
Abstract
A new nucleoside modified by prenylation, simamycin (1), was isolated from the culture broth of a soil-derived Streptomyces sp. Its structure was determined by spectroscopic analysis and chemical synthesis as 5′-O-geranyluridine. Compound 1 induced differentiation of preadipocytes into matured adipocytes.
Similar content being viewed by others
Introduction
Despite the substantial amounts of studies on its secondary metabolites, Streptomyces is still believed to be the most prolific source of novel bioactive compounds. In fact, up to 40% of known microbial metabolites are derived from this group.1 However, it is noteworthy that the number of metabolites isolated is far below the number of gene clusters for secondary metabolite biosynthesis present in the genome of Streptomyces species.2, 3 In our screening for structurally unique or rare secondary metabolites from Streptomyces, we isolated plant hormone-like spiroacetals of polyketide origin,4 a linear polyketide δ-lactone with antimetastatic property,5 macrocyclic polyketides with a biosynthetically unprecedented heterocyclic ring structure6 and methylphenylalanine derivatives with adipocyte differentiation promoting activity.7 Further attempts to obtain new bioactive compounds from Streptomyces, a soil-derived strain TP-A0872, was chosen for chemical investigation, which led to the isolation of a new geranylated nucleoside, simamycin (1; Figure 1).

Structure of simamycin (1).
Results and discussion
The producing strain TP-A0872 was isolated from a soil sample collected in Kochi, Japan, and identified as a member of the genus Streptomyces on the basis of 16S rRNA gene sequence analysis. The whole culture broth of strain TP-A0872 cultured in A-3M liquid medium was extracted with 1-butanol. HPLC/UV analysis of the extract and dereplication using our in-house metabolite database suggested the presence of an unknown peak showing a UV absorption band at 263 nm. HPLC/UV-guided purification from the extract led to the isolation of a new geranylated nucleoside, 1. Subsequent biological evaluation elucidated that 1 induces the differentiation of preadipocytes into matured adipocytes. We herein describe the isolation, structure determination and biological activity of 1.
Compound 1 was obtained as optically active colorless needles that gave an [M+H]+ pseudomolecular ion at m/z 381.2027 appropriate for the molecular formula of C19H29N2O6 (7 degrees of unsaturation). This molecular formula was corroborated by 1H and 13C NMR spectral data (Table 1). The IR absorptions at 1694 and 1664 cm−1 suggested the presence of carbonyl functionalities. One- and 2D NMR data of 1 revealed the presence of 19 carbons, which were assigned to four quaternary sp2 carbons including two oxygenated carbons, four proton-bearing sp2 carbons, six oxygenated sp3 carbons, two aliphatic methylenes and three methyls.
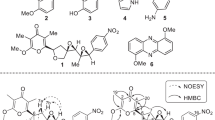
From the 1H-1H COSY spectrum, five proton-bearing fragments were established: H-15/H-16/H-17, H-12/H-13, H-10/H-11, H-7/H-8 and H-5/H-6. The first and the second fragments were joined at C-14 on the basis of HMBC correlations from the methyl protons H-20 to C-13, C-14 and C-15. This fragment was expanded to include a three-carbon fragment C-19/C-18/C-21 on the basis of HMBC correlations from H-19 and H-21 to C-17 and C-18, and to one another, providing a geranyl moiety. The third and the fourth fragments were connected at C-9 by HMBC correlations from H-8 to C-9 and C-10, H-9 to C-8 and C-11, and H-10 and H-11 to C-9, establishing connectivity from C-7 to C-11. Furthermore, HMBC correlations from H-7 to C-10 and H-10 to C-7 connected C-7 and C-10 via an ether linkage, to confirm a pentofuranosyl residue. The last fragment H-5/H-6 was extended to include two carbonyl carbons C-2 and C-4, and the latter carbon was connected to C-5 by HMBC correlations from H-5 and H-6 to C-4. As the molecular formula suggested the presence of two nitrogen atoms in 1, analysis of 1H-15N long-range correlations became necessary to locate their positions. In the 1H-15N HMBC spectrum, both H-5 and H-6 showed a cross-peak to a nitrogen at 156.3 p.p.m. and H-5 to a nitrogen at 155.7 p.p.m. These correlation data, together with 13C and 15N chemical shifts, established this chromophore as pyrimidine-2,4(1H,3H)-dione, namely uracil. Finally, three substructures were joined by analysis of HMBC data. HMBC correlations from H-11 and H-12 to one another connected the geranyl group at C-11 of the pentose unit via an ether linkage, whereas the uracil moiety was connected N-1 to the anomeric carbon (C-7) of the pentose unit by HMBC correlations from H-7 to C-2 and C-6, and H-6 to C-7. Thus, the gross structure of 1 was determined as shown in Figure 2.

COSY and key HMBC correlations for 1.
As the signal overlapping of H-8, H-9 and H-10 in the 1H NMR spectrum did not allow the stereochemical assignment of the pentose unit by NOE and J-based analysis, 1 was derivatized to bis-p-bromobenzoate (2) by the treatment with p-bromobenzoyl chloride in pyridine. The above mentioned proton signals were separated each other in the 1H NMR spectrum of 2. NOESY correlations were detected for H-8/H-9, H-7/H-10, H-2/H-8 and H-9/H-11 (Figure 3). These correlation data allowed the assignment of the pentose unit to possess a ribose configuration, thereby establishing 1 as 5′-O-geranyluridine.

Key NOESY correlations for 2. A full color version of this figure is available at The Journal of Antibiotics journal online.
To determine the absolute configuration of the ribose unit, 1 was synthesized starting from commercially available natural (+)-enantiomer of uridine in three steps. The cis-diol of uridine was protected as dimethylacetonide,8 which was then O-alkylated using geranyl bromide in the presence of NaH to give 4.9 Geranylation at the 5-position of ribose was confirmed by an HMBC correlation from H-12 to C-11. Additional evidence for the alkylation site was the HMBC correlations from 3-NH (δH 9.58) to C-4 (δC 163.6) and C-5 (δC 102.0). The acetonide protective group was removed by treating 4 in HCl to afford 1 (Scheme 1). Optical rotation of the synthetic 1 was [α]D24 +16 (c 0.1, CHCl3), which was in good accordance with that of natural 1, [α]D24 +13 (c 0.1, CHCl3). The absolute configuration of 1 was thus determined as depicted in Figure 1.
Compound 1 was not active against Staphylococcus aureus, Escherichia coli and Candida albicans in the antimicrobial assay (MIC >50 μg ml−1). It was also inactive in cytotoxicity assay (IC50 >50 μM against MCF7 human breast cancer cells). However, 1 was found to induce differentiation of murine ST-13 preadipocyte cells into matured adipocyte cells.10 Maturation of adipocytes leads to the secretion of adiponectin, which downregulates the blood glucose level.11 About 50% of preadipocytes were differentiated to adipocytes with accumulation of lipid droplets by the treatment with 40 μM of 1 for 11 days (Figure 4). JBIR-68 in which the double bond of uridine of 1 is hydrogenated was reported to inhibit influenza proliferation12 but we had no access to this assay.

Adipocyte differentiation induced by 1. A full color version of this figure is available at The Journal of Antibiotics journal online.
Conclusion
In summary, we found 1, a new nucleoside modified by O-prenylation with a geranyl group from a soil-derived Streptomyces by UV-spectroscopic screening. Prenylation of nucleosides is very rare in nature. To date, JBIR-68 (Takagi et al.12) and farnesides A and B13 are known as a secondary metabolite of Streptomyces. JBIR-68 is a dihydrouridine derivative in which the hydroxy group at five-position of ribose is modified with a geranyl group. Farnesides are also dihydrouridine derivatives but an oxygenated farnesyl group is connected to the hydroxy group at five-position of the ribose moiety.
Experimental procedure
General experimental procudures
Optical rotations were measured using a Jasco P-1030 polarimeter (JASCO Corporation, Tokyo, Japan). UV spectra were recorded on a Hitachi U-3210 spectrophotometer (Hitachi-High-Technologies Co., Tokyo, Japan). IR spectra were recorded on a Shimadzu FT-IR-300 spectrophotometer (Shimadzu Corp., Kyoto, Japan). NMR spectra were obtained on a Bruker AVANCE II 500 spectrometer (Bruker Biospin K. K., Yokohama, Japan). HR-FAB-MS and HR-ESI-MS were measured on a JEOL JMS-HX110A spectrometer (JEOL Ltd., Tokyo, Japan) and a Bruker micrOTOF (Bruker Daltonics K. K., Yokohama, Japan), respectively.
Microorganism
The actinomycete, Streptomyces sp. TP-A0872, was isolated from a soil sample collected in Kochi, Japan. The isolated strain was identified as Streptomyces on the basis of 99.9% similarity in the 16S rRNA gene sequence (1463 nucleotides; DDBJ accession number LC192168) to Streptomyces xanthophaeus NRRL B-5414T (accession number JOFT01000080).
Fermentation
Strain TP-A0872 cultured on a slant agar medium was inoculated into 500 ml K-1 flasks, each containing 100 ml of the seed medium consisting of soluble starch 1%, glucose 0.5%, NZ-case 0.3%, yeast extract (Kyokuto Pharmaceutical Industrial, Co., Ltd, Tokyo, Japan) 0.2%, Tryptone (Difco Laboratories, Sparks, MD, USA) 0.5%, K2HPO4 0.1%, MgSO4·7H2O 0.05% and CaCO3 0.3% (pH 7.0). The flasks were cultivated on a rotary shaker (200 r.p.m.) at 30 °C for 4 days. The seed culture (3 ml) was transferred into 500 ml K-1 flasks each containing 100 ml of the production medium consisting of glucose 0.5%, glycerol 2%, soluble starch 2%, Pharmamedia (Traders Protein) 1.5%, yeast extract 0.3% and Diaion HP-20 (Mitsubishi Chemical Co., Yokohama, Japan) 1%. The pH of the medium was adjusted to 7.0 before sterilization. The inoculated flasks were cultured on a rotary shaker (200 r.p.m.) at 30 °C for 7 days.
Extraction and isolation
The whole culture broth of strain TP-A0872 (3 l) was extracted with 1-butanol (100 ml per flasks) on a rotary shaker (200 r.p.m.) for 1 h. The mixture was centrifuged at 6000 r.p.m. for 10 min and the organic layer was separated from the aqueous layer containing the mycelium. Evaporation of the solvent in vacuo provided 5.23 g of extract, which was subjected to silica gel column chromatography with a step gradient of CHCl3-MeOH (16:1, 8:1, 4:1, 2:1, 1:1 and 0:1 v/v). Concentration of fraction 2 (CHCl3-MeOH=8:1) provided 1.1 g of brown powders, which was further purified by HPLC separation with a gradient of MeCN-0.15% KH2PO4 buffer (pH 3.5) (0–6 min: 40%; 6–20 min: 40–50%; 15 ml min−1). Compound 1 was eluted at 17.8 min. Fraction was pooled and concentrated under reduced pressure and the remaining aqueous solution was extracted with EtOAc. The organic layer was concentrated to give 1 (8.8 mg) as a colorless solid.
Simamycin (5-O′-geranyluridine, 1)
Colorless needles; mp 95–96 °C; [α]D24 +13 (c 0.10, CHCl3); UV λmax (log ɛ) 263 nm (3.90); IR (KBr) νmax 3377, 1694, 1664 cm−1; For 1H and 13C NMR data, see Table 1. 15N NMR (CDCl3) δ 156.3 (N-1), 155.7 (N-3) (chemical shifts were indirectly determined from the projection of 1H-15N HMBC spectrum); HR-FABMS [M+H]+ m/z 381.2027 (calcd for C19H30N2O6, 381.2025).
Bis-p-bromobenzoate of 1 (2)
To a solution of 1 (3.0 mg, 7.9 μmol) in dry CH2Cl2 (1.2 ml) and dry pyridine (0.6 ml) were added N,N-dimethyl-4-aminopyridine (a trace amount) and p-bromobenzoyl chloride (3.5 mg, 16 μmol) at 0~5 °C. After stirring for 20 h at room temperature, the reaction mixture was diluted with ice water and extracted with EtOAc. The organic layer was successively washed with sat. CuSO4 solution, sat. NaHCO3 solution and brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane-EtOAc, 10:1~1:1) to give 2 (1.0 mg, 17% yield) as a light yellow solid; 1H NMR (500 MHz, CDCl3) δ 8.16 (1H, s), 5.76 (1H, d, J=8.2 Hz, H-5), 7.99 (1H, d, J=8.2 Hz, H-6), 6.53 (1H, d, J=7.7 Hz, H-7), 5.62 (1H, dd, J=7.7, 5.6 Hz, H-8), 5.73 (1H, m, H-9), 4.48 (1H, br.s, H-10), 3.81 (1H, dd, J=9.4, 1.7 Hz, H-11), 3.72 (1H, dd, J=9.4, 1.7 Hz, H-11), 4.18 (1H, dd, J=11.6, 6.8 Hz, H-12), 4.13 (1H, dd, J=11.6, 7.2 Hz, H-12), 5.37 (1H, t, J=6.5 Hz, H-13), 2.31 (2H, m, H-15), 2.09 (2H, m, H-16), 5.02 (1H, t, J=6.6 Hz, H-17), 1.67 (3H, s, H-19), 1.73 (3H, s, H-20), 1.57 (3H, s, H-21), 7.84 (2H, d, J=8.5 Hz), 7.46 (2H, d, J=8.5 Hz), 7.73 (2H, d, J=8.5 Hz), 7.58 (2H, d, J=8.5 Hz); 13C NMR (125 MHz, CDCl3) δ 150.3 (C-2), 162.4 (C-4), 103.3 (C-5), 140.0 (C-6), 85.9 (C-7), 74.2 (C-8), 73.5 (C-9), 83.1 (C-10), 69.2 (C-11), 67.9 (C-12), 119.4 (C-13), 142.3 (C-14), 39.6 (C-15), 26.4 (C-16), 123.6 (C-17), 131.98 (C-18), 25.7 (C-19), 16.5 (C-20), 17.7 (C-21), 127.3, 127.7, 129.07, 129.13, 131.2, 131.3, 132.0, 132.1, 164.5, 164.8; HR-FABMS [M+Na]+ m/z 767.0580 (calcd for C33H34N2O8Br2Na, 756.0579).
5-O-Geranyl-2,3-O-isopropylidene-1-uracil-β-D-ribofuranoside (4)
To a solution of uridine (700 mg, 2.87 mmol) in dry N,N-dimethylformamide (70 ml) were added 2,2-dimethoxypropane (7 ml, 57 mmol) and p-toluenesulfonic acid monohydrate (70 mg, 0.37 mmol) at room temperature. After stirring for 18 h, the reaction was quenched by adding saturated NaHCO3 solution to the reaction mixture until the pH became 7.0. The reaction mixture was diluted with distilled water and loaded onto an HP-20 column and the column was washed with distilled water. The column was then eluted with MeOH and the eluent was concentrated under reduced pressure to give 2,3-O-isopropylidene-1-uracil-β-D-ribofuranoside (3, 500 mg, 61% yield). Spectroscopic data were in good agreement with reported values.9
To a suspension of 3 (120 mg, 0.42 mmol) in a mixture of dry benzene (1.45 ml) and dry 1,4-dioxane (0.45 ml) was added NaH (40% purity, 60 mg, 2.5 mmol) under argon atmosphere. The mixture was heated at 80~100 °C for 30 min with stirring. After cooling to the ambient temperature, geranyl bromide (0.17 ml, 0.89 mmol) was added and the reaction mixture was heated at 90~95 °C for 3 h. The reaction was quenched by adding water at 0~5 °C and the mixture was extracted with EtOAc. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was applied to silica gel column chromatography (n-hexane:EtOAc=16:1~1.1) to give 4 (39 mg, 22% yield): 1H NMR (500 MHz, CDCl3): δ 9.58 (1H, s), 7.68 (1H, d, J=8.0 Hz), 5.95 (1H, d, J=3.0 Hz), 5.67 (1H, d, J=8.0 Hz), 5.29 (1H, t, J=5.5 Hz), 5.06 (1H, t, J=5.5 Hz), 4.79 (1H, dd, J=6.0, 3.0 z), 4.78 (1H, dd, J=4.0, 2.5 Hz), 4.39 (1H, dd, J=3.5, 3.0 Hz), 4.03 (1H, d, J=7.0 Hz), 3.69 (1H, dd, J=8.0, 2.5 Hz), 3.60 (1H, dd, J=8.0, 3.5 Hz), 2.09 (2H, m), 2.05 (2H, m), 1.68 (3H, s), 1.66 (3H, s), 1.06 (3H, s), 1.59 (3H, s), 1.36 (3H, s); 13C NMR (125 MHz, CDCl3) δ 16.5, 17.7, 25.3, 25.7, 26.3, 27.2, 39.6, 67.8, 69.8, 81.8, 85.1, 85.6, 92.6, 102.0, 114.1, 119.9, 123.7, 131.8, 141.1, 141.3, 150.3, 163.6; HR-ESITOFMS [M+Na]+ 443.2153 (calcd for C22H32N2O6Na, 443.2153).
Synthetic 1
A stirred solution of 4 (9.4 mg, 22 μmol) in 95% EtOH (3 ml) and 0.1 M HCl (1 ml) was heated at 70~80 °C for 4 h. After cooling to the ambient temperature, 0.1 M NaOH solution was added to adjust the pH to 7.0. The reaction mixture was then extracted with EtOAc and the organic layer was dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was purified by preparative HPLC in the same manner described for natural simamycin to yield 6.3 mg of 1 (74% yield): colorless solid; [α]24D +16 (c 0.10, CHCl3); 1H and 13C NMR data were in good agreement with those for natural 1; HR-ESITOFMS [M+Na]+ 403.1840 (calcd for C19H28N2O6Na, 403.1839).
Biological assays
Adipocyte differentiation assay10, antimicrobial assay14 and cytotoxic assay14 were carried out according to the procedures previously described.

Synthesis of 1. Reagents: (a) 2,2-dimethoxypropane, TsOH; (b) geranylbromide, NaH; (c) dil HCl.
References
Bérdy, J. Bioactive microbial metabolites. J. Antibiot. 58, 1–26 (2005).
Ōmura, S. et al. Genome sequence of an industrial microorganism Streptomyces avermitilis: deducing the ability of producing secondary metabolites. Proc. Natl Acad. Sci. USA 98, 12215–12220 (2001).
Ohnishi, Y. et al. Genome sequence of the streptomycin-producing microorganism Streptomyces griseus IFO 13350. J. Bacteriol. 190, 4050–4060 (2008).
Igarashi, Y., Iida, T., Yoshida, R. & Furumai, T. Pteridic acids A and B, novel plant growth promoters with auxin-like activity from Streptomyces hygroscopicus TP-A0451. J. Antibiot. 55, 764–767 (2002).
Igarashi, Y. et al. Absolute configuration of pterocidin, a potent inhibitor of tumor cell invasion from a marine-derived Streptomyces. Tetrahedron Lett. 53, 654–656 (2012).
Igarashi, Y. et al. Alchivemycin A, a bioactive polycyclic polyketide with an unprecedented skeleton from Streptomyces sp. Org. Lett. 12, 3402–3405 (2010).
Igarashi, Y. et al. Jomthonic acid A, a modified amino acid from a soil-derived Streptomyces. J. Nat. Prod. 75, 986–990 (2012).
Zhang, W. et al. Nine enzymes are required for assembly of the pacidamycin group of peptidyl nucleoside antibiotics. J. Am. Chem. Soc. 133, 5240–5243 (2011).
Holý, A. et al. 5′-O-Alkyl-5-fluorouridines: synthesis and biological activity. Collect. Czech. Chem. Commun. 52, 1589–1608 (1987).
Kunimasa, K. et al. Identification of nobiletin, a polymethoxyflavonoid, as an enhancer of an adiponectin secretion. Bioorg. Med. Chem. Lett. 19, 2062–2064 (2009).
Kadowaki, T. et al. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J. Clin. Invest. 116, 1784–1792 (2006).
Takagi, M. et al. Anti-influenza virus compound from Streptomyces sp. RI18. Org. Lett. 12, 4664–4666 (2010).
Ilan, E. Z. et al. Farnesides A and B, sesquiterpenoid nucleoside ethers from a marine-derived Streptomyces sp., strain CNT-372 from Fiji. J. Nat. Prod. 76, 1815–1818 (2013).
Igarashi, Y. et al. Maklamicin, an antibacterial polyketide from an endophytic Micromonospora sp. J. Nat. Prod. 74, 670–674 (2011).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This paper is dedicated to Professor Dr Satoshi Ōmura for his Nobel Prize in Physiology or Medicine 2015.
Rights and permissions
About this article

Cite this article
Igarashi, Y., Kyoso, T., Kim, Y. et al. Simamycin (5′-O-geranyluridine): a new prenylated nucleoside from Streptomyces sp.. J Antibiot 70, 607–610 (2017). https://doi.org/10.1038/ja.2016.163
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2016.163
This article is cited by
-
Engineering nucleoside antibiotics toward the development of novel antimicrobial agents
The Journal of Antibiotics (2019)
