
Abstract
The adenylation domain of nonribosomal peptide synthetase (NRPS) is responsible for the selective substrate recognition and its activation (as an acyl-O-AMP intermediate) during ATP consumption. DhbE, a stand-alone adenylation domain, acts on an aromatic acid, 2,3-dihydroxybenzoic acid (DHB). This activation is the initial step of the synthesis of bacillibactin that is a high-affinity small-molecule iron chelator also termed siderophore. Subsequently, the activated DHB is transferred and attached covalently to a peptidyl carrier protein domain via a thioester bond. Adenylation domains belong to the superfamily of adenylate-forming enzymes including acetyl-CoA synthetase, acyl-CoA synthetase and firefly luciferase. We previously reported a novel N-acylation reaction for an acyl-CoA synthetase (AcsA) that originally catalyzes the formation of a thioester bond between an acid and CoA, yielding acyl-CoA. This novel reaction was also confirmed for acetyl-CoA synthetase and firefly luciferase, but not yet for an adenylation domain. Here, we for the first time demonstrated the synthesis of N-acyl-L-cysteine by a stand-alone adenylation domain, DhbE. When DHB and L-cysteine were used as substrates of DhbE, N-DHB-L-cysteine was formed. A Vmax value of 0.0156±0.0008 units mg−1 and Km values of 150±18.3 mM for L-cysteine and 0.0579±0.0260 mM for DHB were obtained in this novel reaction. Furthermore, DhbE synthesized N-benzoyl-L-cysteine when benzoic acid and L-cysteine were used as substrates. Through the N-acylation reaction of DhbE, we also succeeded in the synthesis of N-aromatic acyl compounds that have never previously been reported to be produced by this enzymatic method.
Similar content being viewed by others

Introduction
Bacillibactin (2,3-dihydroxybenzoate-glycine-threonine trimeric ester) is a high-affinity small-molecule iron chelator also termed siderophore that serves as an iron scavenger to be subjected to iron limitation.1, 2, 3 In many pathogenic microorganisms, siderophores act as virulence factors.4, 5 Bacillibactin is synthesized by nonribosomal peptide synthetases (NRPSs). NRPSs are often involved in the synthesis of numerous secondary metabolites, mainly small peptides that could be antimicrobial, antiviral, immunosuppressant and cytostatic agents of great pharmaceutical potential.6, 7, 8 In contrast to ribosomal peptide synthesis, individual NRPSs produce a single product. NRPSs are often large multienzyme complexes that are organized into modules, each module being further subdivided into a series of domains that catalyze individual steps of nonribosomal peptide synthesis. In general, a minimal module consists of an adenylation domain (that forms the aminoacyl-O-AMP intermediate from an amino acid), a peptidyl carrier protein (PCP) domain (that is, a thiolation domain) (to which the amino acid derived from the corresponding aminoacyl-O-AMP intermediate is tethered to form a thioester intermediate) and a condensation domain (that forms peptide/amide bonds during peptide chain elongation).9
In bacillibactin biosynthesis, 2,3-dihydroxybenzoic acid (DHB) is first activated by the stand-alone adenylation domain (DhbE10), and subsequently the DHB is transferred to the PCP domain of DhbB to yield PCP/DhbB. DHB is covalently attached to 4′-phosphopantetheine in PCP/DhbB via a thioester bond (DHB-S-PCP/DhbB) (Figure 1a). In contrast, glycine and L-threonine also bind to the corresponding PCP domains of DhbF (Gly-S-PCP1/DhbF or Thr-S-PCP2/DhbF) through the actions of the corresponding adenylation domains of DhbF. DHB-Gly-S-PCP1/DhbF and subsequent DHB-Gly-Thr-S-PCP2/DhbF are produced by the condensation domain of DhbF (Supplementary Figure S1).

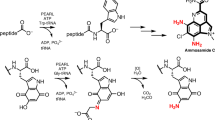
Comparison of original reaction of DhbE involved in the NRPS system (a) with N-DHB-L-Cys synthesis by DhbE (b). In the original reaction, DhbE (adenylation domain) produces 2,3-dihydroxybenzoyl-O-AMP, followed by the formation of DHB-S-DhbB with a covalent thioester bond between DHB and the phosphopantetheinyl prosthetic group of 4′-phosphopantetheine bound to DhbB. DHB, 2,3-dihydroxybenzoic acid; NRPS, nonribosomal peptide synthetase.
The adenylation domains of NRPSs belong to the superfamily of adenylate-forming enzymes.10, 11, 12 We previously reported that acyl-CoA synthetase (AcsA),13 which is also a member of the superfamily of adenylate-forming enzymes, plays an essential role in acid utilization in the nitrile-degrative pathway14, 15, 16 of Pseudomonas chlororaphis B23. The reaction mechanism and structures of acyl-CoA synthetase have been extensively investigated.17, 18, 19 During studies on the characterization of AcsA, however, we discovered a new reaction. When L-cysteine was used as the substrate instead of CoA, N-acyl-L-cysteine was surprisingly detected as the reaction product.20 This finding demonstrated that the enzyme forms a carbon-nitrogen bond involving the carboxyl group of the acid and the amino group of L-cysteine.
Adenylation domains are known to be responsible for selective substrate recognition21 and formation of acyl-AMP as an intermediate during ATP consumption.12 Thus, we here examined the enzyme reaction of one of the NRPS stand-alone adenylation domains, DhbE, using L-cysteine instead of the physiological substrate DhbB. Liu and Bruner22 initially suggested aminoacylation by an adenylation domain. More recently, Maruyama et al.23 reported amide bond formation by the stand-alone adenylation domain involved in streptothricin biosynthesis. However, N-acylation of L-cysteine has never been reported. We discuss the differences in these mechanisms among their reactions reported and our proposed reaction.
Materials and Methods
Materials
L-Cysteine was purchased from Sigma-Aldrich (St Louis, MO, USA). ATP, AMP and 2-hydroxybenzoic acid were purchased from Kishida Chemical (Osaka, Japan). Benzoic acid, 4-methylbenzoic acid, 4-cyanobenzoic acid, phthalic acid, propionic acid, butyric acid, 3-mercaptopropionic acid and L-amino acids (glycine, L-alanine, L-valine, L-leucine, L-isoleucine, L-serine, L-threonine, L-methionine, L-aspartic acid, L-asparagine, L-glutamic acid, L-glutamine, L-arginine, L-lysine, L-histidine, L-phenylalanine, L-tyrosine, L-tryptophan and L-proline) were purchased from Nacalai Tesque (Kyoto, Japan). 3-Hydroxybenzoic acid, 3-cyanobenzoic acid, 2,6-difluorobenzoic acid and pentanoic acid were purchased from Tokyo Chemical Industry (Tokyo, Japan). Other reagents were purchased from Wako Pure Chemical Industries (Osaka, Japan).
Plasmids, strains and medium
Escherichia coli DH10B (Invitrogen, Carlsbad, CA, USA) was used as the host for pUC1824 or pET-24a(+) (Novagen, Madison, WI, USA) derivatives. E. coli BL21-CodonPlus (DE3)-RIL (Stratagene, La Jolla, CA, USA) was used as the host for the pET-dhbE plasmid. E. coli transformants were grown in 2 × YT medium.25
Preparation of DhbE
DNA fragments of the dhbE gene were amplified by PCR using chromosomal DNA of Bacillus subtilis strain 168 as a template. dhbE consists of 1620 nucleotides, encoding 539 amino acid residues with a calculated molecular mass of 59 928 Da. The following pair oligonucleotide primers were used: 5′-AGAATTCCATATGCTGAAAGGATTTACGCCTTG-3′ (NdeI recognition site is underlined) and 5′-TTCTCGAGTTTTTTAAATCCTGCAAGAAGC-3′ (XhoI recognition site is underlined) for dhbE. The PCR products were subcloned into vector pUC18 and checked by DNA sequencing. The resultant plasmid was designated as pUC-dhbE. To produce C-terminally histidine6-tagged DhbE, pUC-dhbE was digested with XhoI completely and then with NdeI partially. The 1.6-kbp fragment harboring the entire coding region of dhbE was also inserted into the corresponding sites of pET-24a(+). The resultant plasmid was designated as pET-dhbE.
E. coli BL21-CodonPlus(DE3)-RIL was transformed with pET-dhbE. The cells transformed with pET-dhbE were grown in 2 × YT medium containing kanamycin (50 μg ml−1) and chloramphenicol (34 μg ml−1) at 37 °C. When A600 nm reached 0.6, the incubation temperature was reduced to 24 °C, and protein expression was induced with 0.01 mM isopropyl-β-D-thiogalactoside. After 24 h of culture, the cells were harvested by centrifugation, washed twice and then suspended in 20 mM sodium phosphate buffer (pH 7.4) containing 5 mM 1,4-dithiothreitol and 1 mM EDTA. The resuspended cells were sonicated, and the cell debris was removed by centrifugation (27 000 g, 30 min). The resulting supernatant was applied to a Ni-chelating column of HisTrap HP (5 ml) (GE Healthcare UK Ltd., Buckinghamshire, UK) and washed with 20 mM sodium phosphate buffer (pH 7.4) containing 0.5 M NaCl. DhbE was eluted with a linear gradient (0.01–0.4 M) of imidazole. The eluted fractions were collected and dialyzed, and then the proteins in the fraction were separated with a HiLoad 16/60 Superdex 200 pg column (120 ml) (GE Healthcare UK Ltd.) equilibrated with 20 mM Tris-HCl buffer (pH 7.5) containing 0.15 M NaCl. The fractions around the 56 kDa size peak were collected and dialyzed with 50 mM HEPES buffer (pH 7.5). The homogeneity of the purified protein was confirmed by SDS–polyacrylamide gel electrophoresis (SDS-PAGE). The NH2-terminal sequence of the purified DhbE determined with a model 490 Procise protein sequencer (Applied Biosystems) was consistent with that deduced from the DNA sequence. This purified DhbE was used in all experiments in this study.
Identification of the reaction products (N-aromatic acyl compounds)
N-aromatic acyl-L-cysteines
The reaction mixture, comprising 10 mM DHB, 400 mM L-cysteine, 5 mM ATP, 8 mM MgCl2, 200 mM (NH4)2SO4 and 0.22 mg ml−1 of the purified enzyme (DhbE) in 200 mM HEPES buffer (pH 7.5), was incubated at 28 °C and the reaction was stopped by adding an equal volume of methanol to the reaction mixture. Then, the mixture was analyzed by HPLC on a Cosmosil 5C18-MS-II column (4.6 × 150 mm; Nacalai Tesque, Kyoto, Japan). The mobile-phase solvent comprised 0.1% formic acid containing 30% (v/v) methanol and chromatographic separation was performed at 30 °C at the flow rate of 1.0 ml min−1. A new product peak (N-2,3-dihydroxybenzoyl-L-cysteine, retention time 9.5 min) other than that of AMP was determined by monitoring the column effluent at 254 nm. The product peak material was fractionated and used for LC-MS/MS analysis. LC-MS/MS analysis data were acquired with the Waters (Milford, MA, USA) Micromass Q-Tof premier mass spectrometer in the positive and negative modes. The capillary voltage was 2.5 kV and the cone voltage was 20 V. The collision energy was adjusted to 12 V. N-benzoyl-L-cysteine was identified by the same method.
S-DHB-3-mercaptopropionic acid
The reaction mixture, comprising 10 mM DHB, 100 mM 3-mercaptopropionic acid, 5 mM ATP, 8 mM MgCl2, 100 mM (NH4)2SO4 and 0.25 mg ml−1 of the purified enzyme (DhbE) in 200 mM Tris-HCl buffer (pH 8.5), was incubated at 28 °C. Then, the mixture was subjected to LC-MS/MS analysis. The analysis was carried out using a Shimadzu (Kyoto, Japan) LCMS-8030 equipped with a Shimadzu Nexera HPLC system and a Develosil RPAQUEOUS column (2.0 × 150 mm; Nomura Chemical Co., Ltd., Aichi, Japan). The mobile phases for elution were as follows: mobile phase A, 10 mM ammonium acetate; and mobile phase B, acetonitrile. Samples were eluted at 25 °C at a flow rate of 0.2 ml min−1 using a linear gradient of 0–20% of mobile phase B over the course of 15 min. The composition of 20% mobile phase B was held for 5 min from 15 to 20 min, and then the final composition of 0% mobile phase B was held for 5 min from 20 to 25 min.
Enzyme assays
Amide bond synthetic activities were assayed by measuring acid-dependent AMP formation. Although the ATP-[32P]PPi exchange assay26 or the colorimetric assay27 has been used for measuring the specific activity of adenylating enzymes (involved in the reaction ATP→AMP+PPi), these assays are based upon indirect quantification of acyl-AMP or PPi, respectively. In our study, therefore, a modified method to measure directly the amount of AMP by HPLC28 was used, because the HPLC method allows easier and more accurate quantification than the methods reported previously. The reaction mixture, comprising 0.05–5 mM DHB, 0.5–500 mM L-cysteine, 0.1–30 mM ATP, 8 mM MgCl2, 200 mM (NH4)2SO4 and 0.22 mg ml−1 of the purified enzyme in 100 mM Tris-HCl buffer (pH 8.5), was incubated at 45 °C for 15 min. The reaction was stopped by adding an equal volume of methanol to the reaction mixture, and then the mixture was analyzed by HPLC. AMP and ATP in each reaction series were separated on a Titansphere TiO HPLC column (4.6 × 150 mm; GL Science, Tokyo, Japan). The mobile-phase solvent comprised 50 mM potassium phosphate buffer (pH 6.0) containing 60% (v/v) acetonitrile and chromatographic separation was performed at 40 °C at the flow rate of 1 ml min−1, with monitoring at 260 nm. One unit was defined as the amount of enzyme that catalyzed the formation of 1 μmol AMP per min under these assay conditions. kcat values were calculated using a Mr of 60 993 for DhbE.
Substrate specificity
The aromatic acids or fatty acids listed in Table 1 (10 mM) and 10 mM L-cysteine were examined as to substrate specificity for the synthesis of N-aromatic acyl- or N-fatty acyl-L-cysteines. The L-amino acids listed in the Materials section or 3-mercaptopropionic acid (10 mM) and 10 mM DHB were examined as to substrate specificity for the synthesis of the corresponding reaction compounds. The increase in the amount of AMP during the enzymatic reaction was determined by HPLC using the assay described under the section Enzyme assays.
Analytical methods
Protein concentrations were measured with a Nacalai Tesque protein assay kit with bovine serum albumin as the standard. SDS-PAGE was performed in an 11.5% polyacrylamide slab gel. The gel was stained with Coomassie brilliant blue R-250. The molecular mass of the enzyme was calculated from the mobilities of the marker proteins, myosin (200 kDa), β-galactosidase (116 kDa), bovine serum albumin (66 kDa), ovalbumin (45 kDa), carbonic anhydrase (31 kDa) and soybean trypsin inhibitor (21.5 kDa).
Results
Expression and purification of DhbE, a stand-alone adenylation domain of bacillibactin synthetase
An expression plasmid (pET-dhbE) was constructed and introduced into E. coli BL21-CodonPlus(DE3)-RIL. After the E. coli transformant had been cultured for 24 h at 24 °C, a cell-free extract was prepared. C-Terminally histidine6-tagged DhbE (547 amino acid residues) was purified by Ni-chelating column chromatography. Although the fraction containing the C-terminally histidine6-tagged DhbE gave a single band corresponding to a molecular mass of 61 kDa on SDS-PAGE, determination of the N-terminal amino acid sequence revealed that the fraction contained two proteins. One protein had an N-terminal sequence of MLKGFTPWP, which is the same as that deduced from the DNA sequence, and the other one of FVESFPQTG. This finding indicated that a part of DhbE was cleaved at amino acid residue 507. Therefore, we examined whether the addition of 1,4-dithiothreitol (5 mM), EDTA (1 mM), diisopropyl fluorophosphates (0.5 mM) and combinations of these compounds to cell-free extracts resulted in inhibition of the enzyme degradation. Because the addition of both 1,4-dithiothreitol and EDTA inhibited the cleavage of DhbE effectively, DhbE was simply purified as described in the Materials and methods section. The purity of DhbE was confirmed by migration of the protein as a single band corresponding to a molecular mass of 61 kDa on SDS-PAGE (Figure 2). The purified DhbE had an N-terminal sequence of MLKGFTPWP.

SDS–polyacrylamide gel electrophoresis (SDS-PAGE) of the purified DhbE protein. Protein bands were detected by staining with Coomassie brilliant blue. Lane M, marker proteins; lane DhbE, purified DhbE (1 μg).
Amide compound synthesis by DhbE
DhbE uses DHB and 4′-phosphopantetheine in DhbB as substrates, and forms a thioester bond involving the carboxyl group of DHB and the thiol group of 4′-phosphopantetheine. Therefore, we examined whether DhbE could catalyze the N-acyl-L-cysteine (amide compound) synthetic reaction or not when L-cysteine was used instead of DhbB as a substrate. The reaction mixture comprising DhbE, ATP, DHB and L-cysteine was incubated, and the reaction was stopped by the addition of methanol to the reaction mixture. The resultant mixture was analyzed by HPLC on a Titansphere TiO HPLC column, a significant increase in AMP with consumption of ATP being observed (Figure 3a). Because DhbE is an adenylating enzyme (involved in the following reaction: ATP→AMP+PPi), the increase in AMP indicates that DhbE catalyzes the above N-acylation reaction. When the same reaction mixture was analyzed on a Cosmosil 5C18-MS-II column, a new product peak other than that of AMP appeared (Figure 3b); no other new peaks were detected. When DhbE was not added to the reaction, on the contrary, the peaks of AMP and the above new product were not detected. We purified the new product according to the procedure described in the Materials and methods section, and analyzed it by LC-MS. The determined molecular mass of the product was 257 Da, indicating that the new product is N-2,3-dihydroxybenzoyl-L-cysteine (N-DHB-L-Cys) or S-2,3-dihydroxybenzoyl-L-cysteine (S-DHB-L-Cys). To determine whether the product is N-DHB-L-Cys or S-DHB-L-Cys, we then performed LC-MS/MS analyses. A fragment ion at m/z 178 (Figure 4a, negative ion mode) derived from the precursor ion at m/z 256 was observed, suggesting that the new product is N-DHB-L-Cys (Figure 1b), but not S-DHB-L-Cys.

HPLC chromatogram of the reaction mixture including 2,3-dihydroxybenzoic acid (DHB), L-cysteine and DhbE. The reaction mixture was separated with a Titansphere TiO HPLC column (a) or a Cosmosil 5C18-MS-II column (b).

MS/MS spectra of N-DHB-L-Cys (a) and N-benzoyl-L-cysteine (b) formed by DhbE in the positive (P) and negative (N) ion modes. The structural formulae show N-DHB-L-Cys (a) and N-benzoyl-L-cysteine (b). The theoretical m/z values of the fragments are provided with the structures. The fragment ion at m/z 178 or 222 (a, negative ion mode) suggests that the product is not S-DHB-L-Cys but N-DHB-L-Cys. The fragment ion at m/z 146 (b, negative ion mode) suggests that the product is not S-benzoyl-L-cysteine but N-benzoyl-L-cysteine. CE, collision energy; DHB, 2,3-dihydroxybenzoic acid.
Characteristics of N-DHB-L-cysteine production by DhbE
The Km and Vmax values of the N-DHB-L-Cys synthetic activity for L-cysteine, DHB and ATP were calculated from Michaelis–Menten kinetics. A Vmax value of 0.0156±0.0008 units mg−1, and Km values of 150±18.3 mM for L-cysteine, 0.0579±0.0260 mM for DHB and 0.320±0.0705 mM for ATP were obtained. There was a significant difference between the Km values for DHB and L-cysteine.
The effects of temperature and pH on N-DHB-L-Cys synthesis were also examined. The reaction mixture comprised 10 mM DHB, 10 mM L-cysteine, 5 mM ATP, 8 mM MgCl2, 200 mM (NH4)2SO4 and 0.22 mg ml−1 of DhbE in 100 mM buffer. The optimal reaction temperature of DhbE for N-DHB-L-Cys synthesis appeared to be 50 °C when the reaction was carried out for 15 min, as shown in Figure 5a. The enzyme exhibited maximum activity at pH 9.0 (Tris-HCl) (Figure 5b).

Effects of pH and temperature on the N-DHB-L-Cys synthesis by DhbE. (a) The reactions were carried out for 15 min at various temperatures. (b) The reactions were carried out for 15 min at 45 °C in the following buffers (0.1 M): citrate/sodium citrate (□), PIPES (■), HEPES (○), Tris-HCl (●), CHES (◊) and glycine/NaOH (♦). Relative activity is expressed as a percentage of the maximum activity attained under the experimental conditions used.
Synthesis of various N-aromatic acyl- or N-fatty acyl-L-cysteines
The ability of DhbE to produce N-aromatic acyl- or N-fatty acyl-L-cysteines from 10 mM various aromatic or fatty acids was examined in a reaction mixture that included 10 mM L-cysteine (Table 1). An increase in AMP was detected when each of the following aromatic acids was utilized as a substrate: 2-hydroxylbenzoic acid (67%), 3-hydroxylbenzoic acid (23%) and benzoic acid (20%) compared with DHB (100%). In the case of the reaction with benzoic acid as an acid substrate, the reaction product other than AMP was separated on a Cosmosil 5C18-MS-II column and analyzed by LC-MS. The molecular mass of the product determined on LC-MS was 225 Da. Furthermore, a fragment ion at m/z 146 (Figure 4b, negative ion mode), which should be derived from only N-benzoyl-L-cysteine, was detected on LC-MS/MS analysis. With regard to fatty acid substrates, an apparent increase in AMP was detected when pentanoic acid (10%), heptanoic acid (7.1%) or octanoic acid (8.2%) was used as a substrate.
Synthesis of S-DHB-3-mercaptopropionic acid
The ability of DhbE to produce the corresponding reaction product from the 10 mM L-amino acids listed under Materials section or 3-mercaptopropionic acid was examined in a reaction mixture containing 10 mM DHB. Although an increase in AMP was not detected when each L-amino acid was utilized as a substrate, an increase in AMP was detected when a thiol compound lacking an amino group (3-mercaptopropionic acid (HS-CH2-CH2-COOH)) was utilized. The activity toward 3-mercaptopropionic acid was 0.00445 units mg−1, being 78% of that toward L-cysteine. Based on that its mass was determined to be 242 Da by LC-MS analysis, and that the fragment ions derived from S-DHB-3-mercaptopropionic acid were detected on LC-MS/MS analysis (Figure 6), we were able to determine the product is S-DHB-3-mercaptopropionic acid.

MS/MS spectra of S-DHB-3-mercaptopropionic acid formed by DhbE in the negative (N) ion mode. The structural formula shows S-DHB-3-mercaptopropionic acid. The theoretical m/z values of the fragments are provided with the structure. CE, collision energy; DHB, 2,3-dihydroxybenzoic acid.
Discussion
Adenylation domains belong to the superfamily of adenylate-forming enzymes, but N-acyl-L-cysteine synthesis by an adenylation domain has never previously been reported. In this study, we initially clarified that DhbE, which was used as one of the adenylation domains, produced N-DHB-L-Cys from DHB and L-cysteine along with ATP consumption and AMP formation. Namely, DhbE was confirmed to catalyze the following reaction:
DHB + L-cysteine + ATP → N-DHB-L-cys + AMP + PPi.
Because N-aromatic acylation by an adenylation domain has never been reported, this is the first report concerning the synthesis of N-aromatic acyl compounds by an adenylation domain.
In bacillibactin synthesis, an amide bond between the carboxyl group of DHB and the amino group of glycine is formed. However, at least three domains (that is, adenylation domain, PCP domain and condensation domain) are required to form the amide bond.29 DhbE, which is the stand-alone adenylation domain, produces 2,3-dihydroxybenzoyl-O-AMP, followed by the formation of DHB-S-PCP/DhbB with a covalent thioester bond between DHB and the phosphopantetheinyl prosthetic group of the PCP domain (Figure 7a). In the same manner, the adenylation domain of DhbF tethers glycine to the PCP domain of DhbF to form Gly-S-PCP1/DhbF. The 2,3-dihydroxybenzoyl group of the thioester intermediate (DHB-S-PCP/DhbB) is attacked by the nitrogen atom (as a nucleophile) of the amino group in Gly-S-PCP1/DhbF, resulting in amide bond formation of DHB-Gly.1, 6 The reaction is catalyzed by the condensation domain of DhbF (Supplementary Figure S1). Therefore, the mechanism for the formation of the amide bond of DHB-Gly by the NRPS system is distinctly different from that of DHB-L-Cys by DhbE that we discovered in this study (Figure 1). Furthermore, for the release of the final product from NRPS multienzyme complexes, a thioesterase domain with hydrolytic cleavage activity is needed.30 Namely, the NRPS system requiring at least four domains to produce an amide compound is very complicated. On the other hand, our new synthesis of amide compounds with only the stand-alone adenylation domain is very simple (Figure 1b), and does not need any thioesterase domains for release of the final product.

Original reactions of members of the superfamily of adenylate-forming enzymes. The substrates of DhbE (adenylation domain) are DHB (carboxylic acid) and PCP domain (a); those of acyl-CoA synthetase are carboxylic acid and CoA (b); and those of luciferase are luciferin and oxygen (c). DHB, 2,3-dihydroxybenzoic acid; ICL, isochorismatase domain; PCP, peptidyl carrier protein.
The Vmax value for the N-DHB-L-Cys synthesis by DhbE (0.0156 units mg−1) was similar to that for the N-acetyl-L-cysteine synthesis by acetyl-CoA synthetase from Saccharomyces cerevisiae (0.0200 units mg−1), but less than one-twelfth that for the N-isobutyryl-L-cysteine synthesis by AcsA (0.197 units mg−1).20 On the other hand, the Km value for L-cysteine of DhbE (150 mM) was quite different from those of both acetyl-CoA synthetase (17.6 mM) and AcsA (71.1 mM). It is possible that the difference of Km values for L-cysteine is due to the structure of the original substrate (4′-phosphopantetheine in PCP domain or CoA).
We found that DhbE utilized not only DHB but also 2- or 3-hydroxybenzoic acid as the substrate (Table 1). Although amide compounds would be synthesized when 2- or 3-hydroxybenzoic acid is used as the substrate, 4-hydroxybenzoic acid has been found to be inert as a substrate. It seems that aromatic acids with a functional group at only the 2 or 3 position are used by DhbE as substrates. Moreover, aromatic acids with a functional group other than a hydroxyl group at the 2 or 3 position were inert as substrates. However, benzoic acid, which is an aromatic acid with no functional group other than a carboxyl group, was utilized (Figure 4b). More interestingly, DhbE showed catalytic activity toward not only aromatic acids but also a straight-chain fatty acid such as pentanoic acid, heptanoic acid or octanoic acid.
We previously reported that acyl-CoA synthetase, AcsA, produces N-acyl-L-cysteine as a product when L-cysteine is used as the substrate instead of CoA.20 Acyl-CoA syntethases originally catalyze the ligation of an acid with CoA through a two-step reaction.19 The initial reaction comprises the formation of an acyl-O-AMP intermediate with the release of PPi, and then the AMP of the acyl-O-AMP intermediate is displaced rapidly by CoA to yield acyl-CoA containing a thioester bond (Figure 7b). Although there had been few reports describing investigation of thiols other than CoA, activity toward L-cysteine as a substrate and identification of the reaction products had never been reported. Firefly luciferase, which belongs to the same superfamily of adenylate-forming enzymes as AcsA and DhbE, has also been shown to produce N-luciferyl-L-cysteine. Luciferase originally catalyzes the oxidation of luciferin in the presence of molecular oxygen and ATP to generate oxyluciferin and light (hν) (Figure 7c).31, 32 Hence, this bioluminescence (monooxygenase) reaction catalyzed by luciferase also differs from the N-luciferyl-L-cysteine synthetic reaction. It is interesting that acyl-CoA synthetase, luciferase and DhbE produced amides from acids and L-cysteine, in spite of the differences between these enzymes in their original reactions (Figure 7).
We have reported the N-acyl-cysteine synthesis by acetyl-CoA synthetase, acyl-CoA synthetase and luciferase, and peptide synthesis by the D-alanine: D-alanyl carrier protein ligase DltA. It has been reported that the three-dimensional structure of DltA resembles that of DhbE,10, 33 but DltA is not an adenylation domain of NRPS. Furthermore, very recently we proposed and confirmed the unprecedented mechanism of amide/peptide synthesis using DltA as follows: (1) the formation of S-acyl-L-cysteine as an intermediate via this ‘enzymatic activity’ and (2) subsequent ‘chemical’ S- to N-acyl transfer in the intermediate, resulting in amide/peptide bond formation.34 Indeed, DhbE synthesized S-DHB-3-mercaptopropionic acid when 3-mercaptopropionic acid lacking an amino group was used as a substrate instead of L-cysteine (Figure 6). Moreover, L-alanine lacking a thiol group was inert as a substrate. These findings suggest that DhbE first synthesizes a thioester intermediate when L-cysteine is used as a substrate. Because DhbE belongs to the superfamily of adenylate-forming enzymes, like acyl-CoA synthetase, luciferase and DltA, the mechanism of N-aromatic acyl-L-cysteine synthesis by DhbE would be identical to theirs.
Liu and Bruner22 first suggested aminoacylation by an adenylation domain. They proposed serine acylation of the amino group in amino-CoA (bound to EntF PCP domain) by an adenylation domain in that report. More recently, Maruyama et al.23 reported amide bond formation by the stand-alone adenylation domain ORF19 of Streptomyces rochei NBRC12908 involved in streptothricin biosynthesis. They showed that ORF19 catalyzes amide bond formation only between L-β-lysine and an L-β-lysine oligopeptide. However, L-cysteine was not utilized as a substrate in the above-mentioned reactions; namely, intramolecular S- to N-acyl transfer of a thioester intermediate does not occur in the reaction mechanisms. Therefore, the mechanism of peptide bond formation by DhbE that we present here is distinctly different from those in the case of the above-mentioned adenylation domains.
We here found the formation of an amide bond involving the α-amino group of an amino acid, and the synthesis of N-aromatic acyl compounds, both of which have never been reported previously, by an enzyme belonging to the superfamily of adenylate-forming enzymes. Some N-acylation compounds are known as biologically valuable agents. For example, N-benzoyl-L-cysteine produced in this study has antioxidant activity,35 and N-acetyl-L-cysteine shows antioxidant activity36 and an antimicrobial effect through destruction of biofilms.37, 38 It is thus possible to synthesize various amides or peptides with biologically active functions by using enzymes belonging to the superfamily of adenylate-forming enzymes.
References
May, J. J., Wendrich, T. M. & Marahiel, M. A. The dhb operon of Bacillus subtilis encodes the biosynthetic template for the catecholic siderophore 2,3-dihydroxybenzoate-glycine-threonine trimeric ester bacillibactin. J. Biol. Chem. 276, 7209–7217 (2001).
Hoffmann, T. et al. High-salinity-induced iron limitation in Bacillus subtilis. J. Bacteriol. 184, 718–727 (2002).
Dertz, E. A., Xu, J., Stintzi, A. & Raymond, K. N. Bacillibactin-mediated iron transport in Bacillus subtilis. J. Am. Chem. Soc. 128, 22–23 (2006).
Barry, S. M. & Challis, G. L. Recent advances in siderophore biosynthesis. Curr. Opin. Chem. Biol. 13, 205–215 (2009).
Miethke, M. & Marahiel, M. A. Siderophore-based iron acquisition and pathogen control. Microbiol. Mol. Biol. Rev. 71, 413–451 (2007).
Finking, R. & Marahiel, M. A. Biosynthesis of nonribosomal peptides. Annu. Rev. Microbiol. 58, 453–488 (2004).
Nguyen, K. T. et al. Combinatorial biosynthesis of novel antibiotics related to daptomycin. Proc. Natl Acad. Sci. USA 103, 17462–17467 (2006).
Galm, U. et al. Comparative analysis of the biosynthetic gene clusters and pathways for three structurally related antitumor antibiotics: bleomycin, tallysomycin, and zorbamycin. J. Nat. Prod. 74, 526–536 (2011).
Hur, G. H., Vickery, C. R. & Burkart, M. D. Explorations of catalytic domains in non-ribosomal peptide synthetase enzymology. Nat. Prod. Rep. 29, 1074–1098 (2012).
May, J. J., Kessler, N., Marahiel, M. A. & Stubbs, M. T. Crystal structure of DhbE, an archetype for aryl acid activating domains of modular nonribosomal peptide synthetases. Proc. Natl Acad. Sci. USA 99, 12120–12125 (2002).
Conti, E., Stachelhaus, T., Marahiel, M. A. & Brick, P. Structural basis for the activation of phenylalanine in the non-ribosomal biosynthesis of gramicidin S. EMBO J. 16, 4174–4183 (1997).
Gulick, A. M. Conformational dynamics in the acyl-CoA synthetases, adenylation domains of non-ribosomal peptide synthetases, and firefly luciferase. ACS Chem. Biol. 4, 811–827 (2009).
Hashimoto, Y. et al. Nitrile pathway involving acyl-CoA synthetase: overall metabolic gene organization and purification and characterization of the enzyme. J. Biol. Chem. 280, 8660–8667 (2005).
Kobayashi, M. & Shimizu, S. Metalloenzyme nitrile hydratase: structure, regulation, and application to biotechnology. Nat. Biotechnol. 16, 733–736 (1998).
Nomura, J. et al. Crystal structure of aldoxime dehydratase and its catalytic mechanism involved in carbon-nitrogen triple-bond synthesis. Proc. Natl Acad. Sci. USA 110, 2810–2815 (2013).
Zhou, Z., Hashimoto, Y., Shiraki, K. & Kobayashi, M. Discovery of posttranslational maturation by self-subunit swapping. Proc. Natl Acad. Sci. USA 105, 14849–14854 (2008).
Starai, V. J., Celic, I., Cole, R. N., Boeke, J. D. & Escalante-Semerena, J. C. Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science 298, 2390–2392 (2002).
Schneider, K. et al. The substrate specificity-determining amino acid code of 4-coumarate:CoA ligase. Proc. Natl Acad. Sci. USA 100, 8601–8606 (2003).
Starai, V. J. & Escalante-Semerena, J. C. Acetyl-coenzyme A synthetase (AMP forming). Cell. Mol. Life Sci. 61, 2020–2030 (2004).
Abe, T., Hashimoto, Y., Hosaka, H., Tomita-Yokotani, K. & Kobayashi, M. Discovery of amide (peptide) bond synthetic activity in acyl-CoA synthetase. J. Biol. Chem. 283, 11312–11321 (2008).
Ehmann, D. E., Shaw-Reid, C. A., Losey, H. C. & Walsh, C. T. The EntF and EntE adenylation domains of Escherichia coli enterobactin synthetase: sequestration and selectivity in acyl-AMP transfers to thiolation domain cosubstrates. Proc. Natl Acad. Sci. USA 97, 2509–2514 (2000).
Liu, Y. & Bruner, S. D. Rational manipulation of carrier-domain geometry in nonribosomal peptide synthetases. Chembiochem. 8, 617–621 (2007).
Maruyama, C. et al. A stand-alone adenylation domain forms amide bonds in streptothricin biosynthesis. Nat. Chem. Biol. 8, 791–797 (2012).
Yanisch-Perron, C., Vieira, J. & Messing, J. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33, 103–119 (1985).
Sambrook, J., Fritsch, E. F. & Maniatis, T. Molecular Cloning: A Laboratory Manual 2nd edn (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, USA, (1989).
Lee, S. G. & Lipmann, F. Tyrocidine synthetase system. Methods Enzymol. 43, 585–602 (1975).
Katano, H., Tanaka, R., Maruyama, C. & Hamano, Y. Assay of enzymes forming AMP+PPi by the pyrophosphate determination based on the formation of 18-molybdopyrophosphate. Anal. Biochem. 421, 308–312 (2012).
Kimura, Y. et al. Microanalysis for MDR1 ATPase by high-performance liquid chromatography with a titanium dioxide column. Anal. Biochem. 326, 262–266 (2004).
Tanovic, A., Samel, S. A., Essen, L. O. & Marahiel, M. A. Crystal structure of the termination module of a nonribosomal peptide synthetase. Science 321, 659–663 (2008).
Trauger, J. W., Kohli, R. M,, Mootz, H. D., Marahiel, M. A. & Walsh, C. T. Peptide cyclization catalysed by the thioesterase domain of tyrocidine synthetase. Nature 407, 215–218 (2000).
Conti, E., Franks, N. P. & Brick, P. Crystal structure of firefly luciferase throws light on a superfamily of adenylate-forming enzymes. Structure 4, 287–298 (1996).
Nakatsu, T. et al. Structural basis for the spectral difference in luciferase bioluminescence. Nature 440, 372–376 (2006).
Osman, K. T., Du, L., He, Y. & Luo, Y. Crystal structure of Bacillus cereus D-alanyl carrier protein ligase (DltA) in complex with ATP. J. Mol. Biol. 388, 345–355 (2009).
Abe, T. et al. Peptide bond synthesis by a mechanism involving an enzymatic reaction and a subsequent chemical reaction. J. Biol. Chem. 291, 1735–1750 (2016).
Fujimoto, A. & Masuda, T. Chemical interaction between polyphenols and a cysteinyl thiol under radical oxidation conditions. J. Agric. Food Chem. 60, 5142–5151 (2012).
Zafarullah, M., Li, W. Q., Sylvester, J. & Ahmad, M. Molecular mechanisms of N-acetylcysteine actions. Cell. Mol. Life Sci. 60, 6–20 (2003).
Dinicola, S., De Grazia, S., Carlomagno, G. & Pintucci, J. P. N-acetylcysteine as powerful molecule to destroy bacterial biofilms. A systematic review. Eur. Rev. Med. Pharmacol. Sci. 18, 2942–2948 (2014).
Ulusoy, A. T., Kalyoncuoğlu, E., Reis, A. & Cehreli, Z. C. Antibacterial effect of N-acetylcysteine and taurolidine on planktonic and biofilm forms of Enterococcus faecalis. Dent. Traumatol. 32, 212–218 (2016).
Acknowledgements
We thank Dr Shinji Kuroda, Dr Shun-Ichi Suzuki and Ms Sachise Eto (Ajinomoto) for the useful discussions and critical advice and Professor Akira Nakamura (University of Tsukuba) for providing the chromosomal DNA of B. subtilis strain 168. This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article

Cite this article
Abe, T., Hashimoto, Y., Sugimoto, S. et al. Amide compound synthesis by adenylation domain of bacillibactin synthetase. J Antibiot 70, 435–442 (2017). https://doi.org/10.1038/ja.2016.117
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2016.117
