
Abstract
Southeast Asian ovalocytosis (SAO) is an erythrocyte abnormality that protects affected individuals from cerebral malaria. This trait is caused by a 27-bp deletion in the SLC4A1 gene, which is lethal when homozygous. We reseqeunced approximately 5 kb of SLC4A1 in an Indonesian population where SAO is prevalent to better understand the evolution of this clinically important trait. The four SAO chromosomes we resequenced share a single haplotype that differs from a sampled non-SAO haplotype only by the 27-bp deletion. Comparison of Indonesian sequence data to that from two other Asian populations (aboriginal Taiwanese and Japanese) shows Indonesian SLC4A1 to be strongly differentiated from the Taiwanese, but not the Japanese. Indeed, the Taiwanese sample contains only chromosomes that are highly divergent from all sampled SAO chromosomes. Because earlier studies have found an association between Austronesian-speakers (who most likely originated in Taiwan) and SAO, our failure to find SAO-like chromosomes in Taiwan is unexpected. Finally, our data find a strong excess of high-frequency derived alleles in all three populations. These alleles include the non-synonymous ‘Memphis’ variant, which is known to affect anion transport across the erythrocyte membrane. Our data suggest a role for recent natural selection acting on Memphis or a linked variant.
Similar content being viewed by others

Introduction
Southeast Asian ovalocytosis (SAO) is a hereditary condition associated with oval-shaped erythrocytes. The trait results from a 9-amino acid (27-bp) deletion in the SLC4A1 gene, which encodes Band 3, a major transmembrane protein of the red blood cell.1 As its name implies, SAO is found mainly in Southeast Asia and the Southwest Pacific; the trait is rare in most localities and reaches maximum frequency of 0.175 in coastal regions of Papua New Guinea.1, 2, 3, 4 SAO is of interest because there is strong evidence that the condition confers a protective effect with respect to cerebral malaria and malaria-related mortality caused by Plasmodium falciparum.5, 6 This protection most likely is mediated by alteration of the cytoadherence properties of Plasmodium-infected SAO erythrocytes relative to wild-type red blood cells.7 As such, it is probable that SAO is maintained in malarial populations by natural selection. Interestingly, the trait appears to act as a balanced polymorphism, with its frequency limited by complete lethality of the homozygous state.8
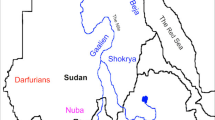
As SAO is a probable target of natural selection, it is important to consider the evolutionary origins and history of the allele. Although no studies to date have examined the molecular population genetics of the locus, the geographical distribution of SAO provides some clues regarding its origins. SAO is limited mainly to populations that have ancestry among speakers of Austronesian languages, including populations throughout the islands of Southeast Asia, coastal regions of Papua New Guinea and even Madagascar in the western Indian Ocean.2, 3, 4, 9 This correlation is also evident at local scales. For instance, among eight populations on the north coast of Papua New Guinea the frequency of the 27-bp deletion that causes SAO has been observed to positively covary with a mitochondrial marker associated with the Austronesian expansion.10 These data suggest that the present distribution of SAO may be shaped strongly by the spread of Austronesian-speaking people in the region beginning some 3500 years BP (see Figure 1).

Map of study area. Numbered dots indicate populations sampling sites (1) Japanese in Tokyo; (2) aboriginal Taiwanese; (3) Indonesians from the island of Sunda. Arrows indicate approximate path of the expansion of Austronesian-speaking peoples (adapted from Gray and Jordan13).
Despite numerous surveys in Southeast Asia aimed at estimating the frequency of SAO, research to date has not characterized the pattern of molecular polymorphism at SLC4A1. Earlier studies have noted that the 27-bp deletion causing SAO is always associated with the rare allele of another SLC4A1 variant known as the ‘Memphis’ polymorphism.1 This association suggests a single origin of the SAO mutation, but does little to clarify allelic relationships between SAO and wild-type chromosomes. Our study here examines the pattern of polymorphism at SLC4A1 in a representative population from Indonesia known to harbor the SAO allele at modest frequency. Our goal is to assess the extent to which SAO chromosomes differ from wildtype (that is, are there sites other than the 27-bp deletion that characterize SAO chromosomes?) and to examine whether the pattern of polymorphism in the surrounding genomic region reflects a history of natural selection. In addition, we compare patterns of sequence variation in Indonesia with samples from two other populations, one from Austronesian-speaking aboriginal Taiwanese individuals, who are suggested by linguistic and genetic evidence to be the source population of the Austronesian expansion,11, 12, 13 and a second population from Japan, which harbors the Memphis polymorphism, but not SAO.
Materials and methods
DNA samples
Nineteen DNA samples were collected in the field from the town of Kodi, on the island of Sumba, East Nusa Tenggara Province, Indonesia. All participating individuals were volunteer donors who provided written informed consent. An earlier genotyping study suggests that the allele frequency of SAO in this population is 0.101 (Sudoyo, unpublished data). Nineteen additional DNA samples from both Japan and Taiwan were obtained from the Coriell Institute for Medical Research (see Supplementary Table S1 for a complete list). Taiwanese samples were collected from the Ami and Atayal aboriginal groups; Japanese samples are part of the HapMap Japanese in Tokyo DNA panel. All sampling locations are shown in Figure 1. At present, falciparum malaria affects only the population from Indonesia.14 However, because Taiwan harbored endemic P. falciparum before its eradication in 1965,15, 16 it is possible that SLC4A1 was affected by malaria-related natural selection in the Ami and Atayal populations.
Sequence analysis
All 19 samples from each of the three study populations were typed for the presence of SAO as described by Jarolim et al.1 In addition, each was resequenced over a 4933 bp region within the SLC4A1 gene. The specific sequence corresponds to bases 39 691 104–39 696 208 of chromosome 17 on the March 2006 assembly of the human genome. We lacked complete sequencing primer coverage for a 174 bp region within the span described above, which we omitted from all analyses. A complete list of amplification and sequencing primers is available from the authors on request. Diploid sequence was edited and assembled using the program Sequencher 4.7 (Gene Codes), and variable sites were identified as either heterozygous positions or as differences between individuals (of alternate homozygous states). In addition to numerous single-nucleotide polymorphisms (SNPs), two single-base insertion/deletion polymorphisms were present in our data (see Results, below). For all analyses, SNPs and insertion/deletions were treated in the same manner.
We analyzed the pattern of polymorphism in our sequence data using the program DNAsp v. 4.50.3 (ref. 17). We estimated the population-mutation parameter (4Neμ, where Ne is the effective population size and μ is the mutation rate per generation) in each population using estimates based on the number of segregating sites, θW,18 and the average number of pairwise differences among sequences, π.19 We used several tests that examine departures of the frequency spectrum of mutations from standard neutral expectations. These included Tajima's D, which is sensitive to excesses of low or intermediate frequency variants;20 Fu and Li's D, which is sensitive to the number of derived singleton mutations;21 and Fay and Wu's H, which is sensitive to the number of high-frequency derived variants.22 The significance of these tests was evaluated by simulation of the coalescent process with population-specific recombination estimates, as implemented by the program ‘ms’.23 Mutation polarity was determined by comparison with outgroup data obtained from the Pan troglodytes (chimpanzee) and Pongo pygmaeus (orangutan) genome sequences. The latter was used for a handful of cases where a gap exists in the chimpanzee genome sequence. We estimated population-recombination parameters (ρ) using the program LDhat.24
We estimated haplotypes for our diploid resequence data using the program fastPHASE v. 1.2.325. These haplotypes were assembled into a network using statistical parsimony, as implemented by the program TCS v. 1.21.26
We examined differentiation between the three sampled populations using two different methods. First, we calculated Fst as per the study of Hudson et al.27 Interpreted under an island model of population subdivision, Fst is an estimator of 1/(1+4Nem), where m is the fraction of migrants per generation.28 In addition, we used Hudson's29 nearest-neighbor statistic (Snn), which can be combined with permutation tests (10 000 permutations performed using DNAsp v. 4.50.3) to evaluate whether DNA sequences show more differentiation than expected under panmixia.
Results
Our resequence survey of approximately 5 kb within the SLC4A1 gene uncovered 28 variable sites, including 25 SNPs and 2 single-base insertion/deletions. In addition, each sample was typed for the 27-bp SAO deletion, which was polymorphic only in the Indonesian population. Of the variable sites in our survey, six have not been described in the literature, nor are they present in the dbSNP database. All variable sites, along with their chromosomal location or dbSNP identifier, are detailed in Figure 2. Of particular note are six non-synonymous variants segregating in our sample, with all other variants occurring in non-coding sequence. One of these non-synonymous variants is the Memphis polymorphism (dbSNP rs5036), which we find in both the Indonesian and Japanese samples, but not among the Taiwanese. Although our sample contains more non-synonymous than synonymous polymorphic sites within coding regions, a MacDonald–Kreitman30 test using orangutan as an outgroup does not suggest a significant difference from expectations based on between-species comparisons (data not shown).

Inferred haplotypes observed in SLC4A1 resequence data. All observed SNPs are listed by either their ‘rs’ identifier or chromosomal position (for those not described earlier). Asterisks indicate non-synonymous variants. The top haplotype shows the ancestral allele for each site. The number of occurrences of each haplotype in the Indonesian, Japanese and Taiwanese samples is listed at right.
Among the three populations we surveyed, the Indonesian sample had the highest genetic diversity according to all mutation-based estimates, as shown in Table 1. The Taiwanese sample ranked second, and the Japanese third. Estimates of recombination varied in rank-order among populations by measure. The minimum number of recombination events (Rm), as estimated by Hudson and Kaplan's31 method, was highest in the Indonesian population (five), followed by the Taiwanese and Japanese (two in each). Estimates of the population-recombination parameter, ρ, followed a different rank-order, with the Taiwanese population having the highest value, followed by the Indonesian, with the Japanese having the lowest value (Table 1).
Tests of neutrality based on the frequency spectrum of mutations revealed no departures from expectations in any population in the case of Tajima's D or Fu and Li's D (Table 1). In contrast, Fay and Wu's H showed significantly negative values in all populations, indicating an excess of high-frequency derived alleles over expectations.
Haplotypes estimated from the diploid resequence data are shown in Figures 2 and 3. Several aspects of the haplotype data are notable. First, the four SAO chromosomes included in our sample share a single haplotype (no. 1 in Figure 2) that differs from a non-SAO haplotype (no. 2 in Figure 2) by only the 27-bp deletion that causes SAO. Interestingly, haplotype no. 2 is found only in the Indonesian and Japanese samples. Also of note is that all four SAO chromosomes carry the rare Memphis allele at rs5036. Chromosomes that carry the Memphis allele at this site occur on five different haplotypes, one of which includes the SAO deletion (haplotypes nos. 1–5, Figure 2). In the Japanese sample, we observe only a single Memphis haplotype (present twice). In contrast, in the Indonesians we observe nine Memphis chromosomes with five different haplotypes. Interestingly, all Memphis chromosomes lie near the estimated root of the haplotype network (Figure 3), and share a number of rare ancestral alleles across several variable sites (Figure 2).

Network showing relationships among observed haplotypes. Smallest circles indicate unobserved haplotypes. When greater than one, the observed number of copies of each haplotype is listed. Purple indicates Indonesian haplotypes, blue indicates Japanese and yellow indicates Taiwanese. The location of the most likely root of the network, determined by comparison with chimpanzee and orangutan sequence, is indicated. An asterisk indicates the haplotype containing the 27-bp SAO deletion.
Population differentiation is pronounced in the survey region among the Indonesians, Japanese and Taiwanese (overall Fst=0.092). This pattern seems largely to be driven by divergence of the Taiwanese from the remaining groups, with a pairwise Fst of 0.130 with the Indonesians and 0.103 with the Japanese. In contrast, there was much less differentiation between the Japanese and Indonesians (Fst=0.036). Despite this heterogeneity in differentiation, Hudson's Snn estimator reveals significant structure among all pairs of populations (Taiwanese/Japanese, Snn=0.682, P<0.0001; Taiwanese/Indonesian, Snn=0.0683, P=0.0001; Japanese/Indonesian, Snn=0.635, P=0.0010).
Discussion
Our examination of DNA sequence variation in the SLC4A1 gene shows a number of interesting patterns. First, chromosomes carrying the SAO deletion mutation are very similar to non-SAO chromosomes segregating within our sample. Indeed, we have uncovered a single rare haplotype that occurs in both Indonesia and Japan that differs from SAO chromosomes only by the 27-bp deletion. This pattern suggests that SAO could be the result of relatively recent mutation occurring on a rare haplotype that still segregates in Asian populations. To better estimate the age of the SAO mutation, additional genotype data examining variation among geographically disparate SAO chromosomes over longer genetic distances will be necessary.
A second interesting facet of our analysis is that the deletion responsible for SAO is most closely related to haplotypes that can be found in Japanese and Indonesian samples, but not those from aboriginal Taiwanese populations. The closest Taiwanese haplotype to an observed SAO chromosome is eight mutational steps away (Figure 3). Although it is possible that we failed to sample Taiwanese alleles that are more similar to SAO alleles in Indonesia, our data show that chromosomes closely related to Indonesian SAO alleles are not a major component of genetic diversity among aboriginal Taiwanese populations. Indeed, estimates of population differentiation (for example, Fst) show the Taiwanese sampled here to be extremely divergent at SLC4A1 from both the Japanese and Indonesian populations. This is most likely due to the absence of haplotypes near the root of the haplotype network in Taiwan, which are quite common in Japan and Indonesia (Figure 3).
The lack of similarity of any Taiwanese chromosomes to SAO is an interesting result with respect to earlier work suggesting that SAO alleles spread across their present range by the Austronesian expansion some 3500 years BP.4, 10 Because Austronesian speakers throughout the Pacific and Indian Ocean are thought to be related to aboriginal Taiwanese,11, 12, 13 it is somewhat surprising that we did not find haplotypes similar to SAO in our Taiwanese sample. There are two possible explanations for this pattern. First, SAO-like chromosomes may simply be rare in aboriginal Taiwanese populations and only increased in frequency in more derived populations of the Austronesian expansion. This could have happened due to simple genetic drift during the spread of Austronesian speakers. Alternatively, it is most likely that P. falciparum was present in many of the regions into which the Austronesian expansion occurred,32 meaning that malaria-related positive natural selection may be responsible for recent increases in SAO frequency. A second scenario that could explain the lack of SAO-like chromosomes in Taiwan is that SAO has its origins in a different geographic area. Recent work has shown that nuclear genome of Austronesian speakers has substantial genetic contributions from other regions of Asia.33 SAO could be a case such as this. Regardless of its origins, SAO confers extraordinary fitness benefits in areas where P. falciparum is highly endemic.6 Because of this, it is interesting to consider the extent to which the SAO mutation itself could have facilitated the spread of Austronesian speakers (or their genes) into malarial environments.
Beyond examination of SAO chromosomes, and their relationship to non-SAO chromosomes, several other aspects of our data are remarkable. First, in our data set we observe five non-synonymous SNPs in addition to the SAO deletion. One of these variants is the Memphis polymorphism (rs5036), which is well documented to be variable among many widespread world populations.34, 35, 36, 37 Remarkably, the ancestral allele at this locus, as determined by comparison with a number of outgroup species,34, 38, 39 is rare (typically less than 0.10) in all populations where the SNP is polymorphic. It is notable that the observed frequency of the ancestral Memphis allele is 0.24 in our Indonesian sample, encompassing five different haplotypes. Maintenance of SAO, which occurs on the Memphis background, by malaria selection most likely accounts for some elevation of Memphis frequency in our sample relative to other populations.
The fact that the ancestral Memphis allele is rarer than the derived allele in our Japanese and Indonesian samples mirrors a pattern seen at SNPs across our data set (and even in the Taiwanese sample, despite the fact that the derived allele at rs5036 is fixed in that population). Examination of Figure 2 shows numerous polymorphic sites where ancestral alleles are much rarer than derived (for example, rs2074108, rs2074107, rs45545233 etc.). This pattern drives the significantly negative values of Fay and Wu's H that we observe in all three populations. Because earlier studies have shown that Memphis is a high-frequency derived allele, it could be argued that our a priori expectation for the value of Fay and Wu's H should be reduced relative to standard neutral expectations.40 To address this, we have followed the approach of Brown et al.41 and re-evaluated the significance of Fay and Wu's H using simulations of the coalescent conditioned on the presence of at least one derived allele present at the same frequency as the Memphis allele in each population. Because the Memphis site is invariant in the Taiwanese sample, we are only able to apply this approach to the Indonesian and Japanese samples. In both cases, the observed value of Fay and Wu's H remains significantly negative. For Indonesia, the associated P-value is 0.0386 for the conditioned coalescent (compared with 0.0359 for the standard neutral coalescent). For Japan, the conditioned coalescent P-value is 0.0247 (compared with 0.0046 for the standard neutral coalescent).
Negative values of Fay and Wu's H are typically associated with genealogies recovering from a complete, or near-complete, selective sweep.22 In addition, several studies have shown that some demographic scenarios can also cause negative skews in Fay and Wu's H.42, 43 To distinguish between these two possible causes of the pattern, it is useful to consider the pattern of polymorphism in the Memphis genomic region in a wider variety of human populations. Examination of HapMap (phase 3) SNP data from our resequenced region shows five overlapping SNPs that have been genotyped in 10 world populations (excluding the Japanese), including three where the derived allele is at high frequency in our sample (Supplementary Table S2). At all three of these SNPs, the ancestral allele is in the minority in nearly every HapMap population, suggesting that other world populations share the excess of high-frequency derived alleles that we observe in the Indonesians, Japanese and Taiwanese. Because these populations have very different demographic histories from those in this study (and from each other),44 we favor recent natural selection as the cause of the skewed frequency distribution of derived alleles in our data set. Whether or not the Memphis site is itself a target of selection is unclear, although variation in Memphis genotype is known to functionally affect anion transport in the red blood cell.37 Regardless, the data we present here suggest that variation closely linked to the region of SLC4A1 that we surveyed has been targeted by natural selection, likely in most world populations.
In conclusion, our survey of polymorphism at the SLC4A1 gene sheds significant light on the origin of SAO, a clinically important trait that is widespread throughout Southeast Asia. SAO chromosomes are similar to a rare haplotype that still segregates in multiple Asian populations, suggesting that the mutation could be of recent origin. Interestingly, however, we find no evidence that SAO chromosomes are closely related to those presently found in aboriginal Taiwanese populations—the most likely source of the Austronesian expansion. This finding does not contradict earlier studies that suggest the distribution of SAO to be shaped by the spread of Austronesian speakers, but it does suggest that an important genetic component of the Austronesian expansion had its source outside of Taiwan. Finally, our data from multiple populations suggests that SLC4A1 has been targeted by recent natural selection, most likely acting on the Memphis polymorphic site (rs5036) or a closely linked variant.
References
Jarolim, P., Palek, J., Amato, D., Hassan, K., Sapak, P., Nurse, G. T. et al. Deletion in erythrocyte band 3 gene in malaria-resistant Southeast Asian ovalocytosis. Proc. Natl Acad. Sci. USA 88, 11022–11026 (1991).
Mgone, C. S., Koki, G., Paniu, M. M., Kono, J., Bhatia, K. K., Genton, B. et al. Occurrence of the erythrocyte band 3 (AE1) gene deletion in relation to malaria endemicity in Papua New Guinea. Trans. R. Soc. Trop. Med. Hyg. 90, 228–231 (1996).
Kimura, M., Shimizu, Y., Settheetham-Ishida, W., Soemantri, A., Tiwawech, D., Romphruk, A. et al. Twenty-seven base pair deletion in erythrocyte band 3 protein gene responsible for Southeast Asian ovalocytosis is not common among Southeast Asians. Hum. Biol. 70, 993–1000 (1998).
Kimura, M., Tamam, M., Soemantri, A., Nakazawa, M., Ataka, Y., Ohtsuka, R. et al. Distribution of a 27-bp deletion in the band 3 gene in South Pacific islanders. J. Hum. Genet. 48, 642–645 (2003).
Allen, S. J., O’Donnell, A., Alexander, N. D., Mgone, C. S., Peto, T. E., Clegg, J. B. et al. Prevention of cerebral malaria in children in Papua New Guinea by southeast Asian ovalocytosis band 3. Am. J. Trop. Med. Hyg. 60, 1056–1060 (1999).
Genton, B., al-Yaman, F., Mgone, C. S., Alexander, N., Paniu, M. M., Alpers, M. P. et al. Ovalocytosis and cerebral malaria. Nature 378, 564–565 (1995).
Cortés, A., Mellombo, M., Mgone, C. S., Beck, H. P., Reeder, J. C. & Cooke, B. M. Adhesion of Plasmodium falciparum-infected red blood cells to CD36 under flow is enhanced by the cerebral malaria-protective trait South-East Asian ovalocytosis. Mol. Biochem. Parasitol. 142, 252–257 (2005).
Liu, S. C., Jarolim, P., Rubin, H. L., Palek, J., Amato, D., Hassan, K. et al. The homozygous state for the band 3 protein mutation in Southeast Asian Ovalocytosis may be lethal. Blood 84, 3590–3591 (1994).
Rabe, T., Jambou, R., Rabarijaona, L., Raharimalala, L., Rason, M. A., Ariey, F. et al. South-East Asian ovalocytosis among the population of the Highlands of Madagascar: a vestige of the island's settlement. Trans. R. Soc. Trop. Med. Hyg. 96, 143–144 (2002).
Tsukahara, T., Hombhanje, F. W., Lum, J. K., Hwaihwanje, I., Masta, A., Kaneko, A. et al. Austronesian origin of the 27-bp deletion of the erythrocyte band 3 gene in East Sepik, Papua New Guinea inferred from mtDNA analysis. J. Hum. Genet. 51, 244–248 (2006).
Trejaut, J. A., Kivisild, T., Loo, J. H., Lee, C. L., He, C. L., Hsu, C. J. et al. Traces of archaic mitochondrial lineages persist in Austronesian-speaking Formosan populations. PLoS Biol 3, e247 (2005).
Friedlaender, J. S., Friedlaender, F. R., Hodgson, J. A., Stoltz, M., Koki, G., Horvat, G. et al. Melanesian mtDNA complexity. PLoS ONE 2, e248 (2007).
Gray, R. D. & Jordan, F. M. Language trees support the express-train sequence of Austronesian expansion. Nature 405, 1052–1055 (2000).
Snow, R. W., Guerra, C. A., Noor, A. M., Myint, H. Y. & Hay, S. I. The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature 434, 214–217 (2005).
Yip, K. Malaria eradication: the Taiwan experience. Parassitologia 42, 117–126 (2000).
Taiwan Provincial Malaria Research Institute & WHO Malaria Team in Taiwan. Malaria control and eradication in Taiwan: progress report, May 1952 to June 1957. Bull. World Health Organ 19, 595–620 (1958).
Rozas, J., Sanchez-DelBarrio, J. C., Messeguer, X. & Rozas, R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19, 2496–2497 (2003).
Watterson, G. A. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7, 256–276 (1975).
Nei, M. & Li, W. H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl Acad. Sci. USA 76, 5269–5273 (1979).
Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595 (1989).
Fu, Y. X. & Li, W. H. Statistical tests of neutrality of mutations. Genetics 133, 693–709 (1993).
Fay, J. C. & Wu, C. I. Hitchhiking under positive Darwinian selection. Genetics 155, 1405–1413 (2000).
Hudson, R. R. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18, 337–338 (2002).
McVean, G., Awadalla, P. & Fearnhead, P. A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics 160, 1231–1241 (2002).
Scheet, P. & Stephens, M. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 78, 629–644 (2006).
Clement, M., Posada, D. & Crandall, K. A. TCS: a computer program to estimate gene genealogies. Mol. Ecol. 9, 1657–1659 (2000).
Hudson, R. R., Slatkin, M. & Maddison, W. P. Estimation of levels of gene flow from DNA sequence data. Genetics 132, 583–589 (1992).
Wright, S. The genetical structure of populations. Ann. Eugenics 15, 323–354 (1951).
Hudson, R. R. A new statistic for detecting genetic differentiation. Genetics 155, 2011–2014 (2000).
McDonald, J. H. & Kreitman, M. Adaptive protein evolution at the Adh locus in Drosophila. Nature 351, 652–654 (1991).
Hudson, R. R. & Kaplan, N. L. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 111, 147–164 (1985).
Joy, D. A., Feng, X., Mu, J., Furuya, T., Chotivanich, K., Krettli, A. U. et al. Early origin and recent expansion of Plasmodium falciparum. Science 300, 318–321 (2003).
Friedlaender, J. S., Friedlaender, F. R., Reed, F. A., Kidd, K. K., Kidd, J. R., Chambers, G. K. et al. The genetic structure of Pacific Islanders. PLoS Genet 4, e19 (2008).
Jarolim, P., Rubin, H. L., Zhai, S., Sahr, K. E., Liu, S. C., Mueller, T. J. et al. Band 3 Memphis: a widespread polymorphism with abnormal electrophoretic mobility of erythrocyte band 3 protein caused by substitution AAG----GAG (Lys----Glu) in codon 56. Blood 80, 1592–1598 (1992).
Ranney, H. M., Rosenberg, G. H., Morrison, M. & Mueller, T. J. Frequencies of Band 3 variants of human red cell membranes in some different populations. Br. J. Haematol. 75, 262–267 (1990).
Palatnik, M., Simoes, M. L., Alves, Z. M. & Laranjeira, N. S. The 60 and 63 kDa proteolytic peptides of the red cell membrane band-3 protein: their prevalence in human and non-human primates. Hum. Genet. 86, 126–130 (1990).
Ideguchi, H., Okubo, K., Ishikawa, A., Futata, Y. & Hamasaki, N. Band 3-Memphis is associated with a lower transport rate of phosphoenolpyruvate. Br. J. Haematol. 82, 122–125 (1992).
Kimura, M. & Ishida, T. Chimpanzee band 3 protein polymorphism and the origin of the band 3 Memphis in humans. Folia Primatol. (Basel). 72, 298–300 (2001).
Schawalder, A., Hue-Roye, K., Castilho, L., Chaudhuri, A. & Reid, M. E. Analysis in non-human primates reveals that the ancestral Band 3 gene encodes Dib and the Band 3-Memphis phenotype. J. Med. Primatol. 35, 144–148 (2006).
Kreitman, M. & Di Rienzo, A. Balancing claims for balancing selection. Trends Genet. 20, 300–304 (2004).
Brown, B. T., Woerner, A. & Wilder, J. A. Ascertainment bias and the pattern of nucleotide diversity at the human ALDH2 locus in a Japanese population. J. Mol. Evol. 64, 375–385 (2007).
Jensen, J. D., Kim, Y., DuMont, V. B., Aquadro, C. F. & Bustamante, C. D. Distinguishing between selective sweeps and demography using DNA polymorphism data. Genetics 170, 1401–1410 (2005).
Przeworski, M. The signature of positive selection at randomly chosen loci. Genetics 160, 1179–1189 (2002).
Keinan, A., Mullikin, J. C., Patterson, N. & Reich, D. Measurement of the human allele frequency spectrum demonstrates greater genetic drift in East Asians than in Europeans. Nat. Genet. 39, 1251–1255 (2007).
Acknowledgements
We thank J Friedlaender and two anonymous reviewers for helpful comments on the manuscript. This work was supported by funds from the National Science Foundation, Williams College, and Northern Arizona University (to JAW) and a Sigma Xi Grant-in-Aid of Research (to EGP).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website (http://www.nature.com/jhg)
Supplementary information
Rights and permissions
About this article
Cite this article
Wilder, J., Stone, J., Preston, E. et al. Molecular population genetics of SLC4A1 and Southeast Asian Ovalocytosis. J Hum Genet 54, 182–187 (2009). https://doi.org/10.1038/jhg.2009.12
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2009.12
