
Abstract
Imputation of genome-wide single-nucleotide polymorphism (SNP) arrays to a larger known reference panel of SNPs has become a standard and an essential part of genome-wide association studies. However, little is known about the behavior of imputation in African Americans with respect to the different imputation algorithms, the reference population(s) and the reference SNP panels used. Genome-wide SNP data (Affymetrix 6.0) from 3207 African American samples in the Atherosclerosis Risk in Communities Study (ARIC) was used to systematically evaluate imputation quality and yield. Imputation was performed with the imputation algorithms MACH, IMPUTE and BEAGLE using several combinations of three reference panels of HapMap III (ASW, YRI and CEU) and 1000 Genomes Project (pilot 1 YRI June 2010 release, EUR and AFR August 2010 and June 2011 releases) panels with SNP data on chromosomes 18, 20 and 22. About 10% of the directly genotyped SNPs from each chromosome were masked, and SNPs common between the reference panels were used for evaluating the imputation quality using two statistical metrics—concordance accuracy and Cohen’s kappa (κ) coefficient. The dependencies of these metrics on the minor allele frequencies (MAF) and specific genotype categories (minor allele homozygotes, heterozygotes and major allele homozygotes) were thoroughly investigated to determine the best panel and method for imputation in African Americans. In addition, the power to detect imputed SNPs associated with simulated phenotypes was studied using the mean genotype of each masked SNP in the imputed data. Our results indicate that the genotype concordances after stratification into each genotype category and Cohen’s κ coefficient are considerably better equipped to differentiate imputation performance compared with the traditionally used total concordance statistic, and both statistics improved with increasing MAF irrespective of the imputation method. We also find that both MACH and IMPUTE performed equally well and consistently better than BEAGLE irrespective of the reference panel used. Of the various combinations of reference panels, for both HapMap III and 1000 Genomes Project reference panels, the multi-ethnic panels had better imputation accuracy than those containing only single ethnic samples. The most recent 1000 Genomes Project release June 2011 had substantially higher number of imputed SNPs than HapMap III and performed as well or better than the best combined HapMap III reference panels and previous releases of the 1000 Genomes Project.
Similar content being viewed by others
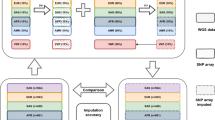

Introduction
Genome-wide association studies (GWAS) have identified numerous loci associated with complex traits.1 Imputation of ungenotyped markers has now become an essential part of GWAS and relies upon the knowledge of linkage disequilibrium from haplotypes in a known reference panel (for example, HapMap2, 3 and the 1000 Genomes Project4) to predict genotypes at the missing or untyped markers. Imputation allows one to accurately evaluate the evidence for association at genetic markers that are not directly genotyped (in addition to the typed markers) and has been shown to improve the power of GWAS to detect disease-associated loci5, 6 and, perhaps more importantly, also facilitates combining studies genotyped on different platforms. Although >15 million common and rare genetic variants exist in the human genome,4 the current (Affymetrix, Santa Clara, CA, USA) and Illumina (San Diego, CA, USA) microarrays only assay up to 2.5 million single-nucleotide polymorphisms (SNPs). Therefore, there is a strong interest in identifying methods to impute high quality genotype data using existing genotypes and reference panels from publicly available resources, such as the International HapMap Project and the 1000 Genomes Project.
Considerable progress has been made in imputing genotype data with high imputation accuracy and reliability in samples of European descent7, 8 and the improvements in imputation of Western Europeans provided by increasing the size of reference panels by combining HapMap III reference panels was assessed in Jostins et al.9
However, progress has been limited in African American samples even though the study of African population structure can be critical for genome-wide association mapping of complex disease susceptibility and pharmacogenetic responses.10, 11, 12, 13 This difficulty is likely due to the complex evolutionary history of the population, high levels of mixed ancestry and admixture, short linkage disequilibrium blocks,14, 15 the lack of population specific genotyping microarray systems and the limited availability of large study populations for evaluation. A previous study that examined imputation quality in African American populations16 used reference panels from HapMap Phase II that are limiting and no longer routinely used and suggested that the best imputation performance was observed using the imputation algorithm MACH separately for the CEU and YRI reference panels, and combining the results. However, their results may well be specific to the Phase II HapMap data. Dependence of statistical power of expression quantitative trait loci discovery on imputation quality was investigated for both Caucasians and African Americans in another study.6 Also, the increasing availability of high-throughput sequencing technologies and 1000 Genomes data has enabled genome-wide association analysis to discover disease-associated rare variants that requires availability of high quality imputed data for analysis of SNPs with low minor allele frequencies (MAF).17, 18
Therefore, given the recent release of data from the 1000 Genomes Project, we set out to (1) systematically investigate the imputation performance of MACH,19 IMPUTE20 and BEAGLE21, 22 (three widely used imputation algorithms based on hidden Markov models) in African Americans using various combinations of reference haplotypes from HapMap Phase III and the 1000 Genomes Project reference panels; (2) determine the optimal reference populations for imputation in African Americans given the current available reference datasets; and (3) explore different imputation quality metrics, specifically on their informativeness for SNPs with low allele frequencies.
Through our experiments we demonstrate that application of our imputation performance metrics stratified by allele frequencies and genotype categories (homozygotes and heterozygotes) can better distinguish imputation performance across various reference panels and imputation methods (particularly for SNPs with low MAF) compared with the traditional metrics used in Hao et al. and Shriner et al.6, 16 We show that the commonly used imputation quality scores are functions of allele frequency and caution should be exercised in using a predetermined and fixed cutoffs for the quality scores for retaining the well-imputed SNPs across all allele frequency ranges for subsequent association analysis, as many studies routinely do. Moreover, we show that inclusion of additional non-African populations in reference panels for imputation results in improved imputation performance compared with purely African reference panels in both HapMap and 1000 Genome datasets. Our results also present performance benchmarks for imputation accuracy in African Americans with various reference panels and imputation algorithms that can provide valuable guidelines to the research community regarding choice of appropriate reference panels for imputation and association analysis.
Materials and methods
Ethics statement
This research involves only the study of existing data with information recorded in such a manner that the subjects cannot be identified directly or through identifiers linked to the subjects.
Study sample
African American Atherosclerosis Risk in Communities Study (ARIC) data consists of 3207 individuals and 839 048 SNPs. The raw data were cleaned using PLINK23 with filters retaining SNPs having MAF⩾0.01, <5% missing data and Hardy–Weinberg equilibrium P-value ⩾0.00001. Further, population stratification was evaluated in two ways. (1) For each individual, the average allele sharing identity-by-state (IBS) with the rest of the study samples was calculated and outliers were identified visually using a density plot. (2) The Eigenstrat software24 was run for 10 iterations and individuals who were >8 s.d. away from the top 10 principal components were marked as genetic outliers. A total of 50 unique individuals were identified as genetic outliers and subsequently removed from the study sample to generate a cleaned data set that was used for imputation experiments. Eigenstrat was also used to visually study the clustering of the several reference populations we have used for imputation in relation to the African Americans from the ARIC cohort. To achieve this, we have combined the study population with the genotypes from the ASW, CEU, YRI, EUR and AFR panels and computed the principal components of the combined data set (Supplementary Figure 12). The shifting of the ARIC African American study samples toward the Europeans indicate European ancestry and population admixture.
Reference panels
We used both HapMap Phase III and 1000 Genomes Project reference samples for our studies. For HapMap Phase III, the panels ASW, CEU and YRI were downloaded from http://hapmap.ncbi.nlm.nih.gov/downloads/phasing/2009–02_phaseIII/HapMap3_r2/ and formatted for MACH, IMPUTE and BEAGLE. The August 2010 release of the 1000 Genomes Project panels of EUR consisted of 90 CEU+92 TSI+43 GBR+36 FIN+17 MXL+5 PUR, and the AFR panel consisted of 78 YRI+67 LWK+24 ASW+5 PUR. The 1000 Genomes Phase I interim release (June 2011) consisted of 1094 samples—381 EUR (87 CEU+93 FIN+89 GBR+14 IBS+98 TSI) and 246 AFR (61+ ASW+97 LWK+88 YRI) and was used with IMPUTE. The 1000 Genome project panels (except June 2011 release) were downloaded from the MACH website (http://www.sph.umich.edu/csg/abecasis/MACH/download/1000G-2010-08.html) and formatted for IMPUTE and BEAGLE, the June 2011 release was downloaded from the IMPUTE website (http://mathgen.stats.ox.ac.uk/impute/data_download_1000G_phase1_interim.html). The different combinations of reference panels used for imputation with each algorithm were prepared by merging the reference haplotypes. These panels are presented in Table 1. The ARIC SNP coordinates are converted from b36 to b37 before imputation using August 2010 and June 2011 releases.
Imputation software
MACH
A two-step imputation procedure was followed with MACH, in which a set of seed samples were used to estimate the genotyping error and recombination rates for every SNP, and those rates were applied to the overall study sample in step two. For each chromosome, the HapMap III reference files are split into chunks spanning 20 Mb with an overlap of 250 Kb between the adjacent chunks. This resulted in 4, 4 and 3 chunks for chromosomes 18, 20 and 22, respectively. For 1000 Genomes Project reference panels, each panel is split into 5-Mb chunks with an overlap of 250 Kb between the adjacent chunks. This resulted in 15, 13 and 7 chunks for chromosomes 18, 20 and 22, respectively. In the first step, a seed set of 451 individuals are obtained by sampling every seventh individual from the study sample. For each of the three chromosomes, Mach was run as :./mach1 -d <mapfile> -p <seed genotype file> –snps <reference panel SNPs> –haps <reference panel haplotypes> –rounds 100 –greedy –autoFlip –prefix outfile. In the second step, the parameters estimated in step one were used to do the actual imputation :./mach1 -d <mapfile> –p <genotype file> −snps <reference panel SNPs> –haps <reference panel haplotypes> –crossoverMap <rec file from step one> –errorMap <erate file from step one> –greedy –mle –mledetails –autoFlip –prefix <output file>.
BEAGLE
BEAGLE version 3.2 was downloaded from http://faculty.washington.edu/browning/beagle/beagle.html. The reference haplotypes from the 1000 Genomes Project reference panels were obtained by splitting each chromosome into 5-Mb chunks with overlap of 250 Kb between adjacent chunks, which resulted in 15, 13 and 7 chunks for chromosomes 18, 20 and 22, respectively. For each chromosome, it was runs as: java –jar beagle.jar unphased=<genotype file> phased=<ref panel haplotypes> markers=<reference panel SNPs>missing=0 out=<output file>.
IMPUTE
Imputation with IMPUTE version 2 (v2.1.2) (released 1 October 2010; http://mathgen.stats.ox.ac.uk/impute/impute_v2.html/) involved a two step procedure for each chromosome—phasing to generate the haplotypes followed by imputation. For both the stages, IMPUTE was run using parameters k=80, iter=30, burnin=10, ne=15 000, where k indicates the maximum number of copying states to be used for diploid phasing updates, iter gives the total number of MCMC iterations to perform, burnin indicates the number of Markov chain iterations to be discarded during the initial burn-in iterations and ne is the effective population size. In addition, each chromosome was split into chunk sizes of 5 Mb with overlap of 250 Kb between adjacent chunks (using input flags –int and –buffer). As a result, 15, 12 and 7 chunks are obtained for chromosomes 18, 20 and 22, respectively, for HapMap panels, and 15, 13 and 7 chunks are obtained for chromosomes 18, 20 and 22, respectively, for panels containing 1000 Genomes reference panels.
Imputation performance
For each of the three chromosomes (18, 20 and 22), randomly chosen 10% of all the SNPs in the study sample were masked by setting and their genotypes to 0 (untyped or missing). The total number of SNPs and count of masked SNPs common across the HapMap and 1000 Genomes Project panels binned by MAF for each chromosome is shown in Table 2. MACH and BEAGLE output the maximum likelihood genotypes using the posterior probability distributions of the imputed genotypes at each SNP position. Using IMPUTE, we obtained the most likely genotypes from the posterior distribution of the imputed genotypes.
Each of the imputation methods generated different statistical measures to ascertain imputation quality for each SNP –  , R2 and Info for MACH, BEAGLE and IMPUTE, respectively. The MACH
, R2 and Info for MACH, BEAGLE and IMPUTE, respectively. The MACH  estimates the correlation between the true allele counts and estimated allele counts from the imputed genotypes. This is evaluated by comparing the variance of the estimated genotype dosages with what would be expected if the dosages were observed without error.5 It is given by
estimates the correlation between the true allele counts and estimated allele counts from the imputed genotypes. This is evaluated by comparing the variance of the estimated genotype dosages with what would be expected if the dosages were observed without error.5 It is given by  , where g denotes SNP dosage and p denotes the estimated frequency of an allele. The quality measures for BEAGLE and IMPUTE are given in details in the review25 (Supplementary S3). We repeat them here for completeness. The BEAGLE imputation quality measure R2 estimates the squared correlation between the best guess genotype and the true allele dosage. For each SNP, it is computed as
, where g denotes SNP dosage and p denotes the estimated frequency of an allele. The quality measures for BEAGLE and IMPUTE are given in details in the review25 (Supplementary S3). We repeat them here for completeness. The BEAGLE imputation quality measure R2 estimates the squared correlation between the best guess genotype and the true allele dosage. For each SNP, it is computed as  . The Info statistic output by IMPUTE is based on measuring the relative statistical information about the population allele frequency θ and is given by
. The Info statistic output by IMPUTE is based on measuring the relative statistical information about the population allele frequency θ and is given by  for
for  , 1 otherwise. Here gi, ei and
, 1 otherwise. Here gi, ei and  denote the observed dosage for a SNP at sample i, the expected allele dosage for a SNP at sample i and sample allele frequency, respectively. If pik denotes the probability that the genotype of the ith sample is k (∈{0,1,2}), fi is given by fi=pi1+4pi2.
denote the observed dosage for a SNP at sample i, the expected allele dosage for a SNP at sample i and sample allele frequency, respectively. If pik denotes the probability that the genotype of the ith sample is k (∈{0,1,2}), fi is given by fi=pi1+4pi2.
For each imputation method and reference panel, imputation performance was measured using the following statistical measures:
-
1)
Concordance accuracy (CA): Degree of concordance between the observed genotypes from the study sample and the imputed genotypes at each masked SNP. It was computed as the proportion of matches between genotype calls from imputed data and the study sample at each masked SNP such that both genotypes are not missing. For a given masked SNP s, it is given by
 . Here, δ denotes the Kronecker delta function, gimp and gtrue denote the maximum likelihood genotypes of the imputed and true genotypes for SNP s, j indexes the genotypes of the individuals and N denotes the count of non-missing genotype calls from imputation for this SNP. Unlike the quality score statistics mentioned above that are measures of correlation, concordance is a more stringent statistic as it is a measure of agreement between the observed and imputed genotypes.
. Here, δ denotes the Kronecker delta function, gimp and gtrue denote the maximum likelihood genotypes of the imputed and true genotypes for SNP s, j indexes the genotypes of the individuals and N denotes the count of non-missing genotype calls from imputation for this SNP. Unlike the quality score statistics mentioned above that are measures of correlation, concordance is a more stringent statistic as it is a measure of agreement between the observed and imputed genotypes. -
2)
Kappa (κ): The κ coefficient26 adjusts the agreement between the original and masked genotypes for the amount of agreement that could be expected under the null hypothesis of independence. For each masked SNP s, it was calculated using the observed and expected frequencies from a square contingency table T as follows. Let SNP s be biallelic with alleles A and a and N be the count of non-missing genotype calls from imputation for this SNP. Then T is a contingency table where T(ij) denotes the frequency of a particular genotype combination of the true and imputed genotypes (
 ) at SNP s. Then the observed proportional agreement between the true and imputed genotypes is given by
) at SNP s. Then the observed proportional agreement between the true and imputed genotypes is given by  , and the expected agreement by chance is given by
, and the expected agreement by chance is given by  , where T(g+) and T(+g) are the totals for the gth row and the gth column, respectively, in T. Using these, the κ is calculated as
, where T(g+) and T(+g) are the totals for the gth row and the gth column, respectively, in T. Using these, the κ is calculated as  .
. -
3)
Power: A quantitative phenotype was simulated for each of the masked SNPs with fraction of variance explained ranging from 0.003 to 0.007 in steps of 0.001 using the genotypes from the study sample. Following masking and imputation for each masked SNP, we regressed the corresponding simulated phenotype on the imputed genotypes of each masked SNP. The statistical power was computed as the fraction of SNPs detected as significant out of the masked SNPs at P-value <0.001. This allowed us to quantitatively evaluate how the imputation performance affect the ability to detect SNPs significantly associated with a given trait.
 . Here, δ denotes the Kronecker delta function, gimp and gtrue denote the maximum likelihood genotypes of the imputed and true genotypes for SNP s, j indexes the genotypes of the individuals and N denotes the count of non-missing genotype calls from imputation for this SNP. Unlike the quality score statistics mentioned above that are measures of correlation, concordance is a more stringent statistic as it is a measure of agreement between the observed and imputed genotypes.
. Here, δ denotes the Kronecker delta function, gimp and gtrue denote the maximum likelihood genotypes of the imputed and true genotypes for SNP s, j indexes the genotypes of the individuals and N denotes the count of non-missing genotype calls from imputation for this SNP. Unlike the quality score statistics mentioned above that are measures of correlation, concordance is a more stringent statistic as it is a measure of agreement between the observed and imputed genotypes. ) at SNP s. Then the observed proportional agreement between the true and imputed genotypes is given by
) at SNP s. Then the observed proportional agreement between the true and imputed genotypes is given by  , and the expected agreement by chance is given by
, and the expected agreement by chance is given by  , where T(g+) and T(+g) are the totals for the gth row and the gth column, respectively, in T. Using these, the κ is calculated as
, where T(g+) and T(+g) are the totals for the gth row and the gth column, respectively, in T. Using these, the κ is calculated as  .
.Intuitively, the SNPs with low allele frequencies were more difficult to impute accurately compared with those with both alleles common. Also at low MAF, the minor allele homozygotes and the heterozygotes were rarer and more prone to prediction errors. We, therefore, computed and reported the above three performance measures for each of the three genotypes at the masked SNPs after binning by four allele frequency ranges: ⩽0.05, 0.05–0.1, 0.1–0.3 and >0.3 (Table 2). Furthermore we computed the distributions for each statistic as the fraction of the masked SNP that have the value of the statistic exceeding a range of cutoffs: 0.0–0.9 in steps of 0.1. These plotted at each allele frequency bin enabled us to visualize the distribution of the imputation quality scores and comprehensively evaluate the imputation methods and the reference panels for the African American study sample.
Results
For each of the three methods MACH, IMPUTE and BEAGLE, we have used the following statistical measures of imputation performance: CA, κ coefficient and power of detecting imputed SNPs significantly associated with simulated phenotypes. The details of each imputation performance metric are given in the Materials and methods section. Previous studies on imputation of African Americans took into account the admixture of European and African genetic components in African American subjects.6, 16, 27 Therefore, we used the following reference panels for our imputation experiments: (1) ASW, CEU and YRI data sets from HapMap III; (2) the pilot 1 YRI data June 2010 release; (3) EUR and AFR data from the August 2010 release; and (4) EUR and AFR data from the June 2011 release of the 1000 Genomes Project. The various combinations of these reference panels investigated in this study are presented in Table 1. As only IMPUTE allows more than one reference panel as input, it was also run with the combined reference panel of YRI samples from the June 2010 release of the 1000 Genomes Project and all HapMap III panels (denoted 1000 G(YRI)+All HapMap III). Also, only IMPUTE was used for imputation on chromosome 18 using the panel from June 2011 release of the 1000 Genomes Project.
For each imputation method and reference panel combination, 10% of the SNPs on chromosomes 18, 20 and 22 in the study sample were set to missing (Table 2) and imputation performance was evaluated following a round of imputation with the masked data and using only the SNPs common across the HapMap and 1000 Genomes Project panels. Henceforth we use the phrase masked SNPs to simply refer to this set of SNPs, unless otherwise mentioned.
In this section, we first present the imputation results for the three methods individually, followed by a comparison using the best common reference panel for each method. To determine the extent to which imputation performance metrics are sensitive to the allele frequencies of the SNPs being imputed, we explored the performance results for the three chromosomes after grouping the masked SNPs into four MAF bins ⩽0.05, 0.05–0.1, 0.1–0.3 and >0.3 for each of the three methods. The imputation performance of the SNPs in the bin MAF ⩽0.05 being most sensitive to allele frequency, we focus on the results for this bin for each method. The results for the other MAF bins are presented in the Supplementary Data.
For each of the three imputation methods, we determined the distribution of the CA for the minor allele homozygotes (denoted CA(aa)), heterozygotes (denoted CA(Aa)) and the major allele homozygotes (denoted CA(AA)) for masked SNPs with MAF ⩽0.05. The distribution of a statistic (CA or κ) was plotted using the fraction of the masked SNP that had the value of the statistic exceeding a range of cutoffs: 0.0–0.9 in steps of 0.1. Thus, cutoff=0.0 for CA(aa) resulted in the most lenient cutoff including all masked SNPs with MAF ⩽0.05 whereas 0.9 resulted in the most stringent cutoff retaining only imputed masked SNPs of highest quality with MAF ⩽0.05 and CA(aa) ⩾0.9. The imputation performance as obtained using the κ combined the agreements across the three genotypes to provide an overall score of imputation concordance. It measured the concordances between the original and masked genotypes and adjusted these values for the amount of agreement that could be expected due to chance alone. In addition to CA for each genotype category, we computed the total CA (denoted CA(Tot)) by simply summing the concordances of the three genotypes as used in previous studies.6, 16 Visualization of the distribution of the concordances and κ demonstrated the trade-off between imputation quality and yield—higher imputation quality threshold resulted in lower yield, that is, fewer SNPs retained after imputation.
Also the ultimate goal of imputation is to boost the power of GWAS by predicting genotypes at markers that are not directly genotyped. Therefore, it is essential to evaluate how imputation quality as measured using CA and κ affected the the ability to detect SNPs associated with a given trait, that is, reference panels achieving higher CA and κ should also attain higher power and vice versa. We therefore computed the statistical power by simulating phenotypes for each masked SNP and regressing the corresponding simulated phenotype on the imputed genotypes (see Materials and methods for details).
Across each imputation method and reference panel used (described in details below), we observed the following:
-
1)
For a given method and a reference panel, the concordances increased in the order: minor allele homozygotes, heterozygotes and major allele homozygotes. For masked SNPs with low MAF, CA(aa) was most sensitive to the choice of the reference panel and effectively distinguished the imputation performance of each reference panel followed by CA(Aa) (Figure 1). At the same time, owing to the presence of a vast majority of major allele homozygotes, CA(AA) and CA(Tot) for the various reference panels differed only slightly from one another (Supplementary Figure 1). This indicates the advantage of estimating the imputation performance for each genotype category separately with focus on the genotypes containing the minor allele, as the performance differences were more visible for minor allele homozygotes and heterozygotes, specially at low MAF. Little or almost no comparative information was provided by the CA(AA) and CA(Tot). Instead, κ is a more informative overall measure (Figure 2a).
Figure 1 
Distribution of concordance accuracy (CA) of minor allele homozygotes and heterozygotes of (a, b) MACH, (c, d) IMPUTE and (e, f) for BEAGLE. (M=MACH, I=IMPUTE and B=BEAGLE).
Figure 2 
Distribution of kappa for (a) MACH, (c) IMPUTE and (e) BEAGLE. Power is shown in (b) for MACH, (d) for IMPUTE and (f) for BEAGLE. (M=MACH, I=IMPUTE and B=BEAGLE).
-
2)
For MACH and BEAGLE, the combined HapMap III reference panel ASW+CEU+YRI III performed better than, or at least as well as, the other HapMap panels whereas the combined 1000 Genomes Project panel EUR+AFR was better than AFR alone. For IMPUTE, EUR+AFR-2011 performs better than or as well as the other panels.
-
3)
The proportion of masked SNPs retained at each cutoff for an imputation quality statistic (that is, the imputation yield) decreased with increasing value of the cutoff (for example, see Figure 3 for the κ statistic). This demonstrates the critical choice a researcher has to face—the cutoff to choose for balancing imputation quality with imputation yield.
Figure 3 
Kappa vs yield for the three algorithms with ASW+CEU+YRI III for minor allele frequencies (MAF) bins (a) ⩽0.05 (b) 0.05–0.1 (c) 0.1–0.3 and (d) 0.3–0.5. (M=MACH, I=IMPUTE and B=BEAGLE).
-
4)
For each reference panel, imputation concordances as measured by CA, κ and power generally increased with increasing MAF, irrespective of the imputation method used (for example, see Figure 4 and Table 3 for reference panel ASW+CEU+YRI III).
Figure 4 
For panel ASW+CEU+YRI III, comparison of (a,b) mean concordance accuracy (CA) for minor allele homozygotes and heterozygotes and (c) mean kappa for each method at different minor allele frequencies (MAF) bins. (M=MACH, I=IMPUTE and B=BEAGLE).
Table 3 Comparison of power of each method using the panel ASW+CEU+YRI III at the four minor allele frequency bins -
5)
MACH and IMPUTE performed equally well and better than BEAGLE.




Next, we discuss the imputation results in details.
Imputation quality and yield using the concordance and κ statistics
Figure 1 shows the CA obtained with each method for the minor allele homozygotes and the heterozygotes for the masked SNPs with MAF⩽0.05 that are common across the HapMap and 1000 Genomes Project panels. Using MACH and BEAGLE, for minor allele homozygotes (Figure 1a and e) and heterozygotes (Figure 1b and f), the combined reference panels ASW+CEU+YRI III (blue) and CEU+YRI III (light green) yielded more SNPs with better CA than the other panels. For both algorithms, the panel ASW+CEU+YRI III (blue) was slightly better than CEU+YRI III (light green) followed by EUR+AFR (turquoise). With IMPUTE (Figure 1c and d), the panels EUR+AFR-2011 (yellow, on chromosome 18) and 1000 G(YRI)+All HapMap III (brown) were the best-performing panels, followed by ASW+CEU+YRI III (blue), CEU+YRI III (light green) and EUR+AFR (turquoise). We found that at MAF ⩽0.05, the minor allele concordance CA(aa) was slightly improved for EUR+AFR-2011 compared with panels containing HapMap haplotypes, and the increase in CA(aa) was greater compared with EUR+AFR. For panel EUR+AFR-2011, 54% of the masked SNPs had CA(aa) >0.8 compared with 52, 53 and 48% achieved by ASW+CEU+YRI III, 1000 G(YRI)+All HapMap III and EUR+AFR, respectively. For all methods, the purely African panels of ASW III (magenta), YRI III (gray) and AFR (dark green) had poor CA(aa) and CA(Aa).
Supplementary Figure 1 shows the distribution of the CA for the major allele homozygotes and that of the total CA, respectively, which do not provide substantial information to distinguish the performance of the reference panels. Only with BEAGLE, CA(AA) and CA(Tot) were able to distinguish the AFR and YRI III panels from the others as BEAGLE discarded 20–25% of the masked SNPs during imputation when used with the AFR and YRI III panels.
The distribution of the overall agreement across the three genotypes as measured by the κ statistic is shown for MACH, IMPUTE and BEAGLE in Figure 2a, respectively. For the methods MACH and BEAGLE, the combined reference panel ASW+CEU+YRI III (blue) outperformed the others whereas the panels ASW III (magenta), YRI III (gray) and AFR (dark green) performed the worst with all three methods. For IMPUTE, the distribution of κ for the panels EUR+AFR-2011 (yellow) and 1000 G(YRI)+All HapMap III panels (brown) closely followed that of ASW+CEU+YRI III (blue). These were in agreement with what we had observed with CA(aa) and CA(Aa). Imputation accuracies with the 1000 G(YRI)+All HapMap III panel also show that the presence of reference populations unrelated to African American ancestry does not adversely affect imputation performance.
Visualization of the distribution of the concordances and κ statistics also highlights the fact that increasing the threshold for each of these metrics would increase the quality of imputation (as measured by κ and CA), but at the same time, would reduce the imputation or yield. Using MACH with the panel ASW+CEU+YRI III (blue), 67% of the masked SNPs (MAF ⩽0.05) had κ statistic >0.8, but dropped to 58% with BEAGLE (Figure 2a and e). Using IMPUTE, at a κ⩾0.8, 67, 66, 70 and 55% of the masked SNPs are retained using EUR+AFR-2011 (yellow), ASW+CEU+YRI III (blue), 1000 G(YRI)+All HapMap III (brown) and EUR+AFR (turquoise), respectively (Figure 2c). These show that the imputation accuracies of EUR+AFR-2011 are improved over EUR+AFR and comparable to that of ASW+CEU+YRI III and 1000 G(YRI)+All HapMap III. Both the 1000 Genomes Project panels EUR+AFR (turquoise) and AFR (dark green) had lower yield in comparison with ASW+CEU+YRI III (blue) indicating poorer quality of imputation with these panels.
Figure 2b show the power of detecting the masked SNPs with simulated phenotypes for each algorithm computed using allele dosage. The performance of the panels follow the same trend as the CA(aa), CA(Aa) and κ statistics, that is, ASW+CEU+YRI III (blue), CEU+YRI III (light green), 1000 G(YRI)+All HapMap III (brown, only for IMPUTE) and EUR+AFR-2011 (yellow, with IMPUTE on chromosome 18) had higher power than the other panels followed by EUR+AFR (turquoise). This demonstrates that reference panels attaining higher concordance and κ computed using maximum likelihood genotypes of the masked SNPs can improve power in subsequent association analysis that uses allele dosage.
The distributions of the CA and κ and the power of each reference panel for the remaining MAF bins 0.05–0.1, 0.1–0.3 and >0.3 are presented in Supplementary Figures 2–7.
Figure 3 depicts the fraction of SNPs retained at each cutoff of the κ statistic for the three methods using the reference panel ASW+CEU+YRI III. Based on the imputation performance using the masked SNPs, we estimated that using MACH and IMPUTE with the panel ASW+CEU+YRI III, 68%, 90%, 96% and 95% of the untyped SNPs in African Americans can be imputed with imputation accuracy κ of 0.92 at the MAF bins ⩽0.05, 0.05–0.1, 0.1–0.3 and >0.3, respectively. With BEAGLE, the percentages dropped to 53%, 77%, 87% and 87% for the above MAF bins, respectively. Using IMPUTE with the combined reference panels 1000 G(YRI)+All HapMap III (Supplementary Figure 8), the yield remained extremely close to that of ASW+CEU+YRI III. This demonstrates that combining reference panels in addition to ASW, CEU and YRI is unlikely to improve the imputation accuracies significantly. Both the 1000 Genomes Project panels EUR+AFR and AFR had lower yield in comparison with ASW+CEU+YRI III due to lower imputation accuracy with these panels.
Comparison between the imputation algorithms
We compared the imputation performances of the three imputation methods using the combined HapMap III panel ASW+CEU+YRI III that achieved high concordance and power with each imputation algorithm. As described in the preceding paragraph, Figure 3 also serves to compare the three methods. Using the κ statistic, MACH (green) and IMPUTE (blue) performs similarly and both are better than BEAGLE (red). Figure 4 compares the imputation performance of the methods using the concordance statistics and κ computed with all the masked SNPs at the different MAF bins. We observed that MACH and IMPUTE consistently achieved higher CA(aa) (Figure 4a), CA(Aa) (Figure 4b) and κ (Figure 4c) than BEAGLE at all allele frequencies. Additional results using the distributions of CA(aa) and CA(Aa) are presented in Supplementary Figure 9.
Using the dosage data, a detailed comparison of the power of each algorithm at all MAF bins using panel ASW+CEU+YRI III is given in Table 3. MACH and IMPUTE performed similarly and steadily outperformed BEAGLE at all MAF bins, the difference in power being higher for masked SNPs with lower MAFs.
Our experiments also indicated that BEAGLE is computationally faster than MACH and IMPUTE whereas IMPUTE is computationally much faster than MACH. The details of the runtimes of each method can be found in Supplementary Data (Supplementary Tables 1 and 2). Although 1000 G(YRI)+All HapMap III has many more haplotypes than AFR and EUR+AFR, the HapMap III panels have fewer SNPs compared with the 1000 G(YRI) panel. At the same time, only 118 haplotypes are present in 1000 G(YRI) compared with 1914 in the combined HapMap III panel. This results in decreased runtime of IMPUTE with the 1000 G(YRI)+All HapMap III reference panel.
Relationship of imputation quality metrics with the concordance and κ statistics
The quality of imputation for each imputed SNP was measured by different statistical metrics for each method. MACH produced the imputation quality measure ( ) that estimates the squared correlation between the estimated allele dosage and true allele dosage. It represents the ratio of the empirically observed variance of allele dosage to the expected binomial variance at Hardy–Weinberg equilibrium.28 BEAGLE generated a similar imputation quality metric (R2) that estimates the squared correlation between the most likely allele dosage and the true allele dosage. The output from IMPUTE contained the Info statistic that represents a measure of the relative statistical information about SNP allele frequency.25 For each method, imputed SNPs with higher values of the corresponding statistic are assumed to be more reliably imputed. Here, we explored the effect of MAF on the imputation quality measures CA and κ when
) that estimates the squared correlation between the estimated allele dosage and true allele dosage. It represents the ratio of the empirically observed variance of allele dosage to the expected binomial variance at Hardy–Weinberg equilibrium.28 BEAGLE generated a similar imputation quality metric (R2) that estimates the squared correlation between the most likely allele dosage and the true allele dosage. The output from IMPUTE contained the Info statistic that represents a measure of the relative statistical information about SNP allele frequency.25 For each method, imputed SNPs with higher values of the corresponding statistic are assumed to be more reliably imputed. Here, we explored the effect of MAF on the imputation quality measures CA and κ when  was used to stratify the SNPs. We show the results only for MACH, as the results of IMPUTE and BEAGLE are similar and presented in the Supplementary Data (Supplementary Figures 10 and 11). Using the panel ASW+CEU+YRI III, Figure 5a–d show how CA and κ are affected by allele frequencies when masked SNPs with
was used to stratify the SNPs. We show the results only for MACH, as the results of IMPUTE and BEAGLE are similar and presented in the Supplementary Data (Supplementary Figures 10 and 11). Using the panel ASW+CEU+YRI III, Figure 5a–d show how CA and κ are affected by allele frequencies when masked SNPs with  a cutoff value were used to compute the corresponding statistics. The four MAF bins are denoted as ⩽0.05 with red, 0.05–0.1 with blue, 0.1–0.3 with green and >0.3 with magenta.
a cutoff value were used to compute the corresponding statistics. The four MAF bins are denoted as ⩽0.05 with red, 0.05–0.1 with blue, 0.1–0.3 with green and >0.3 with magenta.

With MACH (a–c) mean concordance accuracy (CA) for each genotype and (d) mean kappa using masked single-nucleotide polymorphisms (SNPs) exceeding a given  for panel ASW+CEU+YRI III. Four minor allele frequencies (MAF) bins are shown as ⩽0.05 (red), 0.05–0.1 (blue), 0.1–0.3 (green) and 0.3–0.5 (magenta).
for panel ASW+CEU+YRI III. Four minor allele frequencies (MAF) bins are shown as ⩽0.05 (red), 0.05–0.1 (blue), 0.1–0.3 (green) and 0.3–0.5 (magenta).
We observed that  is a poor indicator of imputation quality for SNPs with low MAF. Figure 5a shows the CA(aa) against
is a poor indicator of imputation quality for SNPs with low MAF. Figure 5a shows the CA(aa) against  at each MAF bin. The differences in imputation quality score CA(aa) were more pronounced at lower values of
at each MAF bin. The differences in imputation quality score CA(aa) were more pronounced at lower values of  and gradually converged as
and gradually converged as  increases. The CA(Aa) and CA(AA) vs
increases. The CA(Aa) and CA(AA) vs  are shown in Figure 5b and c, respectively. We observed that the differences in concordance at lower values of
are shown in Figure 5b and c, respectively. We observed that the differences in concordance at lower values of  decreased in the order CA(aa), CA(Aa) and CA(AA), suggesting that imputation accuracy for similar
decreased in the order CA(aa), CA(Aa) and CA(AA), suggesting that imputation accuracy for similar  scores was more affected by allele frequencies in minor allele homozygotes and heterozygotes. The overall concordance score κ (Figure 5d) also highlights this fact and suggests that there is a great deal of uncertainly in imputation quality even when imputed SNPs with
scores was more affected by allele frequencies in minor allele homozygotes and heterozygotes. The overall concordance score κ (Figure 5d) also highlights this fact and suggests that there is a great deal of uncertainly in imputation quality even when imputed SNPs with  0.3 are considered for subsequent analysis as practiced in some studies.29, 30 As a result, we recommend that when maximum likelihood genotypes from imputation are considered, higher
0.3 are considered for subsequent analysis as practiced in some studies.29, 30 As a result, we recommend that when maximum likelihood genotypes from imputation are considered, higher  quality score cutoffs should be used to retain well-imputed SNPs at lower allele frequencies. Alternatively, κ can be used to ascertain imputation quality more reliably, however, a small fraction of the observed genotypes randomly chosen should be masked before imputation to facilitate computation of the κ statistic.
quality score cutoffs should be used to retain well-imputed SNPs at lower allele frequencies. Alternatively, κ can be used to ascertain imputation quality more reliably, however, a small fraction of the observed genotypes randomly chosen should be masked before imputation to facilitate computation of the κ statistic.
Discussion
Although the 1000 Genomes Project panels have far more SNPs than the HapMap panels, no studies to our knowledge have compared the imputation accuracies between the 1000 Genomes Project and HapMap panels for imputation of the African Americans. We have performed a systematic and comprehensive evaluation of the imputation performance of various combinations of reference haplotypes from the HapMap Phase III and the 1000 Genomes Projects using three popular imputation methods—MACH, IMPUTE and BEAGLE in African Americans from the ARIC study. We have used three imputation quality score metrics—CA, κ and power of association analysis to visualize and critically evaluate the performance of the reference panels and imputation methods. The first two statistics use the maximum likelihood genotypes to evaluate the accuracy of imputation, whereas power is computed using the mean genotypes (that is, allele dosage) computed from the posterior distribution of the genotypes.
In previous studies on imputation performance, the metric for comparison of imputation accuracy has been limited to using the proportion of concordance between the original and masked genotype calls across all the study samples and averaged over all the masked SNPs (that is, total concordance).6, 16 Moreover, none of the previous studies had examined the dependency of the imputation accuracy metrics on MAF and evaluated the impact of each of the three genotype classes—minor allele homozygotes, heterozygotes and major allele homozygotes, separately, on the imputation performance. In our study, we have shown that this naive metric is not suitable to adequately differentiate the imputation accuracies between different reference panels and algorithms. Instead, we have computed the concordances for each class of genotypes separately. The overall concordance computed using the κ statistic is found to be far more informative than total concordance. Our results demonstrate that imputation performance is affected by allele frequency, and that imputation of minor allele homozygotes and heterozygotes for SNPs having low MAF is more sensitive when compared with that of the major allele homozygotes. Therefore, imputation accuracies should be evaluated using concordance and κ metrics with minor allele homozygotes and heterozygotes of low MAF SNPs. In addition, visualization of distributions of each quality metric indicates that imputation accuracies for homozygotes and heterozygotes increase, and imputation yields decrease, with increasing imputation quality for each method and reference panel.
We also found that when the commonly used imputation quality score metrics  , R2 and Info are used to estimate imputation accuracy, for SNPs with lower quality scores, the concordances and κ are significantly affected by MAF. This suggests the use of more stringent cutoffs to the imputation quality scores to select well-imputed SNPs at lower allele frequencies for a subsequent GWAS (for example, in Takahashi et al.31), as opposed to the conventional approach of using a single cutoff to select imputed SNPs across all allele frequency ranges. However, no guidelines exist as to what cutoff values should be selected for each MAF range to attain a given imputation accuracy. Masking a small fraction of the observed genotypes before imputation and computing the concordances and κ statistics with the maximum likelihood genotypes allow one to estimate the imputation performance more reliably. Note that we are not advocating the use of maximum likelihood genotypes from imputation for association analysis. Rather, we emphasize that using maximum likelihood genotypes of masked SNPs provides the opportunity to evaluate and compare several imputation methods and reference panels critically and systematically because the best guess genotypes are expected to be very close to the (known) masked genotypes when the imputation accuracies are good.
, R2 and Info are used to estimate imputation accuracy, for SNPs with lower quality scores, the concordances and κ are significantly affected by MAF. This suggests the use of more stringent cutoffs to the imputation quality scores to select well-imputed SNPs at lower allele frequencies for a subsequent GWAS (for example, in Takahashi et al.31), as opposed to the conventional approach of using a single cutoff to select imputed SNPs across all allele frequency ranges. However, no guidelines exist as to what cutoff values should be selected for each MAF range to attain a given imputation accuracy. Masking a small fraction of the observed genotypes before imputation and computing the concordances and κ statistics with the maximum likelihood genotypes allow one to estimate the imputation performance more reliably. Note that we are not advocating the use of maximum likelihood genotypes from imputation for association analysis. Rather, we emphasize that using maximum likelihood genotypes of masked SNPs provides the opportunity to evaluate and compare several imputation methods and reference panels critically and systematically because the best guess genotypes are expected to be very close to the (known) masked genotypes when the imputation accuracies are good.
Our results indicate that imputation in African Americans is challenging in that the imputation accuracy is reduced when compared with that of the Caucasians.6, 7, 8 In the study by Hao et al.,6 the authors imputed both Caucasians and African Americans genome-wide with MACH and BEAGLE using HapMap release 22 CEU and combined CEU and YRI panels, respectively, and explored how the statistical power of expression quantitative trait loci discovery is affected by imputation quality. Imputation accuracy was evaluated using total concordance after masking randomly selected 1% of the SNPs genome-wide. With the Caucasians, they obtained a higher imputation yield of 90% of the HapMap SNPs at 98% imputation accuracy using the CEU panel, whereas for the African Americans imputation yield dropped to 75% of the HapMap SNPs at 96% accuracy using the CEU+YRI panel. Shriner et al.16 imputed genotypes in African Americans from the Washington DC metropolitan area using MACH, BEAGLE and PLINK, with both weighted and un-weighted mixture of the reference panels ASW, CEU and YRI from both HapMap II and III. They found that the weighted mixture approach yielded fewer imputed SNPs than the un-weighted mixture approach and recommended using MACH to perform two imputations, one with CEU as reference and one with YRI as reference, and then merging the results. However, this conclusion was based on their observation that using the HapMap Phase II YRI panel alone as a reference does better than the combined HapMap Phase II CEU+YRI panel for SNPs present in the YRI panel. Thus, these results may well be specific to the Phase II HapMap data. The authors also reported that imputation using MACH with the HapMap Phase III YRI reference panel marginally outperformed the HapMap Phase III ASW panel. In our analysis, across all masked SNPs and MAF bins, at a total concordance value of 0.96, 95% of the masked SNPs were imputed with CEU+YRI and it increased to 98% with the ASW+CEU+YRI III reference panel. However, neither of the above studies stratified the SNPs by MAF or evaluated imputation accuracies for minor allele homozygotes, heterozygotes and major allele homozygotes separately.
Given various reference panels and algorithms for imputation of African Americans, determining the optimal reference panels while balancing imputation accuracy and SNP yield suited to one’s experimental requirements can be an arduous and time consuming task. Therefore, our results establish performance benchmarks for imputation accuracy in African Americans that can guide the researchers in choosing the appropriate reference panels for imputation and association analysis.
The recent June 2011 release from the 1000 Genomes Project contains considerably more SNPs than the previous releases (June 2010 and August 2010) and many times more than that present in HapMap reference panels and resulted in higher imputation coverage (Supplementary Data, Tables 3 and 4). Across both HapMap III and 1000 Genomes reference panels, those consisting of multiple ethnic panels performed better than panels from single ethnic samples alone and the combined HapMap panels had better imputation accuracy compared with the June 2010 and August 2010 releases of 1000 Genomes Project. The most recent June 2011 release performed as well or better than the best combined HapMap III reference panels and previous releases of the 1000 Genomes Project. Because of better genome-wide coverage, greater reference panel size (haplotype count), and greater presumed accuracy with each incremental 1000 Genomes release, we conclude that for imputation of African Americans, users would likely benefit from using a combined multi-ethnic panel from the most current release.
References
Hindorff, L. A., Sethupathy, P., Junkins, H. A., Ramos, E. M., Mehta, J. P., Collins, F. S. et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl Acad. Sci. USA 106, 9362–9367 (2009).
Consortium, I. H., Frazer, K. A., Ballinger, D. G., Cox, D. R., Hinds, D. A., Stuve, L. L. et al. A second generation human haplotype map of over 3.1 million SNPs. Nature 449, 851–861 (2007).
Consortium, I. H., Altshuler, D. M., Gibbs, R. A., Peltonen, L., Altshuler, D. M., Gibbs, R. A. et al. Integrating common and rare genetic variation in diverse human populations. Nature 467, 52–58 (2010).
Consortium, G. P., Durbin, R. M., Abecasis, G. R., Altshuler, D. L., Auton, A., Brooks, L. D. et al. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010).
Li, Y., Willer, C., Sanna, S., Abecasis, G. Genotype imputation. Annu. Rev. Genomics Hum. Genet. 10, 387–406 (2009).
Hao, K., Chudin, E., McElwee, J., Schadt, E. E. Accuracy of genome-wide imputation of untyped markers and impacts on statistical power for association studies. BMC Genet. 10, 27 (2009).
Zhao, Z., Timofeev, N., Hartley, S. W., Chui, D. H., Fucharoen, S., Perls, T. T. et al. Imputation of missing genotypes: an empirical evaluation of IMPUTE. BMC Genet. 9, 85 (2008).
Nothnagel, M., Ellinghaus, D., Schreiber, S., Krawczak, M., Franke, A. A comprehensive evaluation of SNP genotype imputation. Hum. Genet. 125, 163–171 (2009).
Jostins, L., Morley, K. I., Barrett, J. C. Imputation of low-frequency variants using the HapMap3 benefits from large, diverse reference sets. Eur. J. Hum. Genet. 19, 662–666 (2011).
Ma, L., Marmor, M., Zhong, P., Ewane, L., Su, B., Nyambi, P. Distribution of CCR2-64I and SDF1-3’A alleles and HIV status in 7 ethnic populations of Cameroon. J. Acquir. Immune Defic. Syndr. 40, 89–95 (2005).
Williamson, C., Loubser, S. A., Brice, B., Joubert, G., Smit, T., Thomas, R. et al. Allelic frequencies of host genetic variants influencing susceptibility to HIV-1 infection and disease in South African populations. AIDS 14, 449–451 (2000).
Reich, D., Patterson, N., Jager, P. L. D., McDonald, G. J., Waliszewska, A., Tandon, A. et al. A whole-genome admixture scan finds a candidate locus for multiple sclerosis susceptibility. Nat. Genet. 37, 1113–1118 (2005).
Johnson, J. A. Ethnic differences in cardiovascular drug response: potential contribution of pharma-cogenetics. Circulation 118, 1383–1393 (2008).
Bryc, K., Auton, A., Nelson, M. R., Oksenberg, J. R., Hauser, S. L., Williams, S. et al. Genome-wide patterns of population structure and admixture in West Africans and African Americans. Proc. Natl Acad. Sci. USA 107, 786–791 (2010).
Tishkoff, S. A., Reed, F. A., Friedlaender, F. R., Ehret, C., Ranciaro, A., Froment, A. et al. The genetic structure and history of Africans and African Americans. Science 324, 1035–1044 (2009).
Shriner, D., Adeyemo, A., Chen, G., Rotimi, C. N. Practical considerations for imputation of untyped markers in admixed populations. Genet. Epidemiol. 34, 258–265 (2010).
Bansal, V., Libiger, O., Torkamani, A., Schork, N. J. Statistical analysis strategies for association studies involving rare variants. Nat. Rev. Genet. 11, 773–785 (2010).
Asimit, J., Zeggini, E. Rare variant association analysis methods for complex traits. Annu. Rev. Genet. 44, 293–308 (2010).
Li, Y., Willer, C. J., Ding, J., Scheet, P., Abecasis, G. R. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet. Epidemiol. 34, 816–834 (2010).
Howie, B. N., Donnelly, P., Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 5, e1000529 (2009).
Browning, S. R. Multilocus association mapping using variable-length Markov chains. Am. J. Hum. Genet. 78, 903–913 (2006).
Browning, S. R., Browning, B. L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 81, 1084–1097 (2007).
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A. R., Bender, D. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Price, A. L., Patterson, N. J., Plenge, R. M., Weinblatt, M. E., Shadick, N. A., Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006).
Marchini, J., Howie, B. Genotype imputation for genome-wide association studies. Nat. Rev. Genet. 11, 499–511 (2010).
Smeeton, N. C. Early history of the kappa statistic. Biometrics 41, 795 (1985).
Egyud, M. R. L., Gajdos, Z. K. Z., Butler, J. L., Tischfield, S., Marchand, L. L., Kolonel, L. N. et al. Use of weighted reference panels based on empirical estimates of ancestry for capturing untyped variation. Hum. Genet. 125, 295–303 (2009).
de Bakker, P. I. W., Ferreira, M. A. R., Jia, X., Neale, B. M., Raychaudhuri, S., Voight, B. F. Practical as-pects of imputation-driven meta-analysis of genome-wide association studies. Hum. Mol. Genet. 17, R122–R128 (2008).
Zeggini, E., Scott, L. J., Saxena, R., Voight, B. F., Marchini, J. L., Hu, T. et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat. Genet. 40, 638–645 (2008).
Lettre, G., Jackson, A. U., Gieger, C., Schumacher, F. R., Berndt, S. I., Sanna, S. et al. Identification of ten loci associated with height highlights new biological pathways in human growth. Nat. Genet. 40, 584–591 (2008).
Takahashi, Y., Kou, I., Takahashi, A., Johnson, T. A., Kono, K., Kawakami, N. et al. A genome-wide associa-tion study identifies common variants near LBX1 associated with adolescent idiopathic scoliosis. Nat. Genet. 43, 1237–1240 (2011).
Acknowledgements
The Atherosclerosis Risk in Communities Study is carried out as a collaborative study supported by National Heart, Lung, and Blood Institute contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C, and HHSN268201100012C), R01HL087641, R01HL59367 and R01HL086694; National Human Genome Research Institute contract U01HG004402; and National Institutes of Health contract HHSN268200625226C. The authors thank the staff and participants of the ARIC study for their important contributions. Infrastructure was partly supported by Grant Number UL1RR025005, a component of the National Institutes of Health and NIH Roadmap for Medical Research.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Chanda, P., Yuhki, N., Li, M. et al. Comprehensive evaluation of imputation performance in African Americans. J Hum Genet 57, 411–421 (2012). https://doi.org/10.1038/jhg.2012.43
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2012.43
Keywords
This article is cited by
-
A joint use of pooling and imputation for genotyping SNPs
BMC Bioinformatics (2022)
