting of typically developing children of the same age/grade group. Further, they all had normal or corrected to normal vision, and did not have any history of neurological disorders. All of them were right-handed as assessed by Edinburgh handedness test.17
The five probands described in the present paper were between 11–23 years of age. Out of the five, one was a school dropout (case 5) and the others were still attending remedial teaching on regular basis. All of them were administered a battery of neuropsychological tests. In addition to the dyslexia assessment-related tests (such as decoding, oral reading, rapid automatized naming, phonological awareness tests) the battery included Rutter’s Proforma B (to exclude individuals with behavior problems), standard progressive matrices (used as a measure of nonverbal intelligence/abstract reasoning), and non-word repetition (used as a measure of serial recall/verbal short-term memory). Table 1 shows the performance of five dyslexics on these tests. As a reference for comparison, scores of a control individual are provided in the last column.
All the neuropsychological tests were administered individually in a quiet room either at school, at home or at the Department of Studies in Psychology, University of Mysore in several sessions, lasting from 1 to 2 h each. Rapport was built and case history was collected before beginning the actual assessment. Total time taken to complete the tasks on reading measures and rapid automatized naming tests were recorded along with the accuracy, whereas only accuracy scores were recorded for phonological awareness tasks.
For genotyping, 5 ml EDTA blood was collected from all the 38 members (Supplementary Figures 1 and 2) of the family, and genomic DNA was extracted using Promega Wizard Genomic DNA purification Kit (Promega Corporation, Madison, WI, USA). The isolated DNA was quantified by Bio-photometer and gel electrophoresis. This research was approved by the University of Mysore Institutional Human Ethics review committee (IHEC). Written informed consent was obtained from all sample donors and the IHEC approved the sample consent procedure. Written informed consent was obtained from parents/guardians in the cases of participants being minors.
Genotyping
Genome-wide genotyping was performed using an Affymetrix Genome-wide Human SNP Array 6.0 chip, which has 1.8 million combined SNP and CNV markers with the median intermarker distance of 680 bases. This chip provides maximum panel power and the highest physical coverage of the genome. Genotyping quality was assessed using Affymetrix Genotyping Console Software (Affymetrix, Inc., Santa Clara, CA, USA). Briefly, all SNPs that were called using Birdseed v2 algorithm had a quality control call rate of >97% across the subjects and controls in families. All the subjects and members with SNPs that passed SNP quality control procedures were entered into the CNV analysis. Filters were set for ID call rates for the overall SNPs to identify IDs with poor quality DNA. The CNV calls were generated using the Canary algorithm. In Affymetrix Genotyping Console Software contrast quality control has to be >0.4 to be included in the CNV analyses. In this study, contrast quality control was observed to be >2.5 across all the samples showing a robust strength. A description of the CNV methods used in this study is given below. Copy number analysis method offers two types of segmenting methods, univariate and multivariate. These methods are based on the same algorithm, but use different criteria for determining cut-points denoting CNV boundaries.
BirdSuite (v2)
BirdSuite18 is a suite originally developed to detect known common copy number polymorphisms (CNPs) based on prior knowledge, as well as to discover rare CNVs, from Affymetrix SNP 6.0 array data. To do this, it incorporates two main methods; the ‘Birdsuite’ algorithms and the ‘Canary’.19 The Birdsuite algorithm uses a Hidden Markov model approach to find regions of variable copy number in a sample. For the Hidden Markov model, the hidden state is the true copy number of the individual’s genome and the observed states are the normalized intensity measurements of each array probe.
Canary
CNP analysis was performed using the Canary algorithm. Canary was developed by the Broad Institute for making copy number state calls in genomic regions with CNPs. Canary algorithm computes a single intensity summary statistic using a subset of manually selected probes within the CNP region. The intensity summaries are compared in aggregate across all samples to intensity summaries previously observed in training data to assign a copy number state call.
CNV calls from the Canary and Birdsuite algorithms were collated for each sample, and kept as long as they met the following criteria: (i) Birdsuite calls with a log10 of odds score (odds ratio) greater than or equal to 10 (corresponding to an approximate false discovery rate of ∼5%), (ii) Birdsuite calls with copy number states other than 2 were retained; (iii) Canary CNP calls with CN states different from the population mode were retained.
CNVFinder
CNVFinder developed at the Welcome Trust Sanger Institute uses a dynamic, multiple-threshold-based approach to allow robust classification of copy number changes in data of varying qualities. This algorithm makes two main assumptions: (i) that the majority of data points are normally distributed around a log2 ratio of zero and (ii) that data points falling outside of the centralized log2 ratio distribution are representative of a difference in copy number between test and reference genome.
The thresholds are adjusted using a noise factor termed SDe. This factor is equal to the 68th percentile of the complete log2 ratio distribution and is calculated on chromosome basis during copy number detection. The SDe is an inference of the s.d. that is fairly not sensitive to outliers and thus, as long as the two main assumptions hold true, it can be used as a reliable measure of experimental variability.
Data analysis
Genome-wide association and CNV study were carried out using Omicsoft Array Studio20 and Affymetrix Genotyping Console software as prescribed in their manuals. Eigenstrat method21 was used to avoid the possibility of spurious associations resulting from population stratification. This method derives the principal components of the correlations among gene variants and corrects for those correlations in the testing. We removed one individual (1) control mother sample from Family (A) because it was an extreme outlier on one or more significant EIGENSTRAT axes. Bonferroni correction was employed for multiple testing and the corrected data were then used for the association and CNV testing.
Breakpoint validations
CNV breakpoint validations in the regions bearing GABARAP, ACCN1, NEGR1, CNTNAP2 and indel variation of 155 bp DCDC5 were performed on the 270 individuals’ Affymetrix Genome-wide SNP 6.0 samples obtained from the International HapMap Consortium22 and also on the 31 individuals genotyped on the same Affymetrix Genome-wide SNP 6.0 platform from the Tibetan population. The samples for the HapMap come from a total of 270 people: the 30 both-parent-and-adult-child trios from the Yoruba people in Ibadan, Nigeria, 45 unrelated Japanese individuals in Tokyo, 45 unrelated individuals Han Chinese in Beijing, and the 30 both-parent-and-adult-child trios from Center d'Etude du Polymorphisme Humain (CEPH). The raw, unprocessed data from Affymetrix Genome-Wide SNP 6.0 array for the 31 individuals of the Tibetan population were obtained from the ArrayExpress Archive at the European Bioinformatics Institute submitted by Simonson et al.23
Weighted protein interaction network analysis
We used weighted protein network analysis in a first attempt to identify dyslexia-associated modules and their key constituents. Weighted protein network analysis starts from the level of thousands of genes, identifies modules of physically interacting proteins, co-localized proteins, co-expressed proteins and proteins falling under the common pathway, and relates these modules to clinical variables and gene ontology information. We made the use of tools such as GeneMANIA24 BIOGRID and CYTOSCAPE for constructing protein interaction databases. The network generated was then used for conducting network pathway studies to assess the functional consequences of the network topology.
Because gene modules may correspond to biological pathways, focusing the analysis on modules (and their highly connected intramodular hub genes) amounts to a biologically meaningful data reduction scheme. We used the minimal cut sets (MCS) principle to determine the loss of a defined cell function, to study the structural fragility and to identify knocked out regions in reaction networks. We also analyzed functional enrichment for each gene using Gene Ontology and AmiGO to understand the biological significance of module genes and to identify putative disease pathways.
Results
Genome-wide copy number and phenotype assessment of dyslexics
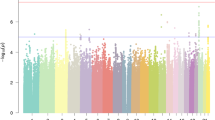
All the 38 members of 10 families were recruited for a thorough diagnostic testing (Supplementary Figures 1 and 2). Out of these, 10 boys and 4 girls with the age range of 11–23 years were found to be dyslexics. All were of average intellectual level and had no major emotional and behavioral problems. Analyzing the collated data from the arrays from those meeting the CNP calls with a log10 of odds score greater than or equal to 10 (corresponding to a false discovery rate of ∼5%) criteria were selected for further investigation. We observed a total of 1198 CNV events across 38 genomes, with 74% duplications and 26% deletions. Various CNVs ranging in size from 100 kb to several Mb were observed in multigene families of alpha-amylase family (AMY1 and AMY2), rRNA genes, immunoglobulins, olfactory receptor genes and in the uridine glucuronosyltransferase genes across the genome of control members and dyslexic subjects. However, CNV analysis revealed the presence of rare de novo microduplications and deletion in five subjects belonging to five different families in regions 17p13.1, 1p31.1, 17q11.21, 11p14.1 and 7q35, respectively (Figure 1). These regions were found bearing GABARAP, ACCN1, NEGR1, CNTNAP2 and an indel variation of 155 bp in DCDC5 (Table 2). These genes were found to express in the brain from the developmental stage. We also found a significant over-representation of these genes in pathways important for brain development. These genes have also been showed to have a role in the learning and cognition making them probable candidate genes for dyslexia. These CNVs were not found in the genomes of the unaffected family members and were limited to only members with dyslexia. Though the consanguineous nature of some of the families (Supplementary Figures 1 and 2) suggested a recessive mode of inheritance, however, careful examination of the CN genotyping revealed otherwise, showing heterozygous deletions and duplications with copy number states being one for the identified deletions and three for the observed duplications in probands.

Schematic representation of log R ratios and copy number state of dyslexia cases. An image of the copy number state (a, b) and log R ratios (c–f) indicates the quantitative assessments of genotyping used to determine CNV in the dyslexic cases. Duplication of GABARAP in case 1 and case 2 shown in (a); CNTNAP2 (b); Partial duplication of NEGR1 in case 3 and case 4 is shown in (c, d); ACCN1 (e); and deletion of DCDC5 in case 5 in (f). A full color version of this figure is available at the Journal of Human Genetics journal online.
Both cases 1 and 2 had gamma-amino butyric acid-A (GABAA) receptor-associated protein (GABARAP) duplications, while Case 2 also had duplication in CNTNAP2 (Table 2). Case 1, a participant who had undergone remedial intervention for more than 6 years, did not show any notable problems with decoding-related tests, but was significantly poor in naming tasks (Table 1). However, case 2, with GABARAP and CNTNAP2 CNVs, exhibited severe problems in all the dyslexia-related reading–spelling tests and also on naming tasks. Both cases 3 and 4 had NEGR1 duplications, while case 4 also had duplication in ACCN1 (Table 2). Both exhibited mild problems with phonological awareness tasks but more severe problems with rapid naming tasks. Relatively, the problems on decoding and naming tasks were more pronounced in case 4 (Table 1). On the other hand, the performance of case 5 with the deletion of DCDC5 performed poorly on all the tasks (Table 1). Thus, the test performances largely reflect the phenotypic differences across different conditions though the genetic effect might have been masked by the remedial interventions of some of the dyslexic individuals assessed. The remaining six cases, 6–11, had duplications and deletion involving PCDH11X in the Xq21.3 region, which will be reported elsewhere (under review elsewhere). Similarly, the remaining three cases, 12–14, carried CNVs in the 17q21.31 region, which was previously reported for dyslexia (under review elsewhere).
Protein interaction network of dyslexia
Network analysis of candidate genes of dyslexia including the genes identified in this study (Figure 2) using GeneMANIA, BIOGRID and Cytoscape has established 48 genes with protein and genetic interaction having more than 125 links. Out of these, 114 interactions involving 30 genes acting in the brain cortex region could be accounted for by the analysis in the present study. These interactions are more specific for dyslexia as they directly participate in axon guidance, neurogenesis and synaptic transmission. The geometrical establishment of dyslexia network shows already identified candidate genes interacting with experimentally identified five genes of the present study, GABARAP, ACCN1, NEGR1, DCDC5 and CNTNAP2. By applying the MCSs concept, we have identified eight other possible candidate genes TUBB3, GRIA2, GRIA7, GPC1, AP2B1, DOCK4, VAPA and PICK1. Each of these genes in the pathway, if disrupted, may result in dyslexia (Figures 3a and b).

Dyslexia neural network establishing the associated gene modules and their key hubs. Network of genes involved in dyslexia with hub genes colored in their respective gene ontology. A full color version of this figure is available at the Journal of Human Genetics journal online.

Schematic representation of the dyslexia molecular signaling pathway for neuronal migration, axon growth, synaptic transmission and cell adhesion. A full color version of this figure is available at the Journal of Human Genetics journal online.
Neurodevelopment pathways
The network analysis of the above genes (Figure 2) and their interconnecting pathways suggest that they are vital in the following six biological functions (Table 3) that include: (1) synaptic transmission, (2) axon guidance, (3) transmission of nerve impulse, (4) neurogenesis, (5) cell migration and (6) protein domain specific binding. Based on this, we constructed two neuronal dyslexia specific pathways: developmental and structural.
The ‘Developmental’ and ‘Structural’ pathways were constructed manually looking into the protein physical interaction from the presently established dyslexia network and available literature to perform a pathway enrichment (over-presenting) analysis separately for the five genes we detected. We stipulated that at least two genes in a pathway must be disrupted, for that pathway to be considered enriched, and used the MCSs concept.
We illustrated in Figure 3a, the possible hypothetical developmental pathway involving the genes in axon guidance and neuronal migration in seven pathways: (1) ROBO family of genes, ROBO1, ROBO2 and ROBO3, are bound by beta-tubulin proteins (TUBB3) act as receptors for SLIT proteins and guides the axons based on attractive and repulsive mechanisms. (2) GRIA2, GRM7, GPC1 and KIAA0319 are transmembrane receptors that are involved in neurotransmission. PICK1 is an adapter protein that binds and organizes these transmembrane receptors through a PDZ protein-binding domain. (3) KIAA0319 also follows a clathrin-mediated endocytic pathway for certain neurotransmitters. KIAA0319 when bound by AP2B1 functions as a transmembrane receptor, but when AP2B1 dissociates, it then follows the clathrin-mediated endocytic pathway. (4) STUB1, an adapter protein, binds to HSP70 and HSC70 by the hydrolysis of ATP and then to DYX1C1 and HSP90 complexes, which finally associates with transmembrane protein VAPA. (5) N-ethylmaleimide-sensitive factor, found ubiquitously in the cytoplasm, has a central role in the transfer of membrane vesicles from one membrane compartment to another, and interacts with VAPA through a signal transduction pathway mediated by YWHAB. (6) NEGR1 is a cell adhesion molecule that regulates the development of neuronal projections when DCDC2 and DCDC5 remain bound to the cytoskeleton, and makes a stable adherence with the postsynaptic cell during synapse. (7) MRPL19, a ribosomal protein, translocates itself to the mitochondria; however, its pathway is yet to be established. The functional role of C2ORF3 is also yet to be identified.
In Figure 3b, the possible five hypothetical structural pathways involving genes involved in neurotransmission and cell adhesion are illustrated: (1) Glutamate is converted to GABA by glutamic acid decarboxylase and gamma-aminobutyric acid, which is transported inside the vesicles by vesicular GABA transporter in the presynaptic cell for efficient transmission into the postsynaptic cell. GABAA and GABAB, which are bound to beta-tubulin proteins, TUBB3, are clustered by GABARAP in the postsynaptic cell, enabling the postsynaptic cell to be ready for the transmission from the presynaptic cell. (2) GABAB inhibits the function of ACCN1, which is a sodium-based ion channel involved in excitatory neurotransmission. The sudden rush of neurotransmitters from ACCN1 during GABAA functioning causes a great flux of both the inhibitory and excitatory neurotransmitters. To avoid such a complex state, GABAB inhibits ACCN1. (3) DYX1C1 alone fails to activate the estrogen receptor elements, but can activate when bound to the ESR1 and ESR2 complex. GABARAP is involved in the trafficking of protein molecules to its target place and clustering them together during synapses by binding to the beta-tubulin family of proteins, TUBB3 and TUBB4. Before even clustering of GABAA and GABAB receptors can take place, GABARAP and DYX1C1 interact through discrete domains helping in the trafficking to membrane. (4) PCDH11X, a calcium-based cell adhesion molecule binds to its counterpart during the establishment of synapse between presynaptic and postsynaptic cell. (5) Although ROBO1 performed as a receptor for SLIT proteins in the developmental stage, in the structural pathway it acts as a cell adhesion molecule with its counterparts in the postsynaptic cell. There are some essential genes that are active in both the developmental and structural pathways. Thus, disruption of the genes involved in these pathways identifies critical regions in cellular networks that can result in dyslexia.
CNV breakpoint validation
The observed de novo CNV breakpoints near/in GABARAP, ACCN1, NEGR1, DCDC5 and CNTNAP2 from this study were checked against 270 HapMap and 31 Tibetan individuals’ genome. The entire 301 non-dyslexic control group did not show such CNV breakpoints in the regions bearing the genes identified in this study. These duplications and deletions were also validated and found to be novel when checked against the online CNV database, Database of Genomic Variants http://projects.tcag.ca/variation.
Discussion
Our study focused CNVs in de novo state in regions of both dyslexia-associated loci and other genes across all chromosomes using Affymetrix Genome-Wide Human SNP Array 6.0 chip having 1.8 million combined SNP and CNV markers with the median intermarker distance of 680 bps. This study revealed five novel loci GABARAP, ACCN1, NEGR1 and DCDC5 and also one already identified candidate gene CNTNAP2 to be candidate genes for dyslexia. However, earlier studies by Girirajan et al.25 using a customized NimbleGen array did not find CNVs in dyslexics probably due to the low resolution of the array adopted (2.6 kb).
The GABARAP clusters GABAA receptors by mediating interaction with cytoskeleton. It is also involved in the trafficking of protein molecules to their target places26, 27 and clustering them together during synapses. GABAA’s association with cognition and memory has been well documented.28, 29, 30, 31 Mohler30 also reported the role of GABAA receptors in the regulation and expression of associative memory in cognitive functions through dendritic spinal plasticity. Nonfunctional GABARAP due to microduplications as we see in cases 1 and 2 of the present study (Table 2) fails to form GABARAP–DYX1C1 complex resulting in the non-trafficking and non-clustering of GABAA into the membranes, further leading to a lack of inhibitory neurotransmission. This creates an uncontrolled cellular environment with sudden unexpected rush of neurotransmitters altering the chemical composition of the postsynaptic neuronal cell. A deletion of chromosome 17p13.1 results in multisystemic manifestations including moderate mental retardation, intractable epilepsy, developmental psychomotor/cognitive deficits and dysmorphic features.32 The cognitive phenotypes observed in the GABARAP-duplicated case 1 and case 2 of the present study is almost similar with the cognitive deficits observed in the 17p13.1 deletion, indicating that GABARAP is involved in the etiology of cognition-related disorders.
NEGR1, a cell adhesion molecule belonging to the Kindred of Iglon molecules (Kilon), regulates the development of neuronal projections via attractive and repulsive mechanisms.33 AsKilon is found chiefly at axons and presynaptic terminals, it can be seen mainly expressing at dendritic postsynaptic spine of neurons.34 A deletion of chromosome 1p31.1–31.3 results in significant speech and language delay, learning disability, developmental co-ordination disorder and attention deficit/hyperactivity disorder.35 Therefore, microduplications of the NEGR1 in the two cases (cases 3 and 4) identified may result in altering the production of dendritic postsynaptic spines in mature neurons.
ACCN1, a key component of acid-activated currents, is implicated in processes underlying synaptic plasticity, learning and memory.36 It is found enriched in synaptosomes at synapses in a pattern that suggests its dendritic localization. Disruption of the ACCN1 impairs hippocampal long-term potentiation and synaptic plasticity. A deletion of chromosome 17q11.23 results in the manifestations of mild-to-moderate psychomotor developmental delay, language delay and cognitive deficits.37, 38 The cognitive deficits associated with the 17q11.23 deletion and the phenotypic deficits observed in the case of microduplication of ACCN1 in the present study suggests the production of a nonfunctional protein that fails to get trafficked into the membrane, resulting in improper neurotransmission resulting in dyslexia.
DCDC5 is localized in the DYX7 region and belongs to the double cortin family of genes that share the microtubule binding double cortin domain. Mutations in these genes cause neuronal migration disorders.39
CNTNAP2, a member of the neurexin family, functions as cell adhesion molecules and receptors. This protein is localized at the juxtaparanodes of myelinated axons and mediates interactions between neurons and glia participating in synaptic plasticity.40, 41, 42 CNTNAP2 is directly bound and regulated by forkhead box protein P2 (FOXP2), a transcription factor related to speech and language development. CNTNAP2 polymorphisms in children with typical specific language impairment had significant association with nonsense word repetition.16 In the present study, duplication in exons 1 and 2 of the CNTNAP2 alters the protein function, and hence the direct interaction of CNTNAP2 with CNTN2, another contactin protein is lost. CNTN2 is a neuronal membrane protein that functions as a cell adhesion molecule and has a critical role in the formation of axon connections in the development of nervous system.43
Genes involved in dendritic spinal synaptic plasticity
Dendritic spines help transmit electrical signals to the neuron’s cell body and undergo changes in the shape, volume and number. This plasticity is implicated in motivation, learning and memory.44, 45, 46, 47 Age-dependent changes in the rate of spine turnover suggest that spine stability impacts developmental learning.34, 48, 49 Alvarez and Sabatini,34 put forth on one hand how structural plasticity, experience and activity leads to the formation of relevant synaptic connections leading to learning and on the other hand, how experience and activity leads to the removal of excessive synaptic connections. It was shown that skill training has lead to the formation and stabilization of new spines while destabilizing old spines, suggests that the learning of a new skill involved a rewiring process of neural circuits. As the extent of spine remodeling correlates with the success of learning, the crucial role of synaptic structural plasticity in learning and memory50, 51 may be inferred. This may perhaps explain how dyslexics start to perform better while going through any effective remediation.
Thus, the five genes identified in the present study showed their involvement in cognition, learning and memory through dendritic spinal synaptic plasticity. Thus, this can explain how dyslexics start to perform better over an extensive learning and skill acquiring period, which can be attributed to the rewiring of dendritic spines leading to stabilized connections.
Molecular network of dyslexia
We propose the dyslexia gene network model that controls six major functions: synaptic transmission, axon guidance, transmission of nerve impulse, neurogenesis, cell migration and protein domain specific binding based on weighted gene protein interaction network. We also identified disease-related networks (modules) and disease-related hub genes, and found four large protein–protein interacting hubs associated with dyslexia: GABARAP, YWHAB, PICK1 and ROBO1. We used MCSs in this network study,52 which helps in studying the structural fragility and to identify knock out strategies in cellular networks. When the MCSs are computed with respect to ‘physical and genetic’ interactions in realistic networks, the experimental block of essential genes will inevitably lead to mutants. Thus, the candidate genes identified in this study are the indicators of major MCSs. The immediate interacting protein partners represents primary level MCSs and the immediate interacting protein partners of the primary level MCSs represent secondary level MCSs. These MCSs are distributed in a fragile interconnected network. This can be demonstrated as follows: GABARAP interacts with DYX1C1 and N-ethylmaleimide-sensitive factor proteins, which is essential for proper trafficking of GABAA receptors, whereas GABAA and GABAB are essential for efficient neurotransmission. Duplication identified in this study in GABARAP disrupts the protein production, which in turn fails to interact with DYX1C1 resulting in the blocked pathway. Similarly, CNTNAP2 interacts with CNTN2, DCDC2, DCDC5, KIAA0319, C2ORF3, PCDH11X and ROBO1 in a pathway regulated by YWHAB through signal transduction. Primary level interactions of YWHAB include two already identified dyslexia candidate genes, VAPA and DOCK4, and one candidate gene, NEGR1, from this current study. CNTNAP2 and NEGR1 have common interaction partners through primary and secondary levels and NEGR1 is found to co-express with GABARAP. CNTNAP2 is also found to co-express with GABARAP, YWHAB, DOCK4, CNTN2 and co-localizes with N-ethylmaleimide-sensitive factor, a protein bridging both GABARAP and YWHAB hubs. GABARAP and YWHAB hubs are in tandem with PICK1 hub that consists largely of glutamate-based transmembrane receptor proteins like GRIA2, GRIP1, GRM7 and GCC1, which are involved in modulating memory performance. ACCN1, a candidate gene from this current study, interacts with KIAA0319, C2ORF3, MRPL19, ACCN2, PCDH11X, PICK1, and is regulated by STOML1. ROBO1 represents another hub consisting of genes involved as cell adhesion molecules and transmembrane receptors. This type of structural fragility in the dyslexia neural network is given by the size distribution over the total set of MCSs. In such networks, a dysfunction can be caused in higher probability, as a malfunction requires only one or a few network elements to fail.
On the basis of our current de novo CNV events, neuronal network and pathway studies, we have made an attempt to visualize the ongoing cellular processes with an aim to understand the functional role of the candidate genes identified in this study and also to study its implication on the other molecules and processes in the cellular network. Our findings on CNVs contribute to the array of dyslexic phenotypes seen in families, which demonstrates the importance of performing a high-resolution assessment of genomic background, even after the detection of a rare and likely damaging CNV. Although dyslexia has long been thought as a neuronal migration disorder, the present study suggests that dyslexia may also as be viewed as a disorder due to defects in neurotransmission and cell adhesion processes.
References
Grigorenko, E. L., Wood, F. B., Meyer, M. S. & Pauls, D. L. Chromosome 6p influences on different dyslexia-related cognitive processes: further confirmation. Am. J. Hum. Genet. 66, 715–723 (2000).
Katusic, S. K. Incidence of reading disability in a population-based birth cohort, 1976-1982, Rochester, Minn. Mayo Clin. Proc. 76, 1081–1092 (2001).
Scerri, T. S. & Schulte-Korne, G. Genetics of developmental dyslexia. Eur. Child Adolesc. Psychiatry 19, 179–197 (2010).
Estivill, X. & Armengo, L. Copy number variants and common disorders: filling the gaps and exploring complexity in genome-wide association studies. PLoS Genet 3, 1787–1799 (2007).
Conrad, D. F., Pinto, D., Redon, R., Feuk, L., Gokcumen, O., Zhang, Y. et al. Origins and functional impact of copy number variation in the human genome. Nature 464, 704–712 (2010).
Freeman, J. L., Perry, G. H., Feuk, L., Redon, R., McCarroll, S. A., Altshuler, D. M. et al. Copy number variation: new insights in genome diversity. Genome Res. 16, 949–961 (2006).
Aitman, T. J., Dong, R., Vyse, T. J., Norsworthy, P. J., Johnson, M. D., Smith, J. et al. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature 439, 851–855 (2006).
Wagenstaller, J., Spranger, S., Lorenz-Depiereux, B., Kazmierczak, B., Nathrath, M., Wahl, D. et al. Copy-number variations measured by single-nucleotide-polymorphism oligonucleotide arrays in patients with mental retardation. Am. J. Hum. Genet. 81, 768–779 (2007).
Sebat, J., Lakshmi, B., Malhotra, D., Troge, J., Lese-Martin, C., Walsh, T. et al. Strong association of de novo copy number mutations with autism. Science 316, 445–449 (2007).
Alarcon, M., Abrahams, B. S., Stone, J. L., Duvall, J. A., Perederiy, J. V., Bomar, J. M. et al. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am. J. Hum. Genet. 82, 150–159 (2008).
Marshall, C. R., Noor, A., Vincent, J. B., Lionel, A. C., Feuk, L., Skaug, J. et al. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 82, 477–488 (2008).
Tam, G. W., Redon, R., Carter, N. P. & Grant, S. G. The role of DNA copy number variation in schizophrenia. Biol. Psychiatry 66, 1005–1012 (2009).
Kirov, G. The role of copy number variation in schizophrenia. Expert Rev. Neurother. 10, 25–32 (2010).
Walsh, T., McClellan, J. M., McCarthy, S. E., Addington, A. M., Pierce, S. B., Cooper, G. M. et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320, 539–543 (2008).
Need, A. C., Ge, D., Weale, M. E., Maia, J., Feng, S., Heinzen, E. L. et al. A genome-wide investigation of SNPs and CNVs in schizophrenia. PLoS Genet. 5, 120–127 (2009).
Peter, B., Raskind, W. H., Matsushita, M., Lisowski, M., Vu, T., Berninger, V. W. et al. Replication of CNTNAP2 association with nonword repetition and support for FOXP2 association with timed reading and motor activities in a dyslexia family sample. J. Neurodevelop Disord. 3, 39–49 (2011).
Oldfield, R. C. The assessment and analysis of handedness: the Edinburgh inventory. Neuropsychologia 9, 97–113 (1971).
Birdsuite Algorithm Http://www.broad.mit.edu/mpg/birdsuite/birdseed.html (2010).
White Paper: AffymetrixCanary Algorithm Version 1.0., 1-7 (2008).
The analysis for this paper was generated using Array Studio software. Array Studio, Array Viewer and Array Server and all other Omicsoft products or service names are registered trademarks or trademarks of Omicsoft Corporation., Research Triangle Park, NC, USA.
Price, A. L., Patterson, N. J., Plenge, R. M., Weinblatt, M. E., Shadick, N. A. & Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006).
The International HapMap Consortium. The International HapMap Project. Nature 426, 789–796 (2003).
Simonson, T. S., Yang, Y., Huff, C. D., Yun, H., Qin, G., Witherspoon, D. J. et al. Genetic evidence for high-altitude adaptation in Tibet. Science 329, 72–75 (2010).
Warde-Farley, D., Donaldson, S. L., Comes, O., Zuberi, K., Badrawi, R., Chao, P. et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 1, 38–45 (2010).
Girirajan, S., Brkanac, Z., Coe, B. P., Baker, C., Vives, L., Vu, T. H. et al. Relative burden of large CNVs on a range of neurodevelopmental phenotypes. PLoS Genet. 7, e1002334 (2011).
Coyle, J. E., Qamar, S., Rajashankar, K. R. & Nikolov, D. B. Structure of GABARAP in two conformations: implications for GABA (A) receptor localization and tubulin binding. Neuron 33, 63–74 (2002).
Chen, Z., Chang, C. S., Leil, T. A. & Olsen, R. W. C-terminal modification is required for GABARAP mediated GABAA receptor trafficking. J. Neurosci. 27, 6655–6663 (2007).
Collinson, N., Kuenzi, F. M., Jarolimek, W., Maubach, K. A., Cothliff, R., Sur, C. et al. Enhanced learning and memory and altered GABAergic synaptic transmission in mice lacking the α5 subunit of the GABAA receptor. J. Neurosci. 22, 5572–5580 (2002).
Mohler, H. Molecular regulation of cognitive functions and developmental plasticity: impact of GABAA receptors. J. Neurochem. 102, 1–12 (2007).
Mohler, H. Role of GABAA receptors in cognition. Biochem. Soc. Trans. 37, 1328–1333 (2009).
Lewis, D. A., Cho, R. Y., Carter, C. S., Eklund, K., Forster, S., Kelly, M. A. et al. Subunit-selective modulation of GABA type A receptor neurotransmission and cognition in schizophrenia. Am. J. Psychiatry 165, 1585–1593 (2008).
Komoike, Y., Shimojima, K., Liang, J. S., Fujii, H., Maegaki, Y., Osawa, M. et al. A functional analysis of GABARAP on 17p13.1 by knockdown zebrafish. J. Hum. Genet. 55, 155–162 (2010).
Hashimoto, T., Yamada, M., Maekawa, S., Nakashima, T. & Miyata, S. IgLON. Cell adhesion molecule Kilon is a crucial modulator for synapse number in hippocampal neurons. J.Brain Res. 1224, 1–11 (2008).
Alvarez, V. A. & Sabatini, B. L. Anatomical and physiological plasticity of dendritic spines. Annu. Rev. Neurosci. 30, 79–97 (2007).
Gillberg, C. & FitzPatrick, D. Case report: further evidence for a recognizable syndrome caused by deletion of 1p31. Adv. Clin. Neurosci. Rehabil. 3, 16–18 (2010).
Wemmie, J. A., Chen, J., Askwith, C. C., Hruska-Hageman, A. M., Price, M. P., Nolan, B. C. et al. The acid activated ion channel ASIC contributes to synaptic plasticity, learning, and memory. Neuron 34, 463–477 (2002).
Riva, P., Corrado, L., Natacci, F., Castorina, P., Wu, B. L., Schneider, G. H. et al. NF1 microdeletion syndrome: refined FISH characterization of sporadic and familial deletions with locus-specific probes. Am. J. Hum .Genet. 66, 100–109 (2000).
Caselli, R., Ballarati, L., Selicorni, A., Milani, D., Maitz, S., Valtorta, C. et al. A 12.4 Mb duplication of 17q11.2q12 in a patient with psychomotor developmental delay and minor anomalies. Eur. J. Med. Genet. 53, 325–328 (2010).
Meng, H., Smith, S. D., Hager, K., Held, M., Liu, J., Olson, R. K. et al. DCDC2 is associated with reading disability and modulates neuronal development in the brain. Proc. Natl Acad. Sci. USA 102, 7053–7058 (2005).
Strauss, K. A., Puffenberger, E. G., Huentelman, M. J., Gottlieb, S., Dobrin, S. E., Parod, J. M. et al. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N Engl. J. Med. 354, 1370–1377 (2006).
Groszer, M., Keays, D. A., Deacon, R. M., de Bono, J. P., Prasad-Mulcare, S., Gaub, S. et al. Impaired synaptic plasticity and motor learning in mice with a point mutation implicated in human speech deficits. Curr. Biol. 18, 354–362 (2008).
Zweier, C., de Jong, E. K., Zweier, M., Orrico, A., Ousager, L. B., Collins, A. L. et al. CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila. Am. J. Hum. Genet. 85, 655–666 (2009).
Yoshihara, Y., Kawasaki, M., Tamada, A., Nagata, S., Kagamiyama, H., Mori, K. et al. Overlapping and differential expression of BIG-2, BIG-1, TAG-1, and F3: four members of an axon-associated cell adhesion molecule subgroup of the immunoglobulin superfamily. J. Neurobiol. 28, 51–69 (1995).
Levitt, P. A monoclonal antibody to limbic system neurons. Science 223, 299–301 (1984).
Pimenta, A. F., Reinoso, B. S. & Levitt, P. Expression of the mRNAs encoding the limbic system-associated membrane protein (LAMP): II. Fetal rat brain. J. Comp. Neurol. 375, 289–302 (1996).
Struyk, A. F., Canoll, P. D., Wolfgang, M. J., Rosen, C. L., D'Eustachio, P., Salzer, J. L. et al. Cloning of neurotrimin defines a new subfamily of differentially expressed neural cell adhesion molecules. J. Neurosci. 15, 2141–2156 (1995).
Funatsu, N., Miyata, S., Kumanogoh, H., Shigeta, M., Hamada, K., Endo, Y. et al. Characterization of a novel rat brain glycosylphosphatidylinositol-anchored protein (Kilon), a member of the IgLON cell adhesion molecule family. J. Biol. Chem. 274, 8224–8230 (1999).
Zuo, A. L., Chang, P. & Gan, W. B. Development of long-term dendritic spine stability in diverse regions of cerebral cortex. Neuron 46, 181–189 (2005).
Holtmaat, A. J., Trachtenberg, J. T., Wilbrecht, L., Shepherd, G. M., Zhang, X., Knott, G. W. et al. Transient and persistent dendritic spines in the neocortex in vivo. Neuron 45, 279–291 (2005).
Yang, G., Pan, F. & Gan, W. B. Stably maintained dendritic spines are associated with lifelong memories. Nature 462, 920–924 (2009).
Xu, T., Yu, X., Perlik, A. J., Tobin, W. F., Zweig, J. A., Tennant, K. et al. Rapid formation and selective stabilization of synapses for enduring motor memories. Nature 462, 915–919 (2009).
Klamt, S. Generalized concept of minimal cut sets in biochemical networks. BioSystems 83, 233–247 (2006).
Rutter, M. Children’s behavior questionnaire for completion by teachers; preliminary findings. J. Child Psy. Pry 8, 1–11 (1967).
Raven, J., Raven, J. C. & Court, J. H. Manual for Raven's Standard Progressive Matrices And Vocabulary Scales, (San Antonio, TX, USA: Pearson, 2000).
Karanth, P. Analysis of acquired disorders of reading in Kannada. JAAISH XV, 65–75 (1984).
Jayabai, K. “Standardization of Kannada Oral Reading Test For Primary School”. An Unpublished MSc. Thesis, (University of Mysore: Mysore, India, 1958).
Rao, C. Reading and Writing in Kannada and English orthographies: A Psycholinguistic Investigation of Processes, Problems and Remedial Measure. Doctoral Thesis, (University of Mysore: Mysore, India, 2004).
Padakannaya, P. Battery of phonological tests. Department of Psychology, (University of Mysore: Mysore, India, 1999).
Denckla, M. B. & Rudel, R. Rapid "Automatized" naming of pictures, objects, colors, and letters, and numbers by normal children. Cortex 10, 186–202 (1974).
Acknowledgements
We thank the funding agency the Department of Science and Technology-Health Science (SR/SO/HS-103/2007), Government of India, New Delhi; the subjects and their families for participating in this study; Prof. H A Ranganath for his help and encouragement; Anand S and Shyamala KV for their help in sample collection; and Mr Surendranath Nishanimutt of Samveda Research and Training Institute, Davangere for providing samples; services of Vimta Labs Ltd., and also the University of Mysore for providing facility to conduct this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Veerappa, A., Saldanha, M., Padakannaya, P. et al. Family-based genome-wide copy number scan identifies five new genes of dyslexia involved in dendritic spinal plasticity. J Hum Genet 58, 539–547 (2013). https://doi.org/10.1038/jhg.2013.47
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2013.47
Keywords
This article is cited by
-
A systematic review of associations between genetic polymorphism and dyslexia in the Indian population
Journal of Biosciences (2022)
-
Neural cell adhesion molecule Negr1 deficiency in mouse results in structural brain endophenotypes and behavioral deviations related to psychiatric disorders
Scientific Reports (2019)
-
In silico approach to identify non-synonymous SNPs with highest predicted deleterious effect on protein function in human obesity related gene, neuronal growth regulator 1 (NEGR1)
3 Biotech (2018)
-
Neurogenetics of developmental dyslexia: from genes to behavior through brain neuroimaging and cognitive and sensorial mechanisms
Translational Psychiatry (2017)
-
Investigating the effects of copy number variants on reading and language performance
Journal of Neurodevelopmental Disorders (2016)

{kind=link}
{kind=link}