
Abstract
Six males in a Chinese family affected by congenital cataracts, cerebellar ataxia, short stature, and mental retardation, which were tentatively named CASM syndrome. Eight female carriers in the family had cataracts alone. Linkage analysis demonstrated that the disease is transmitted through X-linked inheritance, either by setting the syndrome in males as an X-linked recessive trait, or by setting cataracts in the family as an X-linked dominant trait. The gene responsible for the syndrome is mapped to Xpter-Xq13.1, with the highest lod score of 3.91 for DXS1226, DXS991, and DXS1213 at θ = 0. Haplotype analysis identified that the allele harboring the disease gene co-segregated with all female carriers as well as affected males in the family. Clinically and genetically, the disease in this family is different from any known disease. Major features of CASM syndrome that distinguish it from other diseases are X-linked inheritance and cataracts in carrier females.
Similar content being viewed by others

Introduction
As part of our ongoing projects to identify genes for genetic eye diseases, a Chinese family containing 6 males affected by congenital cataracts, cerebellar ataxia, short stature, and mental retardation was investigated. Carrier females show only cataracts without the other signs seen in the affected males. Linkage analysis provided strong evidence localizing the disease locus to the X-chromosome. Clinical manifestation and linkage results indicate that the disease in this family represents a new syndrome, tentatively named the CASM syndrome.
Cataracts, ataxia, mental retardation, and short stature have been documented as being the common features in several hereditary diseases including the Marinesco–Sjögren syndrome (MSS, OMIM 248800), congenital cataracts with facial dysmorphism and neuropathy (CCFDN, OMIM 604168), cataract–ataxia–deafness–retardation syndrome (OMIM 212710), and stomatin-deficient cryohydrocytosis with neurologic defects (OMIM 608885). These diseases are either inherited as an autosomal recessive trait (Begeer et al. 1991; Lagier-Tourenne et al. 2002, 2003; Merlini et al. 2002) or reported as sporadic cases (Fricke et al. 2004). Genes responsible for MSS and CCFDN have recently been identified (Anttonen et al. 2005; Senderek et al. 2005; Varon et al. 2003). Genetically and clinically, the disease in the Chinese family is distinct, and is unlikely to be one of the diseases mentioned above.
Subjects and methods
Family and clinical data
The family described here was ascertained as being from central China (Fig. 1). Twenty-three individuals participated in the study. Informed consent conforming to the tenets of the Declaration of Helsinki and following the Guidance of Sample Collection of Human Genetic Diseases (863-Plan) by the Ministry of Public Health of China was obtained from the participating individuals or their guardians prior to the study. Ocular examinations were performed by an ophthalmologist (Prof. Guo) and neurological evaluations were performed by a neurologist (Prof. Dai). Genomic DNA was prepared from venous blood.

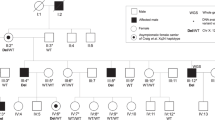
Pedigree and clinical phenotype. a Pedigree of the family and haplotype construction. Black bars represent the disease haplotype. Other bars represent normal alleles. Filled squares indicate individuals affected with CASM syndrome. Circles with a black dot indicate carrier females who only had cataracts. Numbers in parenthesis indicate that genotyping data were inferred from offspring or parents. b Clinical findings. A Short stature of individual IV-1 at the age of 21 years. He was two-thirds the height of his father (right). He could only stand up with the help of his father. B Punctate and pulverulent opacities in lens cortex observed in individual IV-10 (carrier) at 11 years of age just prior to cataract surgery. C Teeth of individual IV-13 (affected) with no sign of screwdriver incisors
Genotyping and linkage analysis
Genotyping and linkage analysis were carried out as previously described (Guo et al. 2006). The full CASM syndrome in this family was analyzed as an X-linked recessive trait, with full penetrance and a disease–gene allele frequency of 0.0001. In order to test if the disease in this family can stand well as an X-linked trait, cataracts in carrier females as well as affected males were also analyzed as an X-linked dominant trait. Markers allele frequencies were arbitrarily assumed to be equal for the initial scan and were calculated from 23 unrelated individuals for markers in the linked region.
Results
CASM syndrome phenotype in affected males
Six males in the family were born with poor visual acuity and were unable to walk by themselves, except 1, IV-13, who could walk after cataract surgery at 7 years of age (Fig. 1a, Table 1). They all showed delayed development of speech and language. So far, none of them has married. Two of the 6, III-1 and III-2, showed the same signs and symptoms as III-3, according to interviews with the family, and died at 14 and 16 years of age respectively. The other 4, III-3, III-8, IV-1, and IV-13, underwent physical examinations including ophthalmological and neurological evaluations. They were much shorter in stature compared with unaffected siblings or parents (Fig. 1b, Table 1) as well as being below 95% of the average stature according to age in the Chinese population (Zhu et al. 1994). Hair and skin were normal. Dental changes of screwdriver incisors were not observed in affected males (Fig. 1b). They did not have difficulty in breathing.
Ophthalmological examination revealed normal cornea, anterior chamber, and iris. The pupillary light reflex was present and total lens opacities were observed in all 4 affected males. Normal fundus appearance was observed in 1 affected male, IV-13, who had undergone cataract surgery at 7 years of age. Visual acuity for all affected individuals could not be measured accurately as the affected males had comprehension and communication difficulties due to mental and linguistic hindrance. Therefore, the visual acuity for all 4 affected males listed in Table 1 is just approximate.
Neurological evaluation of the 4 affected males revealed mild to moderate mental retardation as measured on the Epstein and Wechsler scales (Epstein and Sharma 1998; Wechsler 1974), muscle hypotonia, muscle weakness, dysarthria, and postural tremor. Their stances were broad based with the feet several inches apart. They had difficulty standing with their feet together and were unable to sit or stand without support (astasia). Individuals III-3, III-8, and IV-1 had been unable to walk without assistance since birth, but they could walk a few steps with the help of other people, displaying a wide-based and staggering gait.
IV-13 had better visual acuity after cataract surgery and therefore was available for more detailed neurological evaluation. Apart from the common neurological findings above, he could walk by himself with a wide-based staggering gait and taking a zigzag course. He was positive for the finger–nose test, finger–chase test, and heel–kneel–shin test. Romberg sign was positive with his eyes opened or closed. He showed dysarthric speech. The wide-based staggering gait, positive finger–nose test, positive heel–kneel–shin test, and dysarthric speech found on examination were typical of cerebellar ataxia.
Phenotype in female carriers
Eight females in the family experienced blurred vision starting from early childhood to teenage (Table 1). It is not clear if the female in the first generation (individual I-2) had blurred vision. Seven of the 8 females, II-4, III-5, III-13, III-16, IV-9, IV-10, and IV-12, received ocular examination and the other 1 (II-1) was deceased. Six individuals (III-5, III-13, III-16, IV-9, IV-10, and IV-12) showed either cortical punctate and pulverulent opacities (Fig. 1b) and posterior subcapsular opacities of the lens or had undergone cataract surgery because of congenital cataracts. Individual II-4 had cortical and posterior subcapsular lens opacities in the right eye and a total lens opacity in the left eye at age of 71 years of age. The cornea, anterior chamber, iris, and pupils were normal in all 7 carriers with cataracts. Ophthalmoscopic observation showed a normal fundus in all 7 carriers whose fundus was visible. All affected females had normal gait and muscle strength. Their stature was normal compared with local control population. Mental retardation was not observed in these carriers.
Linkage study
The pedigree is consistent with an X-linked trait partially penetrant in female carriers (Fig. 1). After a complete scan of the X chromosome, the CASM syndrome was mapped to Xpter-q13.1, with maximum lod score of 3.91 for DXS1226, DXS991 and DXS1213 at θ = 0 (Table 2). All 11 markers inside the linked region gave positive lod scores. To test if the disease in the family can stand well as an X-linked trait, cataracts in carrier females as well as affected males in the family were analyzed as an X-linked dominant trait. Linkage analysis mapped cataract to Xpter-q13.1 (Table 2), with the highest lod score of 6.32 for DXS1226 at θ = 0. Similarly, all 11 markers inside the linked region gave positive lod scores. The highest lod score obtained analyzed either under an X-linked recessive mode or under an X-linked dominant mode, reached the theoretical maximum lod score that could be generated in this type of family.
Examination of haplotypes in this family showed the disease allele co-segregated with all female carriers as well as affected males in the family (Fig. 1). Recombination at DXS8052 for individual IV-1 and further recombination at DXS986 for individual IV-13 set the telomeric boundary of the linked region on Xq. The whole short arm was included in the linked region because recombination was not observed. Congenital cataracts when analyzed alone as an X-linked dominant trait in the family co-segregated identically with the entire CASM syndrome analyzed as an X-linked recessive trait. Both lod scores and haplotypes convincingly demonstrated that the CASM in this family is transmitted as an X-linked recessive mode. Having a lod score of 6.23 for X chromosome markers as well as co-segregation of disease with Xpter-q13.1, the chance of an autosomal dominant sex-limited trait should be very low, as opposed to an a priori likelihood of more than 106 of being X-linked (Lander and Kruglyak 1995; Terwilliger and Ott 1994). Mitochondrial DNA inheritance is also unlikely as no offspring of several obligate mtDNA carriers (II-5, II-7, II-9, III-10) had the disease.
Discussion
In this study, a new syndrome presenting with congenital cataracts (whole lens opacities), cerebellar ataxia, short stature, and mental retardation is described in a Chinese family. This syndrome was present in 6 males in the family. Eight female carriers only had cataracts (partial lens opacities). Linkage analysis not only demonstrated X-linked recessive transmission of the syndrome, but also mapped the disease loci to Xpter-q13.1. Major features of the CASM syndrome that distinguish it from other diseases (with cataracts, ataxia, short stature, and mental retardation) are X-linked recessive inheritance of the full trait and congenital cataracts in carrier females. More detailed clinical data, such as brain CT scans and muscle biopsy, would be desirable, but were not available.
The X-linked recessive CASM syndrome described here is different from other known diseases mapped to Xpter-q13.1. In this region, X-linked dominant cataract was reported in Nance-Horan syndrome (NHS; Burdon et al. 2003), oculofaciocardiodental syndrome (OFCD; Ng et al. 2004), and X-linked dominant chondrodysplasia punctata 2 (CDPX2; Derry et al. 1999). Cerebellar ataxia has not been observed in these three diseases. In addition, individuals with these syndromes show other typical phenotypes that are not present in the Chinese family with the CASM syndrome.
In NHS, affected males and carrier females have teeth changes similar to screwdriver incisors (Burdon et al. 2003), which are not observed in the CASM syndrome. Short stature was not reported in NHS. For OFCD syndrome, females usually manifest congenital cataract, microphthalmia, facial abnormalities, cardiac anomalies, and dental abnormalities. The disease may be lethal in males, as it has not been observed in any males (Gorlin et al. 1996). Most CDPX2 patients are female, as it may be lethal for hemizygous males. Other signs seen in females with CDPX2, such as skin defects, hyperkeratosis, coarse lusterless hair and alopecia, and craniofacial defects (Derry et al. 1999), were not observed in carrier females of the Chinese family with CASM syndrome. Patients with CCT do not have other symptoms seen in the CASM syndrome except for cataracts.
X-linked mental retardation and short stature have been reported to be associated with a large number of gene variations (Chelly and Mandel 2001), but none has been reported to be associated with cataract and cerebellar ataxia.
In summary, an X-linked recessive CASM syndrome was identified in a Chinese family and was mapped to Xpter-q13.1 with the highest lod score of 3.91 at θ = 0. When cataracts alone were analyzed as an X-linked dominant trait, a lod score of 6.32 was obtained. Recruitment of additional families to narrow down the linked region will accelerate identification of the causative gene.
References
Anttonen AK, Mahjneh I, Hamalainen RH, Lagier-Tourenne C, Kopra O, Waris L, Anttonen M, Joensuu T, Kalimo H, Paetau A, Tranebjaerg L, Chaigne D, Koenig M, Eeg-Olofsson O, Udd B, Somer M, Somer H, Lehesjoki AE (2005) The gene disrupted in Marinesco–Sjögren syndrome encodes SIL1, an HSPA5 cochaperone. Nat Genet 37:1309–1311
Begeer JH, Scholte FA, van Essen AJ (1991) Two sisters with mental retardation, cataract, ataxia, progressive hearing loss, and polyneuropathy. J Med Genet 28:884–885
Burdon KP, McKay JD, Sale MM, Russell-Eggitt IM, Mackey DA, Wirth MG, Elder JE, Nicoll A, Clarke MP, FitzGerald LM, Stankovich JM, Shaw MA, Sharma S, Gajovic S, Gruss P, Ross S, Thomas P, Voss AK, Thomas T, Gecz J, Craig JE (2003) Mutations in a novel gene, NHS, cause the pleiotropic effects of Nance–Horan syndrome, including severe congenital cataract, dental anomalies, and mental retardation. Am J Hum Genet 73:1120–1130
Chelly J, Mandel JL (2001) Monogenic causes of X-linked mental retardation. Nat Rev Genet 2:669–680
Derry JM, Gormally E, Means GD, Zhao W, Meindl A, Kelley RI, Boyd Y, Herman GE (1999) Mutations in a delta 8-delta 7 sterol isomerase in the tattered mouse and X-linked dominant chondrodysplasia punctata. Nat Genet 22:286–290
Epstein MH, Sharma J (1998) Behavioral and emotional rating scale: a strength-based approach to assessment. PRO-ED, Austin, TX
Fricke B, Jarvis HG, Reid CD, Aguilar-Martinez P, Robert A, Quittet P, Chetty M, Pizzey A, Cynober T, Lande WF, Mentzer WC, During M, Winter S, Delaunay J, Stewart GW (2004) Four new cases of stomatin-deficient hereditary stomatocytosis syndrome: association of the stomatin-deficient cryohydrocytosis variant with neurological dysfunction. Br J Haematol 125:796–803
Gorlin RJ, Marashi AH, Obwegeser HL (1996) Oculo-facio-cardio-dental (OFCD) syndrome. Am J Med Genet 63:290–292
Guo X, Li S, Jia X, Xiao X, Wang P, Zhang Q (2006) Linkage analysis of two families with X-linked recessive congenital motor nystagmus. J Hum Genet 51:76–80
Lagier-Tourenne C, Chaigne D, Gong J, Flori J, Mohr M, Ruh D, Christmann D, Flament J, Mandel JL, Koenig M, Dollfus H (2002) Linkage to 18qter differentiates two clinically overlapping syndromes: congenital cataracts-facial dysmorphism-neuropathy (CCFDN) syndrome and Marinesco–Sjögren syndrome. J Med Genet 39:838–843
Lagier-Tourenne C, Tranebaerg L, Chaigne D, Gribaa M, Dollfus H, Silvestri G, Betard C, Warter JM, Koenig M (2003) Homozygosity mapping of Marinesco–Sjögren syndrome to 5q31. Eur J Hum Genet 11:770–778
Lander E, Kruglyak L (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11:241–247
Merlini L, Gooding R, Lochmuller H, Muller-Felber W, Walter MC, Angelicheva D, Talim B, Hallmayer J, Kalaydjieva L (2002) Genetic identity of Marinesco–Sjögren/myoglobinuria and CCFDN syndromes. Neurology 58:231–236
Ng D, Thakker N, Corcoran CM, Donnai D, Perveen R, Schneider A, Hadley DW, Tifft C, Zhang L, Wilkie AO, van der Smagt JJ, Gorlin RJ, Burgess SM, Bardwell VJ, Black GC, Biesecker LG (2004) Oculofaciocardiodental and Lenz microphthalmia syndromes result from distinct classes of mutations in BCOR. Nat Genet 36:411–416
Senderek J, Krieger M, Stendel C, Bergmann C, Moser M, Breitbach-Faller N, Rudnik-Schoneborn S, Blaschek A, Wolf NI, Harting I, North K, Smith J, Muntoni F, Brockington M, Quijano-Roy S, Renault F, Herrmann R, Hendershot LM, Schroder JM, Lochmuller H, Topaloglu H, Voit T, Weis J, Ebinger F, Zerres K (2005) Mutations in SIL1 cause Marinesco–Sjögren syndrome, a cerebellar ataxia with cataract and myopathy. Nat Genet 37:1312–1314
Terwilliger JD, Ott J (1994) Handbook of human genetic linkage. Johns Hopkins University Press, Baltimore
Varon R, Gooding R, Steglich C, Marns L, Tang H, Angelicheva D, Yong KK, Ambrugger P, Reinhold A, Morar B, Baas F, Kwa M, Tournev I, Guerguelcheva V, Kremensky I, Lochmuller H, Mullner-Eidenbock A, Merlini L, Neumann L, Burger J, Walter M, Swoboda K, Thomas PK, von Moers A, Risch N, Kalaydjieva L (2003) Partial deficiency of the C-terminal-domain phosphatase of RNA polymerase II is associated with congenital cataracts facial dysmorphism neuropathy syndrome. Nat Genet 35:185–189
Wechsler D (1974) Wechsler intelligence scale for children—revised. Psychological Corporation, New York
Zhu F, Wu R, Hu Y (1994) Practical pediatrics, 3rd edn. People’s Health Press, Beijing
Acknowledgements
The authors thank all patients and family members for their participation. This study was supported in part by the National 863 Plan of China (04AA104092 to XG; Z19-01-04-02 to QZ), Guangdong Natural Science Foundation (04009335 to XG), National Natural Science Foundation of China (30572006 to QZ), and Foundation from the Ministry of Education of China (20050558073 to QZ).
Author information
Authors and Affiliations
Corresponding author
Additional information
Xiangming Guo, Huangxuan Shen, Xueshan Xiao, Qilin Dai, Fielding Hejtmancik, and Qingjiong Zhang contributed equally to this work
Rights and permissions
About this article
Cite this article
Guo, X., Shen, H., Xiao, X. et al. Cataracts, ataxia, short stature, and mental retardation in a Chinese family mapped to Xpter-q13.1. J Hum Genet 51, 695–700 (2006). https://doi.org/10.1007/s10038-006-0009-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-006-0009-1
Keywords
This article is cited by
-
KITLG Mutations Cause Familial Progressive Hyper- and Hypopigmentation
Journal of Investigative Dermatology (2011)
