
Abstract
The methanol extract of the Vietnamese freshwater cyanobacterium Nostoc sp. CAVN2 exhibited cytotoxic effects against MCF-7 and 5637 cancer cell lines as well as against nontumorigenic FL and HaCaT cells and was active against methicillin-resistant Staphylococcus aureus (MRSA) and Streptococcus pneumoniae. High-resolution mass spectrometric analysis indicated the presence of over 60 putative cyclophane-like compounds in an antimicrobially active methanol extract fraction. A paracyclophanes-focusing extraction and separation methodology led to the isolation of 5 new carbamidocyclophanes (1–5) and 11 known paracyclophanes (6–16). The structures and their stereochemical configurations were elucidated by a combination of spectrometric and spectroscopic methods including HRMS, 1D and 2D NMR analyses and detailed comparative CD analysis. The newly described monocarbamoylated [7.7]paracyclophanes (1, 2, 4 and 5) differ by a varying degree of chlorination in the side chains. Carbamidocyclophane J (3) is the very first reported carbamidocyclophane bearing a single halogenation in both butyl residues. Based on previous studies a detailed phylogenetic examination of cyclophane-producing cyanobacteria was carried out. The biological evaluation of 1–16 against various clinical pathogens highlighted a remarkable antimicrobial activity against MRSA with MICs of 0.1–1.0 μM, and indicated that the level of antibacterial activity is related to the presence of carbamoyl moieties.
Similar content being viewed by others

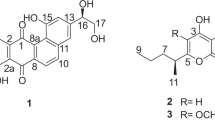
Introduction
Since the first synthesis of di-p-xylylene by Brown and Farthing1 in 1949, and the first description of [m.n]paracyclophanes by Cram and Steinberg2 in 1951, cyclophanes have become a widespread and well-known class of organic molecules in nearly all fields of chemistry.3 Yet, it took almost another four decades until the first naturally occurring [7.7]paracyclophanes with cytotoxic effects against different cancer cell lines, nostocyclophane D and cylindrocyclophane A, have been isolated from the cyanobacterial strains Nostoc linckia (Roth) Bornet (UTEX B1932) and Cylindrospermum licheniforme (ATCC 29204), respectively.4 Subsequently, numerous molecules with a varying substitution pattern of the slightly modified [7.7]paracyclophane skeleton have been isolated and reported from several terrestrial cyanobacteria belonging to the order Nostocales. Besides cylindrocyclophanes A−F,5 A1−A4, C1−C4, F4 and AB4,6 nostocyclophanes A−D7 and merocyclophanes A and B8 with diverse biological effects, cytotoxically and antimicrobially active carbamidocyclophanes A−G9, 10 have been described from the cyanobacteria Nostoc spp. CAVN10 and UIC 10274. In comparison with the cylindrocyclophane/carbamidocyclophane carbon skeleton, the nonhalogenated merocyclophanes possess α-branched methyls at C-1/C-14, and lack in the presence of β-branched methyl groups at C-2/C-15, respectively. Nostocyclophanes contain neither α- nor β-branched methyls, but including exclusively chlorine atoms at C-3 and C-16. Furthermore, nostocyclophane A and B are glycosylated derivatives (see Figure 1a and Supplementary Information S0). The carbamidocyclophane subgroup is characterized by the presence of carbamoyl moieties attached to C-1/C-14 of the [7.7]paracyclophane scaffold. Both mono- and dicarbamoylated carbamidocyclophanes exhibited cytotoxic activity against several tumor cell lines and antimicrobial activity against Gram-positive bacteria; for example, Mycobacterium tuberculosis, Entercoccus faecalis and Staphylococcus aureus.9, 10 A cytotoxicity-guided evaluation of different extracts from various filamentous cyanobacteria revealed a new [7.7]paracyclophane-biosynthesizing strain, the Vietnamese freshwater cyanobacterium Nostoc sp. CAVN2. In this article, we describe an optimized extraction and separation procedure for the detection and isolation of closely related [7.7]paracyclophanes as well as the complete structure elucidation of novel carbamidocyclophanes with a partly hitherto unknown halogen atom distribution and the biological evaluation of all isolates.

Structurally diverse paracyclophanes biosynthesized by Nostoc sp. CAVN2. (a) Carbamidoyclophane and cylindrocyclophane core structure and (b) HPLC-UV chromatogram (λ=228 nm) of the cyclophane-rich solid-phase extraction (SPE) fraction. (c) UV reference spectrum of carbamidocyclophane A.9 The table describes structures and structural proposals belonging to selected peaks of (b).
Results and Discussion
Cytotoxicity screening
Initially, 53 dried extracts from 14 cyanobacterial strains, belonging to the orders Nostocales and Oscillatoriales, were evaluated for cytotoxic activity against several cell lines (Supplementary Information S1–S3). A total of 30 extracts were inactive (IC50 >500 μg ml−1) and 21 exhibited only marginal cytotoxicity (20 μg ml−1 ≤IC50 ≤500 μg ml−1). Merely the ethyl acetate extract of the culture medium and the methanolic biomass extract from Nostoc sp. CAVN2 were found to have significant inhibitory activity against breast adenocarcinoma MCF-7 cells (IC50 <13.5 μg ml−1). In addition, the MeOH extract was active against the human urinary bladder carcinoma cell line 5637 (IC50=6 μg ml−1), but also exhibited moderate cytotoxicity against nontumorigenic FL (IC50=11.6 μg ml−1) and HaCaT (IC50=14 μg ml−1) cells. Furthermore, it was strongly active against methicillin-resistant Staphylococcus aureus (MRSA) and Streptococcus pneumoniae with an MIC of 0.8 and 3.2 μg ml−1, respectively. No activity was observed against Gram-negative bacteria such as Escherichia coli, Klebsiella pneumoniae and Pseudomonas aeruginosa.
Therefore, 93 mg of the methanol extract from Nostoc sp. CAVN2 were subjected to bioactivity-guided fractionation utilizing solid-phase extraction (SPE) and S. aureus ATCC 6538 as indicator organism. An aliquot of the bioactive fraction, eluted with 80% MeOH in H2O, was subjected to analytical HPLC-DAD-ESI-TOF-HRMS analysis. Using a pentafluorophenyl endcapped core-shell column, we were able to distinguish over 60 compounds with UV spectra comparable to that of carbamidocyclophane A (15).9 Extensive HRMS data examination of each compound, including critical evaluation of its isotopic distribution pattern and predicted degree of double-bond equivalents, indicated the presence of already described compounds (carbamidocyclophanes A–F; cylindrocyclophanes A, A1–A4, C, C1–C4 and F) alongside unknown putative cyclophanes differing in the level of esterification and halogenation; that is, from nonhalogenated to trichlorinated molecules with only one carbamoyl moiety on C-1 or C-14 (see Figure 1). On the basis of obtained MS data, we assumed that the second carbamoyl group might be substituted by a hydroxyl group or just by a hydrogen atom. Furthermore, the LC-MS data also indicated several glycosylated cyclophanes in the retention time range from 4.5 to 11.5 min (data not shown) as it was reported for nostocyclophanes A and B.7 In some cases, several peaks were detected at different retention times when analyzing distinct monoisotopic mass selected ion chromatograms. This might indicate the presence of different constitutional isomers, depending on the substituent distribution of the cyclophane core structure, confirming Nostoc sp. CAVN2 as a producer of a high diversity of cyclophanes.
Isolation procedure and structure elucidation
For isolation of cyclophanes, the methanol extract of the Nostoc sp. CAVN2 biomass was also fractionated via the SPE procedure. The cyclophane-containing fraction, eluting with 80% methanol in water, was collected. This sample was subjected to semi-preparative reversed-phase HPLC on a polar endcapped ether-linked phenyl phase to obtain five fractions: P1 to P5. Each of these contained paracyclophanes with an equal degree of halogenation, but the level of chlorination continuously increased from P1 to P5. The rich diversity of closely related cyclophane analogs in Nostoc sp. CAVN2 made this prepurification step necessary to achieve a proper separation, for example of the binary mixtures 8/9, 10/3, 12/13 and 16/19, as the compounds differ only slightly in their physicochemical properties. Novel cyclophane derivatives 1–5 were isolated besides the previously described cyclophanes (6–16) by a second round of semi-preparative HPLC, this time using a pentafluorophenyl stationary phase (Figure 2).

Separation and isolation procedure of novel and previously reported [7.7]paracyclophanes. (a) First semi-preparative HPLC of cyclophane-containing fraction utilizing an ether-linked phenyl phase. (b) Second semi-preparative HPLC of fractions P1 to P5 using a pentafluorophenyl stationary phase. A full color version of this figure is available at The Journal of Antibiotics journal online.
Analytical HPLC-DAD-MS analysis of fraction P1 indicated the presence of six compounds with UV spectra comparable to carbamidocyclophane A (15) (Figure 1c) and in negative mode [M–H]− ions at m/z 933.3, 933.3, 646.4, 669.4, 626.4 and 583.4. The compounds occurred in a relative ratio (%) of 4.4:4.7:1.9:100:21.0:1.4 (Figure 2).
The final semi-preparative RP-HPLC separation of P1 yielded three white, amorphous substances successively eluting in the following order: 7 (4.3 mg, 0.18% of dry biomass) as the major compound, and 1 (1.0 mg, 0.04% of dry biomass) and 8 (0.2 mg, 0.01% of dry biomass) as minor metabolites. According to their spectroscopic data and by comparison with reported values, compound 7 was identified as the known nonhalogenated carbamidocyclophane E, and 8 as its bidescarbamoyl analog cylindroyclophane A.4, 5, 9
The negative-mode HR-ESI-TOF-MS analysis of 1 agreed with a molecular formula of C37H56NO4 (found m/z 626.4070, calculated 626.4062 for [M–H]−, Δ 1.28 p.p.m.). The isotope distribution indicated, in accordance with 7 and 8, the absence of halogen atoms in 1. The observed molecular mass of 1 is 43 Da lower than the molecular mass of 7, and 43 Da higher compared with 8, suggesting the presence of only one carbamoyl moiety in 1.
As presented in Table 1, the 1H-NMR data of 1 and the two known derivatives, carbamidocyclophane E (7)9 and cylindrocyclophane A (8),4, 5 were very similar even though the spectra of 1 showed the double set of signals because of the loss of symmetry compared with 7 and 8 (Figure 1). The 1H spectra included signals for aromatic protons (δ 6.0–6.5), methine proton signals at δ∼3.2 (H-7 and H-20), and in the range δ∼1.5–1.8 (H-2 and H-15), methyl groups (δ 0.7–1.2) and numerous methylene proton signals in the range δ 0.5 to δ 2.1. In addition, the 1H NMR spectrum of 1 showed one oxymethine signal at δ 4.81, comparable to the signal for H-1/H-14 in 7, and one oxymethine signal at δ 3.74, similar to that exhibited for the signal of H-1/H-14 in 8, indicating an unsymmetrical substitution pattern for C-1 and C-14 as described for carbamidocyclophane F (6).10 Analysis of homo- and heteronuclear 2D NMR data (COSY, TOCSY, HSQC and HMBC) revealed typical correlations for carbamidocyclophanes.9, 10 The 2D NMR signals and correlations corresponding to the core structure (C-1–C-26) matched perfectly with those described for 6, whereas the substituents (C-27–C-30 and C-31–C-34) showed the typical signal pattern for alkyl chains and matched with the values described for 7 and 8. HMBC correlation of H-1 (δ 4.81) to a quaternary carbon atom with a chemical shift value of δ 159.5 (C-37) corroborated the monocarbamoylation indicated by MS analysis. Taking into account all analytical data, compound 1 was identified as the nonhalogenated congener of 6 and was named carbamidocyclophane H (Figure 3).

Monocarbamoylated carbamidocyclophanes 1, 2, 4, 5 and 6*. *The structure was published by Luo et al.10 during preparation of this article.
Fraction P2 contained five compounds that showed UV spectra comparable to the compounds in P1 and monoisotopic [M–H]− ions at m/z 703.4, 723.3, 660.4, 683.4 and 617.4, occurring in given order with a relative ratio in % to the most abundant substance of 100:<0.1:20.6:0.6:1.5 (Figure 2).
Compounds 9, 2 and 10 were also obtained as white, amorphous powders after semi-preparative HPLC with a quantity of 3.4 mg (0.15% of dry biomass), 1.5 mg (0.06% of dry biomass) and 0.4 mg (0.02% of dry biomass). Comparing the analytical data of compounds 9 and 10 with data published by Bui et al.9 and Chlipala et al.,6 9 was revealed as carbamidocyclophane D and 10 as cylindrocyclophane A1. HR–ESI-TOF-MS of 2 in negative mode showed an isotopic pattern for a monochlorinated molecule and suggested the elemental composition of C37H55ClNO7 (found m/z 660.3680, calculated 660.3673 for [M–H]−, Δ 1.06 p.p.m.). Again, the observed mass difference of 43 Da for 2 compared with 9 and 10 indicated the presence of only one carbamoyl group within the molecule, which could be proven by NMR spectroscopic analysis. Like for carbamidocyclophane H (1), the NMR spectra of 2 showed one oxymethine signal with chemical shifts of δH 4.80/δC 83.4, correlating to a carbonyl C-atom at δC 159.9, and a second one with shifts of δH 3.74/δC 81.5. All other signals from 2D NMR analysis matched perfectly with those of 1, with the only difference in one side chain (C-27–C-30), where signals were shifted downfield compared with those of 1. As C-30 is represented by a signal corresponding to a methylene group with a chemical shift of δH 3.44/δC 45.6, monochlorination of C-30 was demonstrated.
The analytical HPLC-DAD-MS investigation of fraction P3 indicated the presence of six compounds with similar UV spectra as described above (Figure 1c) and [M–H]− ions at m/z 703.4, 737.3, 694.3, 737.3, 694.3 and 651.3 in a percentage ratio of 0.5:07.6:1.8:100:19.8:1.3 based on the integrated peak areas in the UV chromatogram at λ=228 nm (Figure 2). For each substance, the observed isotope distribution was in good agreement with the presence of two chlorine atoms within the molecule. Because of the observed isotopic patterns and the detection of identical monoisotopic mass peaks at different retention times, the presence of constitutional isomers was assumed once again, and hence the combination of fractions containing compounds with the same m/z values was avoided.
The processing of P3 resulted in four pure compounds 3, 11, 4 and 12 as white, amorphous powders. Compound 3 was collected as the first peak of fraction P3 (0.7 mg, 0.05% of dry biomass), showing the same high-resolution monoisotopic mass ion m/z 737.3339 [M–H]− (calculated 737.3341 for [M–H]−, Δ −0.27 p.p.m.) in negative mode and an equal isotopic pattern as the known compound carbamidocyclophane C (11). This resulted in the proposed elemental composition of C38H55Cl2N2O8 that is identical to the determined one for 11. However, differing retention times suggested 3 being a constitutional isomer of 11. This assumption was confirmed by NMR analysis. The analysis of 1D and 2D NMR spectra led to a symmetrical structure, similar to carbamidocyclophane A (15), because of only 19 detected carbon signals compared with the MS analysis, suggesting 38 carbon atoms in the molecular formula. NMR signals and correlations corresponding to the core structure (C-1–C-26) matched those described for 15 and therefore demonstrating bicarbamoylation of 3. Differences were detected for the chemical shift values of the side chains (C-27–C-30 and C-31–C-34). The monochlorination of chain end C-30, respectively C-34, in 3 was undoubtedly proven by the combination of HSQC and COSY/TOCSY experiments, showing a CH2-group (C-30/C-34) with chemical shifts of δH 3.44, δC 45.4 attached to a C3H6 unit bound to C-7, respectively C-20 of the core structure (Table 1). These results corroborated the suggested structure from HRMS analysis, and thus compound 3 was named carbamidocyclophane J and is shown in Figure 4.

Carbamidocyclophane J (3) with a novel halogenation pattern of the paracyclophane core structure.
To the best of our knowledge, carbamidocyclophane J (3) is the first reported naturally occurring C-30/C-34-dihalogenated [7.7]paracyclophane. Having identified this new variant as a constitutional isomer of 11, the LC-HRMS data analysis of the initial cyclophane-containing fraction (Figure 1) indicated further putative constitutional isomers of structurally confirmed, geminally dichlorinated cyclophanes. At least for the extracted ion chromatogram (EIC) for the [M–H]− ion of carbamidocyclophane K (4) as well as of cylindrocyclophane A2 (12), several peaks were detected at different retention times, always showing a congruent isotopic pattern consistent with the structurally elucidated dichlorinated congeners (see Figure 1). Misinterpretations due to unwanted ESI fragmentations could be excluded by comparison of retention times in the EICs of relevant [M–H]− ions of fraction P3 (see Supplementary Information S4). Compound 12, the latest eluting compound of this fraction with a yield of 0.2 mg (0.01% of dry biomass), and 11, the major molecule in this fraction with a yield of 9.2 mg (0.60% of dry biomass), were identified by comparison with previously reported data as cylindrocyclophane A26 and carbamidocyclophane C,9 respectively. Compound 4 eluted between 11 and 12 and was obtained as white, amorphous solid in a yield of 1.4 mg (0.09% of dry biomass). The HR-ESI-TOF-MS analysis of 4 indicated the molecular formula of C37H54Cl2NO4 (found m/z 694.3293, calculated 694.3283 for [M–H]−, Δ 1.44 p.p.m.) with an isotopic pattern consistent with the predicted degree of chlorination and differing from 11 and 12 by 43 Da, suggesting to also contain only one carbamoyl moiety. Evidence for the monocarbamoylation could be received via NMR spectroscopy. Similar to what could be shown for 1 and 2, again for derivative 4 two different sets of chemical shifts for C-1 and C-14 are present (δH 4.81/δC 83.4 and δH 3.75/δC 81.6). Also in this case, the signal appearing more downfield shows a HMBC correlation with a carbonyl C-atom at δC 159.6, proving connectivity to the carbamoyl moiety. Dichlorination at C-30 could be shown by the chemical shift-pair δH 5.82/ δC 75.0 for a CH group attached to the alkylic side chain (C-27–C-29).
The analytical HPLC-DAD-MS data of fraction P4 uncovered a set of three main compounds with corresponding cyclophane-related UV spectra (Figure 1c) and [M–H]− ions at m/z 771.3, 728.3 and 685.3 that occurred in a ratio (%) of 100:18.8:1.4 (Figure 2). The compounds 13, 5 and 14 were obtained in mentioned order as white, amorphous powders after isolation with yields of 7.8 mg (0.32% of dry biomass), 1.2 mg (0.05% of dry biomass), and 0.2 mg (0.01% of dry biomass). According to reported data by Bui et al.9 and Chlipala et al.,6 13 was revealed as carbamidocyclophane B, and 14 as cylindrocyclophane A3. HRMS data of 5 in negative mode showed the isotopic pattern of a trichlorinated molecule and agreed with the elemental composition C37H53Cl3NO7 (found m/z 728.2879, calculated 728.2893 for [M–H]−, Δ−1.92 p.p.m.). Once again, a mass difference of 43 Da indicated only one carbamoyl residue in compound 5 compared with 13 and 14, and was in agreement with the investigations of the already described cyclophane clusters in P1 to P3. Similar to 1, 2 and 4, the monocarbamoylation could be proven by NMR spectroscopic analysis. Again, two different sets of signals corresponding to oxymethine groups are present in the spectra of 5. One of them shows chemical shifts of δH 4.81/δC 83.4 and correlates (HMBC) to a carbonyl C-atom with a chemical shift of δC 159.6. The other oxymethine-related signal shows chemical shifts of δH 5.83/δC 74.9 that are typical for CHOH groups. Evidence for the chlorination pattern (dichlorination in C-30 and monochlorination in C-34) was received via analysis of chemical shifts for one methine signal (δH 5.83/δC 74.9) and one methylene signal (δH 3.43/δC 45.6) in combination with COSY and HMBC correlations.
Fraction P5 comprised three compounds with similar UV spectra and monoisotopic ions of m/z of 805.2, 762.3 and 719.2 [M–H]− in negative MS mode in a relative ratio to the most abundant derivative (calculated in %) of 100:15.4:0.9 (Figure 2). The final semi-preparative isolation yielded three white, amorphous solids eluted in following order: 15 (10.8 mg, 0.64% of dry biomass), 6 (1.6 mg, 0.09% of dry biomass) and 16 (0.1 mg, 0.01% of dry biomass). For each compound, the observed isotopic pattern indicated a tetrachlorinated molecule. Based on their recorded spectroscopic data, compounds 15 and 16 were identified as the previously reported carbamidocyclophane A9 and cylindrocyclophane A4,6 respectively.
Recently, Luo et al.10 described the isolation of carbamidocyclophane G besides the carbamidocyclophanes A–C (15, 13 and 11) from the freshwater cyanobacterium Nostoc sp. (UIC 10274). The comparison of NMR and MS data with literature data identified 6 as carbamidocyclophane F, also produced by Nostoc sp. UIC 10274. However, in contrast to already published HRESIMS data, the monoisotopic mass of 6 (m/z 762.2502 for [M–H]−) and the resultant isotopic distribution pattern were used to deduce the elemental composition of 6 and to confirm its degree of chlorination, as it was previously reported for tetrachlorinated paracyclophanes.6, 9 Taken together, NMR analysis revealed 1, 2, 4, 5 and 6 as monocarbamoylated cyclophane structures with different degrees of chlorination in the substituents named carbamidocyclophane H (1), I (2), K (4), L (5) and F (6)10 (Figure 3 and Table 1). The chlorination pattern was deciphered via analysis of 2D NMR data (TOCSY, COSY and HMBC). Key correlations are shown exemplary for 5 in Figure 5.

Selected TOCSY/COSY and HMBC correlations for carbamidocyclophane L (5).
Stereoconfigural analysis
The stereochemical configuration of isolated cyclophanes was established by careful analysis of NMR and CD spectroscopic data. As previously reported for the monocarbamoylated carbamidocyclophane F(6) as well as for the dicarbamoylated carbamidocyclophanes A–E (15, 13, 11, 9 and 7), large 3J coupling constants were determined for 1–5 in the range 10.2–10.3 Hz for H-1/H-2 and 9.5–10.1 Hz for H-14/H-15.9, 10 These data are congruent with an anti-conformation of H-1/H-2 and H-14/H-15 and a pseudoequatorial position of the methyl residues on C-2 and C-15, respectively.4, 5, 6, 9, 10 The recorded CD spectra of 1, 2, 4 and 5 were comparable with recently described data of carbamidocyclophane F (6) as well as similar to previously reported cylindrocyclophanes A, A4, C, F, nostocyclophanes A–D and merocyclophanes A and B, showing a negative Cotton effect at ∼217–219 nm (Δɛ −0.55 to −1.21) with a negative shoulder extending from 220 to 230 nm and a weaker negative broad peak in the region between 265 and 280 nm with a second negative Cotton effect at ∼278–281 nm (Δɛ −0.34 to −1.06).5, 6, 7, 8, 10 In addition, we examined CD data for cylindrocyclophanes A1–A3 (10, 12 and 14), as they were not described in the literature. Derivatives 10, 12 and 14 showed comparable CD spectra with negative Cotton effects at ∼217–218 nm (Δɛ −1.42 to −2.87) and at ∼278–280 nm (Δɛ −0.34 to −0.52) to those of 1, 2, 4, 5 and 6, underlining the results of the configurational analysis by Chlipala et al.6 To corroborate our results, we measured the CD spectrum of cylindrocyclophane A (8) as a reference, because its absolute stereochemical configuration has been confirmed by Mosher’s method.5 Identical CD behavior of 1, 2, 4, 5, 6, 10, 12 and 14 compared with 8 suggests the same absolute stereochemical configurations. In contrast to this, carbamidocyclophane J (3), a dicarbamoylated paracyclophane, displayed a significantly different CD spectrum compared with the derivatives described above. A broad positive peak could be detected in the range from 220 to 240 nm with a positive Cotton effect at 233 nm (Δɛ 2.78) in addition to the familiar negative Cotton effect at 280 nm of Δɛ −0.94. These values are in good agreement with the data reported by Moore et al.5 for cylindrocyclophane D, the 1,14-diacetylated cylindrocyclophane A, that is the most similar known structure to carbamidocyclophane J (3). Furthermore, CD data for dicarbamoylated carbamidocyclophanes A–E (15, 13, 11, 9 and 7) were recorded and could confirm the previously suggested absolute configurations.9 All five compounds showed comparable CD spectra with a positive Cotton effect at ∼233–235 nm (Δɛ 1.88–2.31) and the negative Cotton effect at ∼279–281 nm (Δɛ −0.74 to −1.03) to those of 3 and cylindrocyclophane D, which was also available as a reference standard during this study. The parallel biosynthesis of the configurationally determined cylindrocyclophane A and various carbamidocyclophane derivatives in Nostoc sp. CAVN2 supports the assumption that these paracyclophanes widely share the same biogenetically coded pathway. Therefore, an identical absolute configuration of all stereogenic carbon atoms is reasonable and promoted by all recorded data. As structurally typical for this class of paracyclophanes, we conclude that 1–16 do have the same absolute configuration at C-1, C-2, C-14 and C-15; namely, 1R, 2S, 14R and 15S. In addition, it is assumed that the compounds do not differ in their absolute configuration at C-7/C-20. However, only the stereo descriptors have to be altered because of a priority change of the residues from S to R in case of any halogenation of C-30/C-34.
Biological evaluation of compounds 1–16
The initial screening results (see cytotoxicity screening) of the MeOH extract from CAVN2 are in good agreement with previously reported antimicrobial and cytotoxic data of [7.7]paracyclophane-containing extracts.4, 5, 6, 7, 8, 9, 10 Bui et al.9 reported an inhibition of MRSA strains 535 and 847 by the methanol extract obtained from the Vietnamese Nostoc sp. strain CAVN10 in addition to the cytotoxicity of the pure carbamidocyclophanes. Because of the increase of nosocomial and community-acquired infections with antibiotic-resistant staphylococci, especially of MRSA and vancomycin-resistant S. aureus, the search for novel pharmaceutical leads has become of crucial importance.11, 12, 13, 14 Having all these facts in mind, compounds 1−16 were examined for antibacterial activity against selected, clinically relevant pathogens.
As presented in Table 2, all tested isolates exhibited remarkable effects against S. pneumoniae (MICs of 0.25–2.10 μM) and even better results against MRSA (MICs of 0.10–1.02 μM). Furthermore, 1−16 displayed stronger antibacterial activity against both strains than commercially available antibiotics such as vancomycin (MIC 1.35 μM) or fusidic acid (MIC 3.77 μM) that were used as positive controls, and the pathogenic strains tested are not described to be intermediate or resistant to these antibiotics. No significant correlation between bioactivity and the degree of halogenation could be observed. However, slightly higher antibacterial activity was found for cyclophanes containing one or two carbamoyl moieties compared with noncarbamoylated derivatives with a 5- to 10-fold higher MIC of the latter. In accordance with the report of Luo et al.,10 no activity against Gram-negative bacteria could be found up to a concentration of 50 μg ml−1.
Based on the fact that initial colonizations of MRSA and S. pneumoniae usually affect the nose atrial, the throat or other areas of the skin, we have chosen immortal human keratinocytes, HaCaT cells, to evaluate the cytotoxicity of 1−16 by the CellTiter-Blue cell viability assay. Not surprisingly, as numerously reported by previous investigations, the IC50 values of 1−16 (2.8–11.5 μM) indicated a moderate cytotoxicity. The values are in the same range as those previously published for other naturally occurring paracyclophanes against various tumorigenic cell lines (0.5–5 μM) as well as nontransformed FL cells (IC50s of 3.3–5.1 μM).5, 6, 8, 9, 10 In summary, all compounds 1−16 possess stronger activity against Gram-positive pathogens, especially against MRSA, than cytotoxicity against HaCaT keratinocytes. However, some carbamoylated cyclophanes exhibited larger distances between determined in vitro concentrations for antibacterial activity and unwanted cytotoxic effects. Of these, carbamidocyclophane H (1) and D (9) are the most promising derivatives revealing MICs at least 50-fold lower than their corresponding IC50 values.
Taxonomic identification
The initial phenotypical characterization of the filamentous Vietnamese freshwater strain CAVN2 was conducted by microscopy observation. Based on examined morphological features,15, 16, 17, 18, 19, 20 CAVN2 was supposed to be a member of the genus Nostoc just as the other two carbamidocyclophane-producing cyanobacteria, Nostoc sp. UIC 10274 and Nostoc sp. CAVN10.9, 10 For further taxonomic identification and molecular phylogenetic analysis, a 1.4-kb fragment of the 16S rRNA gene from CAVN2 was sequenced. A primary online BLAST21 search (http://blast.ncbi.nlm.nih.gov) and comparison of the partial CAVN2 16S rDNA nucleotide sequence with available GenBank sequence data revealed high homologies to various Nostoc and Anabaena strains with best hits for Nostoc sp. PCC 7423, KK-01 and CENA61 or Anabaena variabilis ATCC 29413. In order to infer the phylogenetic relationship of Nostoc sp. CAVN2 and these strains as well as previously reported paracyclophane-producing cyanobacteria, a phylogenetic tree was constructed on the basis of available 16S rDNA data using the maximum likelihood method (Figure 6). To make this report more comparable to previous studies, 16S rRNA gene sequences of at least 1 kb from Bergey’s reference strains and other related or former investigated species were added to the sequence alignment. In addition, the so far unknown partial 16S rRNA gene sequence of the first reported carbamidocyclophane producer, Nostoc sp. CAVN10, was also investigated.

Phylogenetic tree based on a secondary structure alignment of cyanobacterial 16S rRNA gene sequences. The tree was inferred using a maximum likelihood method. Numbers given on the branches display bootstrap proportions as percentage of 1000 replicates for values ≥50%. The investigated cyanobacterial strains Nostoc sp. CAVN2 and Nostoc sp. CAVN10 are shown in bold. Filled circles (•), squares (▪) and triangles (▴) denote cylindro-, carbamido- and merocyclophane producers, respectively. Reference strains according to Bergey’s Manual of Systematic Bacteriology19 are marked with an asterisk (*). The INSDC accession numbers are given in brackets. For entries that represent a whole genome, the genomic location of the considered sequence is provided additionally.
The resulting phylogenetic tree in Figure 6 revealed that Nostoc sp. CAVN2 is a member of the same monophyletic clade, including the cylindrocyclophane- and carbamidocyclophane-producing strains Nostoc spp. UIC 10022A and UIC 10274, previously reported and designated as Nostoc cluster 3.3 by Chlipala et al.6 and Luo et al.10 The assignment of the genus Nostoc to strain CAVN2 by the initial phenotypic characterization is in accordance with the presence of reference strain Nostoc sp. PCC 7423 in this group. Although CAVN2 shows a sequence homology of 98% to Nostoc sp. UIC 10274 as well as to Nostoc sp. UIC 10022A based on primary structure information, the phylogenetic tree based on a secondary structure alignment could elucidate that CAVN2 and UIC 10022A share a more recent common ancestor.
To our surprise, the 16S rDNA sequence data of CAVN2 and CAVN10 were completely identical. Based on the generally high conservation of the 16S rRNA, it is not an adequate phylogenetic marker gene when studying taxonomic relations among closely related species. Therefore, we examined several other molecular markers such as hetR, rbcLX intergenic spacer, the phycocyanin intergenic spacer (PC-IGS) and the 16S-(tRNAIle-tRNAAla)-23S rRNA internal transcribed spacer that are assumed to reveal more explanatory significance between strains at the intraspecific level. These markers all share either a unique distribution among filamentous cyanobacteria or at least a partial relatively high sequence variation.22, 23, 24, 25, 26 As with the 16S rDNA sequence data, we could find no nucleotide differences between both strains in the aforementioned marker genes. Nevertheless, we recommend both strains CAVN2 and CAVN10 to be understood as independent and individual Nostoc sp. strains as they differ in the diversity of cyclophanes they are producing. This could undoubtedly be shown by a comparison of the chromatograms of carbamidocyclophanes A–E containing fraction F2 of CAVN10 and paracyclophanes 1–16 containing fraction F2CAVN2 of CAVN2. When using the same cultivation, extraction and separation procedure as well as equal HPLC conditions described by Bui et al.,9 only in F2CAVN2 distinct double peaks could be detected, indicating the absence of mono- and nonesterified paracyclophanes in CAVN10 (see Supplementary Information S5). In addition, to exclude that 1–5 are only artifacts derived from dicarbamoylated cyclophanes during separation or isolation procedures, all compounds could be detected by LC-MS analysis of the crude extract from Nostoc sp. CAVN2 (see Supplementary Information S6).
Methods
General experimental procedures
Optical rotations were determined on a P-2000 polarimeter (JASCO, Gross-Umstadt, Germany) at 20 °C and 589 nm. UV spectra were measured on a BioPhotometer plus (Eppendorf AG, Hamburg, Germany) in the wavelength range from 190 to 320 nm. CD spectra were recorded on a J-810 CD spectropolarimeter (JASCO), measuring the ellipticity θ in dependence of the wavelength from 200 to 300 nm at 20 °C and were analyzed with Spectra Manager Software (version 1.53.01; JASCO). Used concentrations are given in μmol l−1. Cylindrocyclophane A and D were used as references. Attenuated total reflexion-IR (ATR-IR) spectra were recorded using a Nicolet IR 200 Fourier transform-IR spectrometer (Thermo Scientific, Bremen, Germany) at 22 °C. Raw data were processed with OMNIC Spectra Software (Thermo Scientific). NMR spectra were recorded in MeOH-d4 on a 500 MHz Avance III (UltraShield) spectrometer (Bruker BioSpin, Rheinstetten, Germany) or on a 700 MHz Avance III (Ascend), each one equipped with a cryoplatform, at 298 K, if not specified differently. Chemical shift values δ of 1H- and 13C-NMR spectra are reported in p.p.m. relative to the residual solvent signal given as an internal standard.27 Multiplicities are described using the following abbreviations: s=singlet, d=doublet, t=triplet, q=quartet, m=multiplet, b=broad; corrected coupling constants are reported in Hz. HPLC-UV-MS analysis was conducted on a Shimadzu LC20A Prominence comprising a CBM-20A controller, a DGU-14A degasser, LC20A pumps, a SIL-AC HT auto sampler, a CTO-10-ASvp column oven, SPD-M20A Diode Array Detector (DAD) coupled to a LCMS-8030 triple quadrupole (QqQ) mass spectrometer (Shimadzu, Kyoto, Japan). High-resolution mass spectra were recorded on an ion trap-time of flight-MS (IT-TOF-MS, Shimadzu) equipped with an ESI source and attached to the above-mentioned HPLC set-up. Semi-preparative HPLC was performed on a Shimadzu HPLC system consisting of SCL-10Avp system controller, a LC-10ATvp liquid chromatograph, a FCV-M10Avp low pressure gradient unit, a SPD-M10Avp DAD (Shimadzu) and a JETSTREAM 2 PLUS column thermostat (Goebel Instrumentelle Analytik, Hallertau, Germany).
Cyanobacterial strains and culture conditions
The cyanobacterial strains investigated in this research included 13 freshwater and 1 brackish water strain belonging to the orders Nostocales and Oscillatoriales (Supplementary Information S1 and S2).15, 16, 17, 18, 19 The freshwater cyanobacteria were originally isolated from samples of rice fields and shallows in Northern Vietnam (Thanh Hoa, Thai Binh, Nam Dinh and Hanoi) and established as unialgal laboratory cultures by Dr Nhi V Tran (Institute for Biotechnology, Hanoi, Vietnam). The strains were incorporated into the culture collection of the Institute of Pharmacy, University of Greifswald. The brackish water cyanobacterial strain was isolated from the Baltic Sea near the coast of Grabow (Ruegen island) by B Cuypers (University of Greifswald).
For the preparation of extracts for bioactivity screening, strains were cultured in 500 ml aliquots of BG11 medium28 in 1.8 l Fernbach Flasks under continuous fluorescent light (8 μmol m−2 s−1) and the temperature was maintained at 20±1 °C. Only Nostoc sp. CAVN2 was cultivated in modified WC medium29 (MBL medium) without the described vitamin mix, but buffered with 0.5 mM TES to a pH of 7.2, under otherwise identical conditions. After 6–8 weeks of growth, the cyanobacterial cells were harvested and separated from the medium by centrifugation. Biomasses were lyophilized and supernatants were evaporated to dryness and finally stored at −20 °C until use.
For isolation of paracyclophanes, Nostoc sp. CAVN2 was cultured in a glass column containing 35 l of MBL medium under continuous fluorescent light (20 μmol m−2 s−1), the temperature was maintained at 22±1 °C and pH was adjusted to 8.5 using CO2 supplementation.30 After 28 days, the cells were harvested by centrifugation, lyophilized and stored at −20 °C. The yield of freeze dried biomass was 286 mg l−1.
The axenic cultures of Nostoc sp. CAVN2 and CAVN10 were achieved by a combined use of traditional microalgae isolation techniques.31 Both strains were cultivated in BG11 medium at 28±1 °C under continuous light (15 μmol m−2 s−1) and aerated with 0.5% CO2 in air. Then, 50 ml aliquots (log-phase) were harvested and centrifuged. Biomass pellets were washed with sterilized distilled water, centrifuged again and stored at −20 °C until use.
For comparison of [7.7]paracyclophane biodiversity between Nostoc sp. CAVN10 and Nostoc sp. CAVN2, culture, extraction and separation conditions reported by Bui et al.9 were used.
Morphological characterization and identification
Phenotypic characterization of investigated cyanobacteria was based on literature data.15, 16, 17, 18, 19
Different parameters were used for the identification of the isolates; for example, shape (length and width) and relative size of vegetative and end cells, presence and distribution of heterocysts and akinetes, trichome polarity, level of branching and general morphological structure of the filaments. Microscopic examinations were carried out in culture media, mostly BG11 medium, using an inverted light microscope (Axioskop 2 plus, Carl Zeiss, Oberkochen, Germany) with coupled digital camera (AxioCam MRc camera, software Axiovision version 4).
DNA isolation, PCR amplification and sequencing
The DNA extraction procedure was performed according to Franche and Damerval,32 modified as follows: biomass pellets of Nostoc sp. CAVN2 and CAVN10 were thawed and aliquots were resuspended in 1 ml TE buffer (50 mM EDTA, 50 mM Tris–HCl, pH 8). Cell wall breakage was performed by a Sonopuls UW 2200 ultrasound-homogenizer (BANDELIN Electronic, Berlin, Germany). The centrifuged pellet (20 000 g, 20 °C, 10 min) was combined with 300 μl STET buffer (8% (w/v) sucrose, 5% (v/v) Triton X-100, 50 mM EDTA, 50 mM Tris–HCl, pH 8), 15 μl chloroform/isoamyl alcohol (Roti-C/I, Carl Roth, Karlsruhe Germany) and 35 μl Lytic Enzyme Solution (QIAGEN, Hilden, Germany), and the samples were incubated at 37 °C for 1 h. Then, 100 μl 10% SDS and 100 μl 5 M NaCl were added and samples were treated at 65 °C until cell lysis was completely achieved. After adding 200 μl of 1 M NaCl and incubation for another 15 min, the aqueous phase was rid of proteins by chloroform/isoamyl alcohol addition and genomic DNA was precipitated in a new 1.5 ml centrifuge tube with isopropyl alcohol at −20 °C. After incubation for 2 h, the centrifuged DNA pellet (20 000 g, 4 °C, 15 min) was washed with 500 μl EtOH and separated from the supernatant again. The briefly air-dried DNA was dissolved in a proper volume of TE buffer and finally subjected to RNA digest at 37 °C for 1 h by adding 1 μl RNAse (20 μg μl−1, Sigma-Aldrich, Hamburg, Germany). The solution was heated to 65 °C for 10 min to inactivate the RNAse and then stored at −20 °C until use. PCR amplification was carried out with a MJ Mini Personal Thermal Cycler (Bio-Rad Laboratories, Richmond, CA, USA) utilizing Opti Taq DNA polymerase (5 U μl−1, Roboklon, Berlin, Germany) and listed oligonucleotides (see Table 3) according to the manufacturer’s recommendations. PCR products were verified and separated by electrophoresis on 1.5% agarose gels in 1 × TBE buffer. Excised PCR products were purified with the QIAquick Gel Extraction Kit (QIAGEN, Hilden Germany) as described in the manual provided by the manufacturer. Gel-purified amplicons were sequenced by MWG Operon (Ebersberg, Germany) using primers in Table 3. The resulting sequence data were deposited with GenBank under accession numbers KJ511227–KJ511238 (Table 4).
Phylogenetic tree construction
Single sequencing reads were assembled in Geneious version 6.1.7 (available from http://www.geneious.com). The 16S rDNA sequences of Nostoc sp. CAVN2 and Nostoc sp. CAVN10 were automatically aligned according to the SILVA SSU Ref NR99 r115 database (available from http://www.arb-silva.de)33 using the Silva INcremental Aligner (SINA) version 1.2.11.34 SINA-aligned sequences and an additional sequence (AB075983) from the SILVA web release r117 database were imported into the ARB software package version 5.5.35 The alignment of 52 sequences, also including previously used reference strains and available [7.7]paracyclophane-producing cyanobacterial strains,10 was manually refined taking into account the secondary structure information of the rRNA. Phylogenetic reconstruction was performed with 51 sequences (CAVN2 and CAVN10 share 100% identity and were only considered once during reconstruction) using a maximum likelihood method. The final tree was calculated with RAxML version 8.0.14 (GTRGAMMA model)36 and based on 638 distinct alignment patterns. The best tree out of 1000 independent inferences (Figure 6) is presented without outgroup sequences of Bacillus subtilis DSM 10 (AJ276351) and E. coli ATCC 25922 (DQ360844). The sequences of strains CAVN2 and CAVN10 are available from the INSDC (International Nucleotide Sequence Database Collaboration) databases; that is, DDBJ, EMBL and GenBank, under accession numbers KJ511229 and KJ511235 (Table 4).
Cytotoxicity assays
The cytotoxicity screening of crude extracts from investigated cyanobacteria against a breast adenocarcinoma cell line (MCF-7), human amniotic epithelial Fibroblast-Like (FL) cells and a human urinary bladder carcinoma cell line (5637) were performed by using either the crystal violet or the neutral red uptake assay as previously described.9, 37 The cell viability investigation for the cytotoxic evaluation of 1–16 was done by using the CellTiter-Blue assay (Promega, Mannheim, Germany) and HaCaT cells (human adult low calcium high temperature keratinocytes). HaCaT cells were obtained from German Cancer Research Center DKFZ (Heidelberg, Germany) and were cultured in calcium-free Gibco DMEM medium (Life Technologies, Vienna, Austria) with 10% fetal calf serum (PAA Laboratories, Pasching, Austria). HaCaT cells were plated at 3 × 104 cells per ml in 96-well plates (100 μl per well) for 18 h before stimulating them with different concentrations (serial dilution from 100 to 0.002 μg ml−1) of crude extract or compounds, dissolved in DMSO, or left untreated (negative control) for 24 h. Subsequently, cells were incubated for 3 h with 10% (v/v) of CellTiter-Blue Reagent (Promega), and cell viability was assessed at an excitation wavelength of 530 nm and an emission wavelength of 590 nm in a multiplate reader (Infinite 200 PRO, Tecan, Gröding, Austria). Tests were performed twice in triplicates and values are reported as average of the determined IC50 values.
Determination of MIC
Tests of Nostoc sp. CAVN2 crude extract and isolated compounds were carried out according to the EUCAST criteria38, 39, 40 in a 96-well dilution assay with compound concentrations, for crude extract or purified substances, between 50 and 0.01 μg ml−1 and incubated with bacteria solution in a concentration of 104 bacteria per ml. After 24 h, the MIC of a substance was determined. Tests were done in triplicates and reported values are shown as average of multiple MIC tests. All tested bacteria belong to the SeaLife Pharma MDR pathogen collection and have been isolated from infected patients. The screening included MRSA, S. pneumoniae, E. coli, K. pneumoniae and P. aeruginosa.
Extraction, separation and isolation of [7.7]paracyclophanes
Initial screening for cytotoxic activity was performed with crude n-hexane, methanol and water extracts of biomasses and the ethyl acetate extracts of media from investigated cyanobacterial strains (Supplementary Information S3). For this purpose, 1 g dried biomass of each strain was successively extracted with n-hexane, followed by methanol and water (each three times for 1 h under stirring) to yield three crude extracts after removal of the solvents. The corresponding ethyl acetate extract was obtained by a threefold extraction of 3 l medium from the culture supernatant with a 1:1 mixture of EtOAc/H2O over 24 h, followed by subsequent combination of the upper phases and evaporation to dryness. For analytically scaled structural investigations of Nostoc sp. CAVN2 biomass, portions of ∼100 mg freeze dried cells (in total 400 mg) were extracted five times with 25 ml MeOH under stirring (750 r.p.m.) for 30 min. Cell wall breakage was provided by sea sand grinding and usage of ultrasonication. The methanolic supernatants were separated from the residues by centrifugation (3300 g, 10 min, 15 °C) and filtration through Whatman filter papers No. 1 (GE Healthcare, Buckinghamshire, UK), pooled and evaporated to yield altogether 93.3 mg of crude extract. This methanol extract was subjected to SPE utilizing a Strata C18-E 10 g/60 ml (55 μm, 70 Å) SPE cartridge (Phenomenex, Torrance, CA, USA) and a MeOH/H2O step gradient of 0, 10, 25, 50, 80 and 100% (v/v) MeOH in water. Eluates were concentrated to dryness and 500 μg of each fraction were tested for antibacterial activity against S. aureus ATCC 6538 using previously described agar diffusion assay.41, 42 Bioactive fraction (37.5 mg), eluting with 80% MeOH, was subjected to HPLC-DAD/ESI-IT-TOF-HRMS. Separation of paracyclophanes was performed with a Kinetex PFP column (100 × 4.6 mm, 2.6 μm, 100 Å; Phenomenex) and a gradient of MeOH in deionized water with a flow rate of 0.6 ml min−1 from 60 to 77.5% MeOH in 35 min at 40 °C. Structural formulas and structure proposals were made on the basis of monoisotopic mass ions and corresponding isotopic distribution patterns from obtained high-resolution mass spectra of detected cyclophanes with ChemBioDraw Ultra software version 12.0 (CambridgeSoft, Cambridge, MA, USA) and the Formula Predictor tool provided by LabSolutions software version 3.60.361 (Shimadzu).
A 2.71 g biomass aliquot from the 35 l cultivation of Nostoc sp. CAVN2 (in total 10.02 g of lyophilized biomass) was successively extracted with methanol as already described for analytical investigations to yield 491 mg crude extract. This extract was also subjected to SPE under above-mentioned conditions to obtain 169.7 mg of cyclophane-containing fraction. A portion (156 mg) of cyclophane-rich fraction, eluted with 80% methanol, was separated by semi-preparative HPLC with a Synergi Polar RP column (250 × 10.0 mm, 4 μm, 80 Å; Phenomenex) and a binary gradient of MeOH in deionized water with a flow rate of 3.1 ml min−1 from 62 to 85% MeOH in 32 min at 25 °C. Multiple rounds of isolation yielded fractions P1 (9.4 mg), P2 (9.9 mg), P3 (24.1 mg), P4 (12.5 mg) and P5 (23.4 mg). Initial analytical HPLC-DAD-QqQ-MS analysis of P1 to P5 as well as the purity control of isolated compounds from P1 to P5 and the detection of 1–5 in the crude extract (Supplementary Information S6) was performed by using a Luna PFP(2) column (250 × 4.6 mm, 5 μm, 100 Å, Phenomenex) and a MeOH–H2O gradient with a flow rate of 0.8 ml min−1 from 60 to 85% MeOH in 32 min at 25 °C. The relative abundance of detected compounds in P1−P5 was calculated based on obtained areas at 226 nm in the UV chromatogram. The area of the major peak of every fraction was set to 100%. For final isolation of compounds 7, 1 and 8, fraction P1 (9.0 mg) was subjected to semi-preparative HPLC utilizing a Luna PFP(2) column (250 × 10.0 mm, 5 μm, 100 Å, Phenomenex) and the same conditions previously described for the separation of cyclophane-rich SPE fraction, using a flow rate of 3.5 ml min−1. Several rounds of separation yielded 4.3 mg of 7, 1 mg of 1 and 0.2 mg of 8 in described order. A portion (9.2 mg) of P2 yielded 9 (3.4 mg), 2 (1.5 mg) and 10 (0.4 mg) when utilizing the same HPLC column, but with a binary methanol water gradient from 63 to 86% MeOH in 32 min at 25 °C. Pure compounds 3 (0.7 mg), 11 (9.2 mg), 4 (1.4 mg) and 12 (0.2 mg) were obtained by separation of fraction P3 (14.9 mg) carried out with a MeOH–H2O gradient from 59 to 82% MeOH in 32 min at 30 °C and a flow rate of 3.5 ml min−1. The semi-preparative HPLC of P4 (12.4 mg) and P5 (15.9 mg) was performed as already described for P1. Compounds 13 (7.8 mg), 5 (1.2 mg), 14 (0.2 mg) were obtained from P4 and 15 (10.8 mg), 6 (1.6 mg), and 16 (0.1 mg) from fraction P5. The percentaged yields of obtained compounds 1–16 were calculated based on initially used 2.71 g lyophilized Nostoc sp. CAVN2 biomass. Because of the small amounts of 8, 10, 12, 14 and 16, additional rounds of mentioned extraction and separation procedures were performed. Only focusing on the isolation of these minor derivatives, finally, 8, 10, 12, 14 and 16 yielded 0.5, 0.8, 0.4, 0.7 and 0.5 mg, respectively.
Carbamidocyclophane H (1)
White, amorphous powder; [α] +12.5 (c=0.8 g per 100 ml; MeOH); UV (c=5 μM; MeOH) λmax (log ɛ) 205 (5.250), 208 (5.225), 210 (5.164), 226 (4.580), 275 (3.903); CD (c=30 μM; MeOH) λmax (Δɛ) 211 (4.09), 218 (−0.92), 233 (0.24), 261 (−0.30), 278 (−0.64) nm; ATR-IR (film) max 3384 (br), 2927, 2856, 1699, 1590, 1431, 1375, 1331, 1017, 834 cm−1; for 1H and 13C NMR data see Table 1; HRMS (ESI): calcd for C37H56NO7 [M–H]−: 626.4062, found m/z 626.4070.
+12.5 (c=0.8 g per 100 ml; MeOH); UV (c=5 μM; MeOH) λmax (log ɛ) 205 (5.250), 208 (5.225), 210 (5.164), 226 (4.580), 275 (3.903); CD (c=30 μM; MeOH) λmax (Δɛ) 211 (4.09), 218 (−0.92), 233 (0.24), 261 (−0.30), 278 (−0.64) nm; ATR-IR (film) max 3384 (br), 2927, 2856, 1699, 1590, 1431, 1375, 1331, 1017, 834 cm−1; for 1H and 13C NMR data see Table 1; HRMS (ESI): calcd for C37H56NO7 [M–H]−: 626.4062, found m/z 626.4070.
Carbamidocyclophane I (2)
White, amorphous powder; [α] −6.7 (c=0.9 g per 100 ml; MeOH), UV (c=6 μM; MeOH) λmax (log ɛ) 204 (5.181), 208 (5.054), 229 (4.426), 274 (3.824) nm; CD (c=65 μM; MeOH) λmax (Δɛ) 211 (1.72), 219 (−0.55), 229 (−0.07), 238 (0.66), 258 (0.22), 278 (−0.34) nm; ATR-IR (film) max 3366 (br), 2927, 2854, 1699, 1591, 1431, 1375, 1017, 832, 650, 619 cm−1; for 1H and 13C NMR data see Table 1; HRMS (ESI): calcd for C37H55ClNO7 [M–H]−: 660.3673, found m/z 660.3680.
−6.7 (c=0.9 g per 100 ml; MeOH), UV (c=6 μM; MeOH) λmax (log ɛ) 204 (5.181), 208 (5.054), 229 (4.426), 274 (3.824) nm; CD (c=65 μM; MeOH) λmax (Δɛ) 211 (1.72), 219 (−0.55), 229 (−0.07), 238 (0.66), 258 (0.22), 278 (−0.34) nm; ATR-IR (film) max 3366 (br), 2927, 2854, 1699, 1591, 1431, 1375, 1017, 832, 650, 619 cm−1; for 1H and 13C NMR data see Table 1; HRMS (ESI): calcd for C37H55ClNO7 [M–H]−: 660.3673, found m/z 660.3680.
Carbamidocyclphane J (3)
White, amorphous powder; [α] +2.0 (c=0.5 g per 100 ml; MeOH); UV (c=3 μM; MeOH) λmax (log ɛ) 205 (5.355), 207 (5.342), 210 (5.315), 229 (4.669), 275 (4.938) nm; CD (c=30 μM; MeOH) λmax (Δɛ) 211 (4.90), 224 (1.79), 233 (2.78), 251 (0.43), 280 (−0.94), 297 (0.09) nm; ATR-IR (film) max 3363 (br), 2929, 2854, 1699, 1590, 1432, 1374, 1334, 1040, 1018, 832, 648 cm−1; for 1H and 13C NMR data see Table 1; HRMS (ESI): calcd for C38H55Cl2N2O8 [M−H]−: 737.3341, found m/z 737.3339.
+2.0 (c=0.5 g per 100 ml; MeOH); UV (c=3 μM; MeOH) λmax (log ɛ) 205 (5.355), 207 (5.342), 210 (5.315), 229 (4.669), 275 (4.938) nm; CD (c=30 μM; MeOH) λmax (Δɛ) 211 (4.90), 224 (1.79), 233 (2.78), 251 (0.43), 280 (−0.94), 297 (0.09) nm; ATR-IR (film) max 3363 (br), 2929, 2854, 1699, 1590, 1432, 1374, 1334, 1040, 1018, 832, 648 cm−1; for 1H and 13C NMR data see Table 1; HRMS (ESI): calcd for C38H55Cl2N2O8 [M−H]−: 737.3341, found m/z 737.3339.
Carbamidocyclophane K (4)
White, amorphous powder; [α] −2.5 (c=1.2 g per 100 ml; MeOH); UV (c=4 μM; MeOH) λmax (log ɛ) 205 (5.456), 208 (5.385), 211 (5.342), 228 (4.686), 275 (3.933) nm; CD (c=82 μM; MeOH) λmax (Δɛ) 211 (2.09), 219 (−1.21), 238 (0.47), 252 (−0.05), 279 (−1.06) nm; ATR-IR (film) max 3370 (br), 2928, 2856, 1683, 1590, 1431, 1376, 1335, 1016, 833, 743, 653, 620 cm−1; for 1H and 13C NMR data see Table 1; HRMS (ESI): calcd for C37H54Cl2NO7 [M–H]−: 694.3283, found m/z 694.3293.
−2.5 (c=1.2 g per 100 ml; MeOH); UV (c=4 μM; MeOH) λmax (log ɛ) 205 (5.456), 208 (5.385), 211 (5.342), 228 (4.686), 275 (3.933) nm; CD (c=82 μM; MeOH) λmax (Δɛ) 211 (2.09), 219 (−1.21), 238 (0.47), 252 (−0.05), 279 (−1.06) nm; ATR-IR (film) max 3370 (br), 2928, 2856, 1683, 1590, 1431, 1376, 1335, 1016, 833, 743, 653, 620 cm−1; for 1H and 13C NMR data see Table 1; HRMS (ESI): calcd for C37H54Cl2NO7 [M–H]−: 694.3283, found m/z 694.3293.
Carbamidocyclophane L (5)
White, amorphous powder; [α] −6.0 (c=1.0 g per 100 ml; MeOH); UV (c=5 μM; MeOH) λmax (log ɛ) 206 (5.301), 209 (5.246), 210 (5.220), 229 (4.602), 275 (3.903) nm; CD (c=46 μM; MeOH) λmax (Δɛ) 208 (2.00), 217 (−0.78), 228 (−0.44), 242 (0.70), 251 (0.11), 258 (0.42), 281 (−0.75) nm; ATR-IR (film) max 3360 (br), 2927, 2855, 1678, 1586, 1430, 1393, 1375, 1336, 1012, 988, 829, 746, 648, 620−cm−1; for 1H and 13C NMR data see Table 1; HRMS (ESI): calcd for C37H53Cl3NO7 [M–H]−: 728.2893, found m/z 728.2879.
−6.0 (c=1.0 g per 100 ml; MeOH); UV (c=5 μM; MeOH) λmax (log ɛ) 206 (5.301), 209 (5.246), 210 (5.220), 229 (4.602), 275 (3.903) nm; CD (c=46 μM; MeOH) λmax (Δɛ) 208 (2.00), 217 (−0.78), 228 (−0.44), 242 (0.70), 251 (0.11), 258 (0.42), 281 (−0.75) nm; ATR-IR (film) max 3360 (br), 2927, 2855, 1678, 1586, 1430, 1393, 1375, 1336, 1012, 988, 829, 746, 648, 620−cm−1; for 1H and 13C NMR data see Table 1; HRMS (ESI): calcd for C37H53Cl3NO7 [M–H]−: 728.2893, found m/z 728.2879.
Accession codes
References
Brown, C. J. & Farthing, A. C. Preparation and structure of di-p-xylylene. Nature 164, 915–916 (1949).
Cram, D. J. & Steinberg, H. Macro rings. I. Preparation and spectra of the paracyclophanes. J. Am. Chem. Soc. 73, 5691–5704 (1951).
Morisaki, Y. & Chujo, Y. Cyclophane-containing polymers. Prog. Polym. Sci. 33, 346–364 (2008).
Moore, B. S. et al. [7.7]Paracyclophanes from blue-green algae. J. Am. Chem. Soc. 112, 4061–4063 (1990).
Moore, B. S., Chen, J. L., Patterson, G. M. L. & Moore, R. E. Structures of cylindrocyclophanes A-F. Tetrahedron 48, 3001–3006 (1992).
Chlipala, G. E. et al. Cylindrocyclophanes with proteasome inhibitory activity from the Cyanobacterium Nostoc sp. J. Nat. Prod. 73, 1529–1537 (2010).
Chen, J. L., Moore, R. E. & Patterson, G. M. L. Structures of nostocyclophanes A-D. J. Org. Chem. 56, 4360–4364 (1991).
Kang, H.-S. et al. Merocyclophanes A and B, antiproliferative cyclophanes from the cultured terrestrial Cyanobacterium Nostoc sp. Phytochemistry 79, 109–115 (2012).
Bui, H. T. N., Jansen, R., Pham, H. T. L. & Mundt, S. Carbamidocyclophanes A-E, chlorinated paracyclophanes with cytotoxic and antibiotic activity from the Vietnamese cyanobacterium Nostoc sp. J. Nat. Prod. 70, 499–503 (2007).
Luo, S. et al. Carbamidocyclophanes F and G with anti-Mycobacterium tuberculosis activity from the cultured freshwater cyanobacterium Nostoc sp. Tetrahedron Lett. 55, 686–689 (2014).
Appelbaum, P. C. MRSA-the tip of the iceberg. Clin. Microbiol. Infect. 12, 3–10 (2006).
Boucher, H. W. & Corey, G. R. Epidemiology of methicillin-resistant Staphylococcus aureus. Clin. Infect. Dis. 46 (Suppl 5), 344–349 (2008).
Furuno, J. P. et al. Methicillin-resistant Staphylococcus aureus and vancomycin-resistant enterococci co-colonization. Emerging Infect. Dis 11, 1539–1544 (2005).
Schito, G. C. The importance of the development of antibiotic resistance in Staphylococcus aureus. Clin. Microbiol. Infect. 12, 3–8 (2006).
Komarek, J. The modern classification of cyanoprokaryotes (cyanobacteria). Oceanol. Hydrobiol. St. XXXIV, 5–17 (2005).
Komarek, J. Cyanobacterial taxonomy: current problems and prospects for the integration of traditional and molecular approaches. Algae 21, 377–392 (2006).
Komarek, J. Modern taxonomic revision of planktic nostocacean cyanobacteria: a short review of genera. Hydrobiologia 639, 231–243 (2010).
Rajaniemi, P. et al. Phylogenetic and morphological evaluation of the genera Anabaena, Aphanizomenon, Trichormus and Nostoc (Nostocales, Cyanobacteria). Int. J. Syst. Evol. Microbiol. 55, 11–26 (2005).
Castenholz, R. W. in Bergey's Manual of Systematic Bacteriology. Phylum BX. Cyanobacteria eds Boone D. R., Castenholz R. W., 473–599 Springer-Verlag: New York, NY, USA, (2001).
Franche, C. & Reynaud, P. A. Characterization of several tropical strains of Anabaena and Nostoc: morphological and physiological properties, and plasmid content. Ann. Inst. Pasteur/Microbiol 137 (Suppl A), 179–197 (1986).
Altschul, S. F. et al. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Han, D., Fan, Y. & Hu, Z. An evaluation of four phylogenetic markers in Nostoc: implications for cyanobacterial phylogenetic studies at the intrageneric level. Curr. Microbiol. 58, 170–176 (2009).
Rudi, K., Skulberg, O. M. & Jakobsen, K. S. Evolution of cyanobacteria by exchange of genetic material among phyletically related strains. J. Bacteriol. 180, 3453–3461 (1998).
Neilan, B. A., Jacobs, D. & Goodman, A. E. Genetic diversity and phylogeny of toxic cyanobacteria determined by DNA polymorphisms within the phycocyanin locus. Appl. Environ. Microbiol. 61, 3875–3883 (1995).
Janson, S. & Graneli, E. Phylogenetic analyses of nitrogen-fixing cyanobacteria from the Baltic Sea reveal sequence anomalies in the phycocyanin operon. Int. J. Syst. Evol. Microbiol. 52, 1397–1404 (2002).
Janson, S., Matveyev, A. & Bergman, B. The presence and expression of hetR in the non-heterocystous cyanobacterium Symploca PCC 8002. FEMS Microbiol. Lett. 168, 173–179 (1998).
Gottlieb, H. E., Kotlyar, V. & Nudelman, A. NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 62, 7512–7515 (1997).
Rippka, R. et al. Generic assignments, strain histories and properties of pure cultures of cyanobacteria. J. Gen. Microbiol. 111, 1–61 (1979).
Guillard, R. R. L. & Lorenzen, C. J. Yellow-green algae with chlorophyllide c. J. Phycol. 8, 10–14 (1972).
Mundt, S., Kreitlow, S., Nowotny, A. & Effmert, U. Biochemical and pharmacological investigations of selected cyanobacteria. Int. J. Hyg. Envir. Heal 203, 327–334 (2001).
Andersen, R. A. & Kawachi, M. in Algal Culturing Techniques. Traditional Microalgae Isolation Techniques (ed Andersen R. A)., 83–100 Elsevier, Acad. Press: Amsterdam, The Netherlands, (2005).
Franche, C. & Damerval, T. Tests on nif probes and DNA hybridizations. Methods Enzymol. 167, 803–808 (1988).
Yilmaz, P. et al. The SILVA and “all-species living tree project (LTP)” taxonomic frameworks. Nucleic Acids Res. 42, 643–648 (2013).
Pruesse, E., Peplies, J. & Glöckner, F. O. SINA: accurate high throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829 (2012).
Ludwig, W. et al. ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371 (2004).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Bäcker, C. et al. Triterpene glycosides from the leaves of Pittosporum angustifolium. Planta Med. 79, 1461–1469 (2013).
European Committee for Antimicrobial Susceptibility Testing (EUCAST). Determination of minimum inhibitory concentrations (MICs) of antibacterial agents by agar dilution. Clin. Microbiol. Infect. 6, 509–515 (2003).
Kahlmeter, G. et al. European Committee on Antimicrobial Susceptibility Testing (EUCAST) Technical Notes on antimicrobial susceptibility testing. Clin. Microbiol. Infect. 12, 501–503 (2006).
Kahlmeter, G. et al. European harmonization of MIC breakpoints for antimicrobial susceptibility testing of bacteria. J. Antimicrob. Chemother. 52, 145–148 (2003).
Collins, C. H. et al. Collins and Lyne's Microbiological Methods 168–186 Arnold: London, UK, (2004).
Kreitlow, S., Mundt, S. & Lindequist, U. Cyanobacteria-a potential source of new biologically active substances. J. Biotechnol. 70, 61–63 (1999).
Muyzer, G., Teske, A., Wirsen, C. O. & Jannasch, H. W. Phylogenetic relationships of Thiomicrospira species and their identification in deep-sea hydrothermal vent samples by denaturing gradient gel electrophoresis of 16S rDNA fragments. Arch. Microbiol. 164, 165–172 (1995).
Wilmotte, A., Van der Auwera, G. & De Wachter, R. Structure of the 16 S ribosomal RNA of the thermophilic cyanobacterium Chlorogloeopsis HTF ('Mastigocladus laminosus HTF') strain PCC7518, and phylogenetic analysis. FEBS Lett. 317, 96–100 (1993).
Brosius, J., Palmer, M. L., Kennedy, P. J. & Noller, H. F. Complete nucleotide sequence of a 16S ribosomal RNA gene from Escherichia coli. Proc. Natl Acad. Sci. USA 75, 4801–4805 (1978).
Muyzer, G. et alin Molecular Microbial Ecology Manual. Denaturing Gradient Gel Electrophoresis (DGGE) in Microbial Ecology eds Akkermans A. D., van Elsas J. D., de Bruijn F. J., 1–27 Kluwer Academic Publishers: Dordrecht, The Netherlands, (1998).
Acknowledgements
Michael Preisitsch was financially supported in part by a grant of Landesgraduiertenförderung MV, Ernst-Moritz-Arndt-University, Greifswald. The Vietnam National Foundation for Science and Technology Development is thanked for supporting the work of Dr Hang TL Pham. We thank Professor Philip Williams (Department of Chemistry, University of Hawaii, Honolulu, USA) and Professor Yoshiharu Iwabuchi (Graduate School of Pharmaceutical Sciences, Tohoku University, Sendai, Japan) for providing (-)-cylindrocyclophane A and the synthetic analog. General support from Cyano Biotech GmbH, Berlin, Germany, is acknowledged. We are also grateful to Mrs Jana Kumpfmüller (Institute of Pharmacy, Ernst-Moritz-Arndt-University, Greifswald, Germany) for her assistance with the molecular biological investigations as well as Mrs Monika Beerbaum (Leibniz-Institute of Molecular Pharmacology (FMP), Berlin, Germany) for recording 1H-NMR spectra of the known compounds, Professor Klaus Weisz (Institute of Biochemistry, Ernst-Moritz-Arndt-University, Greifswald, Germany) for kindly providing the CD spectrometer and Dr Olaf Morgenstern and Janine Technau (Institute of Pharmacy, Ernst-Moritz-Arndt-University, Greifswald, Germany) for measuring the IR spectra.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article

Cite this article
Preisitsch, M., Harmrolfs, K., Pham, H. et al. Anti-MRSA-acting carbamidocyclophanes H–L from the Vietnamese cyanobacterium Nostoc sp. CAVN2. J Antibiot 68, 165–177 (2015). https://doi.org/10.1038/ja.2014.118
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2014.118
This article is cited by
-
The impact of culture conditions on growth and metabolomic profiles of freshwater cyanobacteria
Journal of Applied Phycology (2018)
-
A new strategy for aromatic ring alkylation in cylindrocyclophane biosynthesis
Nature Chemical Biology (2017)
-
Impact of temperature on the biosynthesis of cytotoxically active carbamidocyclophanes A–E in Nostoc sp. CAVN10
Journal of Applied Phycology (2016)
