
Abstract
A rapid and simple method was developed and validated for the determination of colistin A and B in Mueller–Hinton broth using LC-tandem MS. Both analyte and internal standard (IS) (polymyxin B1) were determined using ESI. The MS data were obtained via the selected reaction monitoring in positive-ion mode. A linear regression (weighted 1/concentration) was used to fit calibration curves over the concentration range of 0.0241–2.41 μg ml−1 for colistin A and 0.0439–4.39 μg ml−1 for colistin B. No interference peaks were found in the blank Mueller–Hinton broth tested. Inter- and intraday precision and accuracy were within 85–115% (coefficient of variation). Colistin was stable in the autosampler for at least 24 h at 4 °C and in Mueller–Hinton broth for at least 120 h at 35 °C. This assay has been successfully used to determine colistin A and B in Mueller–Hinton broth for in vitro pharmacodynamic model studies. Accurate determination of colistin in bacterial growth medium has a vital role in the studies examining dosage regimen of and bacterial resistance to colistin.
Similar content being viewed by others

Introduction
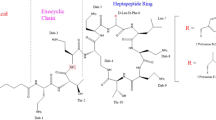
Polymyxins are increasingly used as the only therapeutic option for multidrug-resistant Gram-negative Pseudomonas aeruginosa, Acinetobacter baumannii and Klebsiella pneumoniae, which are resistant to all other antibiotics.1 Colistin, also known as polymyxin E, is an important member of the polymyxin class antibiotics. It was first isolated in 1949 from Bacillus polymyxa var. colistinus and became available for clinical use in 1959. The two major components are colistin A (polymyxin E1) and colistin B (polymyxin E2), accounting for more than 85% of total weight of the raw material.2 The two substances differ in the length of fatty acid side chain by one methylene group (Figure 1). Colistin methanesulfonate, an inactive pro-drug of colistin,3 is used by intravenous administration in clinical practice. It converts to partially sulfomethylated derivatives and colistin base in aqueous solution. Colistin is responsible for the antibacterial and toxic effects after the administration of colistin methanesulfonate.1

Chemical structure of colistin A and colistin B. Dab=L-α, γ-diaminobutyric acid. α and γ indicate the respective -NH2 involved in the peptide linkage. Polymyxin E1 (colistin A): FA=(+)-6-methyloctanoate, X=D-Leu; polymyxin E2 (colistin B): FA=(+)-6-methylheptanoate, X=D-Leu; polymyxin B1: FA=(+)-6-methyloctanoate, X=Phe.
Over the past decade, significant progress has been made in understanding their chemistry, pharmacokinetics (PKs) and pharmacodynamics (PDs). However, the rapid emergence of resistance to colistin urges us to pursue rational approaches as soon as possible, to maximize efficacy while minimizing the development of resistance. Identification of the most predictive PK/PD index is essential for optimizing dosing strategies of antibiotics in patients, and validated in vitro PK/PD models, which can serve as key tools in optimizing the regimen of antibiotics.4, 5, 6
Accurate simulation of antibiotic PKs in patients is a key prerequisite to establish in vitro PK/PD models. This highlights the importance of a robust analytical method for measurement of its concentrations in bacterial culture media. Unfortunately, no such robust method is available for colistin. A number of LC assays with different detection methods, including fluorescence and tandem MS (MS/MS), have been developed for quantification of colistin A and B in animal plasma, urine, milk and tissues, fishery products, and human plasma and urine,7, 8, 9, 10, 11, 12, 13 including one LC-tandem MS (LC-MS/MS) assay for colistin measurement in bacterial culture medium using just a simple precipitation step.14 However, all the previous methods cannot be considered very satisfactory for various reasons: (i) sample pretreatment was tedious or (ii) run times were relatively long or very long or (iii) lack of application in vitro PK/PD study on different bacteria. Above all, the absence of matrix effect study in all methods is of major significance, as solid-phase extraction (SPE) or protein precipitation is not considered to provide clean extracts and interference from matrix elements in MS/MS determination of colistin is very possible, for the method described by Jansson et al.14 Another major limitation is the absence of IS, as simple protein precipitation is not sufficient for bacterial culture media when using MS/MS to quantitate colistin. In addition, an IS should be used to overcome the potential recovery and instrumental variations. The use of such a precaution (netilmicin13 or polymyxin B1 as IS9) was not retained by Jansson et al.14
An HPLC method was ever employed to measure colistin in Mueller–Hinton broth.15 However, the HPLC method has a longer run time (15 min) and derivatization is labor-intensive and time-consuming.
In the present study, a simple, rapid, specific and reliable LC-MS-MS method was developed and validated for measuring colistin A and colistin B in the bacterial culture medium used in in vitro PK/PD model studies, which provides a solid foundation for the research of colistin resistance and dosage regimen.
Results and discussion
LC and MS
The primary amine groups of colistin led to considerable adsorption to the stationary phase of the LC column and caused significant peak tailing and obvious residue. A number of different types of chromatographic columns were tested. Atlantics d C18 (3 μm, 2.1 × 50 mm2) was finally selected for its good peak performance and robustness when running a large number of samples from in vitro PK/PD studies.
A gradient elution procedure (Supplementary Table 1) using a mixture of 0.1% formic acid in water and 0.1% formic acid in acetonitrile was used to gain a better peak performance. As colistin A, colistin B and polymyxin B have similar chemical structures and properties, it is not surprising that they have very similar retention times, about 4.8–5.0 min. Although overlapping of analytical peaks occurred in the total ion current mode, individual polypeptides under selected reaction monitoring experimental conditions did not impose any interference with each other for confirmation and quantification purposes.
Colistin A and B can form multiple charged ions in ionization process. The most common were triply and doubly charged ions. In this experiment, tuning solution (base, 0.829 mg ml−1) containing colistin A and B was introduced into the electrospray source by direct infusion (5 μl min−1) during the automatic tuning process. As the triply charged ions had better ion response than doubly charged ions in the experiment, the triply charged ions were used as the precursors for establishing selected reaction monitoring analysis (Supplementary Table 2). The main ions produced in MS and MS/MS were identified in the positive ionization mode. All MS parameters were optimized to achieve the highest sensitivity.
Sample preparation
The above LC-MS/MS conditions worked well for the standard solutions, but additional clean-up procedures were required for Mueller–Hinton broth samples. Jansson et al.14 have developed a method for the detection of colistin in Mueller–Hinton broth. However, when we tried to reproduce that in our laboratory, there was significant endogenous interference observed for colistin A, colistin B and the IS. Several conventional sample extraction methods such as protein precipitation, liquid–liquid extraction and solid-phase extraction techniques were also considered during the method development. A number of reported methods for plasma or urine samples were also carried out,9, 10, 12 but none could be applied to Mueller–Hinton broth samples because of significant interference. It is speculated that the physicochemical property of some medium composition was so close to colistin that it could not be removed by simple protein precipitation or one-step solid-phase extraction, and the liquid–liquid extraction method was beyond consideration for its low recovery (about 10%). Therefore, we developed an improved clean-up program. Samples were directly subjected to cleanup with solid-phase extraction, varieties of ingredients of the medium, and microorganisms and their metabolites were cleaned by three steps of washing program. Although the required sample amount of this method was higher than that of the Jansson’s method (50 μl), it was not an issue for the in vitro PK/PD experiment.
Results of method validation
Under the above condition, the retention times of colistin A, colistin B and the IS in Mueller–Hinton broth samples were very close, about 4.8–5.0 min. There was no interference with the analyte and IS. The representative chromatograms of blank Mueller–Hinton broth, spiked Mueller–Hinton broth sample (LLOQ) with colistin A, colistin B and the IS, and Mueller–Hinton broth samples from in vitro PK/PD model are shown in Figure 2.

Representative chromatograms of colistin A, colistin B and the IS (polymyxin B1) in Mueller–Hinton broth medium. (a) Blank Mueller–Hinton broth sample; (b) spiked Mueller–Hinton broth sample at the lower limit of quantification 0.024 μg ml−1 for colistin A and 0.044 μg ml−1 for colistin B; (c) a Mueller–Hinton broth sample collected at 5 h from an in vitro pharmacokinetic/pharmacodynamic (PK/PD) model study after administration of a single dose of colistin.
Calibration curves were linear in the range 0.0241–2.41 μg ml−1 for colistin A and 0.0439–4.39 μg ml−1 for colistin B, with r2>0.99. The LLOQ was 0.0241 μg ml−1 for colistin A and 0.0439 μg ml−1 for colistin B; the typical chromatograms of LLOQ samples are shown in Figure 2b.
The intraday precision ranged from 3.0 to 6.3% for colistin A and from 3.4 to 6.9% for colistin B, whereas the interday precision for colistin A and colistin B ranged from 2.9 to 6.6% and from 6.1 to 7.2%, respectively. The intraday accuracy ranged from 1.49 to 2.10% for colistin A and from −6.51% to 2.24% for colistin B. Interday accuracy ranged from −2.96% to 2.70% for colistin A and from −5.88% to 3.32% for colistin B (Supplementary Table 3).
The recoveries were consistent. The absolute recovery at the three concentration levels (±relative standard deviation) was 50.9±13.0, 55.9±4.07 and 51.9±5.48% for colistin A (n=3) and 50.4±6.22, 52.9±5.13 and 48.8±5.79% for colistin B (n=3). As described above, there were severe interference peaks from Mueller–Hinton broth for colistin A, colistin B and the IS using some reported methods. This problem has been resolved successfully by our three-step washing program. The only regret is the relatively low recovery of this method, because part of the analyte could be washed away.
In terms of matrix factor, the average values of three concentrations were in the range of 101−122%, 95.1–107%, 96.5–112% and 98.5–108% for colistin A in Mueller–Hinton broth without bacteria, with ATCC 19606, ATCC 27853 and ATCC 700603, respectively. The corresponding values for colistin B were 101.0–108, 98.2–104, 96.9–104 and 97.2–102% (Table 1). Our results show that the presence of various microorganisms had little effect on the LC-MS/MS assay.
Stability assessments were carried out to demonstrate that colistin was stable under typical sample storage and processing conditions (Table 2). Colistin was stable for at least 6 h at room temperature in Mueller Hinton broth, and 24 h in the matrix after preparation. In the in vitro PK/PD model at 35 °C, it was stable for at least 120 h. It was also stable after two freeze–thaw cycles from −40 °C to room temperature and for 1-month storage at −40 °C. The stock solution of colistin sulfate was stable for 4 months at −40 °C.
Application of the assay in in vitro PK/PD model studies
A steady-state colistin concentration 2.5 μg ml−1 was simulated in the model to mimic the PK of formed colistin in critically ill patients with infections caused by Gram-negative bacteria.16 The samples at different time points were assayed by the above method, and the average concentration at each point was plotted in Figure 3. Concentration trends of both colistin A and colistin B were similar to the target values.

Pharmacokinetic (PK) simulation of colistin A and B in the in vitro PK model study (mean±s.d., n=3).
Conclusion
A simple LC-MS/MS method was developed and validated for quantification of colistin in bacterial culture medium. This LC-MS/MS method overcomes the challenges due to interferences from the matrix in Mueller–Hinton broth. It is very sensitive, selective, accurate and reproducible. Importantly, our assay has been successfully used in the analysis of Mueller–Hinton broth samples from in vitro PK/PD studies.
Materials and methods
Chemicals and reagents
Colistin sulfate and polymyxin B sulfate were purchased from Sigma Chemicals (St Louis, MO, USA). Methanol, acetonitrile and formic acid (HPLC grade) were obtained from Sigma-Aldrich Laborchemikalien GmbH (Seelze, Germany). Mueller–Hinton broth was purchased from Difco BD (Franklin Lakes, NJ, USA). Milli-Q water (Millipore, Billerica, MA, USA) was used in all experiments.
Instrumentation
The LC-MS/MS was performed using a Waters Alliance 2690 high-performance liquid chromatography system (Milford, MA, USA) coupled to a TSQ Quantum triple quadrupole mass spectrometer equipped with an electrospray ionization (ESI) probe (Thermo Finnigan, San Jose, CA, USA). Data acquisition and processing were performed by Xcalibur 2.0.7 software (Thermo Finnigan, USA). Positive pressure-96 processor and Oasis HLB SPE C18 cartridges (30 mg, 1 ml) were obtained from Waters (Milford, MA, USA).
Chromatographic and mass spectrometric conditions
Chromatography was performed on an Atlantis column (50 × 2.1 mm2, i.d. 3 μm; Waters) maintained at room temperature and the injection volume was 10 μl. The mobile phase comprised a mixture of (A) 0.1% formic acid in water and (B) 0.1% formic acid in acetonitrile, and was delivered at a flow rate of 0.20 ml min−1 under a gradient elution procedure. The gradient used was 0–0.5 min, 5% B; 0.8–1.8 min, 90% B; 1.8–2.8 min, 5% B; 9 min, stop. The mass spectrometer was operated in the positive-ion mode. The optimized conditions were nitrogen sheath and auxiliary gas at 40 and 12 psi, respectively, with a spray voltage of 4.0 kV and vaporizer temperature of 270 °C. Quantification was performed using selected reaction monitoring of the transitions of m/z 101.2 from triply charged precursors of colistin A (m/z 391.0); m/z 380.6 from triply charged precursors of colistin B (m/z 386.0); and m/z 101.2 from triply charged precursors of the IS polymyxin B1 (m/z 402.0) with a scan time of 0.2 s per transition. Argon gas was used as the collision gas at a pressure of 1.5 mTorr, and the optimized collision energies of 18, 11 and 18 eV were chosen for colistin A, colistin B and the IS, respectively.
Preparation of stock and standard solutions
Two stock solutions of colistin (0.829 mg ml−1, the concentration given as that of the colistin base, i.e., without the sulfate moiety) were prepared independently in Milli-Q water. One stock solution was used for the standards and the other for the quality controls (QCs). The IS (polymyxin B sulfate) was weighed separately and a solution (1 mg ml−1) was prepared using Milli-Q water.
As no pure colistin A and B reference standards were available, colistin A and B had to be estimated. The percentage of colistin A and colistin B in the reference substance were estimated using a reported method.2 The flow rate was 1 ml min−1 and 20 μl of colistin solution (base, 0.829 mg ml−1) was injected (n=6). The batch of colistin was estimated to contain 29.1±0.26% of colistin A (±s.d.) and 53.0±0.29% of colistin B.
Blank Mueller–Hinton broth was spiked to prepare standard samples in the range of 0.0241–2.41 and 0.0439–4.39 μg ml−1 for colistin A and B, respectively, and QC samples for colistin A (0.0720, 0.483 and 1.93 μg ml−1) and for colistin B (0.132, 0.878 and 3.51 μg ml−1). Stock solutions, medium standards and QCs were stored at −40 °C. All dilution steps were carried out in 1.5 or 2 ml polypropylene tubes (Watson, Tokyo, Japan).
Sample pretreatment
An aliquot (600 μl) of Mueller–Hinton broth from the in vitro PK/PD model was vortex-mixed with 30 μl internal standard (10 μg ml−1 polymyxin B sulfate in water) in a polypropylene tube. The mixture was directly loaded onto an Oasis HLB SPE cartridge preconditioned with methanol (1 ml) followed by Milli-Q water (1 ml) on a positive pressure-96 processor (Waters). The first wash was conducted with 500 μl 60% methanol, followed by 500 μl of 70% methanol and 500 μl of 80% methanol. After eluting analytes with 2 × of 300 μl methanol–water–formic acid (80/20/0.2 (v v−1)), the combined eluents were injected directly into LC-MS/MS.
To ensure the samples injected into the LC-MS/MS contained no bacteria, the Mueller–Hinton broth samples were first centrifuged (12 000 r.p.m. for 15 min), and 100 μl of the supernatant was injected into LC-MS/MS. Colonies were counted after 48 h of incubation at 35 °C. Results showed that no bacteria were detected on the Mueller–Hinton agar plates, which demonstrated that our cleanup program was effective for the detection of colistin in Mueller–Hinton broth, regardless of whether there are bacteria.
Method validation
The LC-MS/MS method for the determination of colistin was validated with reference to the Guidance for the Industry: Bioanalytical Method Validation.17
The selectivity of the medium was examined by comparing chromatograms of the blank Mueller–Hinton broth spiked with colistin standards, and samples from in vitro PK/PD model after the administration of colistin.
Two linear calibration curves were generated using the ratio of the intensities of colistin A or B to polymyxin B1 vs concentration of colistin A or B. Calibration curves were obtained by least-squares linear regression with a weighting factor (1/x). Intra- and interday accuracy and precision were determined by duplicate analysis of six sets of samples spiked with three different concentrations of analyte, within a day and on six consecutive days. Accuracy was expressed as a relative error of the measured concentration over the targeted concentration (RE, %). Precision was expressed as the relative standard deviation (%). The acceptance criteria for the intra- and interday precision and accuracy were within 15% for QC samples at three different levels.
The absolute recoveries of colistin A and colistin B in Mueller–Hinton broth was determined by comparing the peak area of spiked Mueller–Hinton broth samples with the samples prepared with mobile phase. Recovery was determined at 0.0720, 0.483 and 1.93 μg ml−1 of colistin A and at 0.132, 0.878 and 3.51 μg ml−1 of colistin B in three replicates. As multidrug-resistant Gram-negative bacteria P. aeruginosa, A. baumannii and K. pneumoniae were examined in most in vitro PK/PD studies on colistin, the matrix effect of Mueller–Hinton broth samples were assessed in the presence and absence of different bacteria. The type strains P. aeruginosa ATCC 27853, A. baumannii ATCC 19606 and K. pneumoniae 700603 (up to108 CFU ml−1) were used in this experiment. To evaluate the matrix effect on the ionization of the analyte, six blank Mueller–Hinton broth samples from six different batches of blank Mueller–Hinton broth (with or without bacteria) were extracted and reconstituted with colistin in the mobile phase at three QC concentrations. The corresponding peak areas (A) were then compared with those of the standard samples in mobile phase (B). The ratio (A/B × 100%) is defined as the matrix factor, indicating the magnitude of the matrix effect.
The stability of colistin under different conditions was assessed by using the low-, medium- and high-level QC samples during all stages of this assay, including the short-term stability, postpreparative stability at autosampler (4 °C), freeze–thaw stability, long-term stability, 35 °C stability in Mueller–Hinton broth and stock solution stability.
Application of the assay in in vitro PK/PD studies
As there is a lack of PK data of colistin in Chinese subjects, a one-compartment PK/PD model was developed based on the recently published population PK model.16 For colistin, a one-compartment model was sufficient to describe the data, and the estimated half-life was 14.4 h. We constructed the in vitro PK/PD infection model as described previously with modifications.18 For the experiments, the entire system (except the computer-controlled peristaltic pump) was placed in an incubator at 35 °C with 5% CO2. The central compartment holds 200 ml Mueller–Hinton broth. To simulate colistin PK with this system; that is, to set the T1/2 as 14.4 h, the flow rate was 0.16 ml min−1. The drug was injected into the system through the dosing port at the zero time point. The dose added to the central compartment was 0.600 mg of colistin sulfate as a 0.5 ml solution, which is equivalent to 2.5 μg ml−1 colistin base, to mimic the PK of formed colistin in critically ill patients with infections caused by Gram-negative bacteria. After dosing, a 700 μl sample was obtained from the central compartment to determine concentration of colistin A and colistin B at the following time points: 1, 2, 4, 6, 8, 10, 12, 16, 20 and 24 h.
References
Bergen, P. J . et al. Pharmacokinetics and pharmacodynamics of 'old' polymyxins: what is new? Diagn. Microbiol. Infect. Dis. 74, 213–223 (2012).
Orwa, J. A ., Van Gerven, A ., Roets, E . & Hoogmartens, J . Liquid chromatography of polymyxin B sulphate. J. Chromatogr. A 870, 237–243 (2000).
Bergen, P. J ., Li, J ., Rayner, C. R . & Nation, R. L . Colistin methanesulfonate is an inactive prodrug of colistin against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 50, 1953–1958 (2006).
Andes, D ., Anon, J ., Jacobs, M. R . & Craig, W. A . Application of pharmacokinetics and pharmacodynamics to antimicrobial therapy of respiratory tract infections. Clin. Lab. Med. 24, 477–502 (2004).
Martinez, M. N ., Papich, M. G . & Drusano, G. L . Dosing regimen matters: the importance of early intervention and rapid attainment of the pharmacokinetic/pharmacodynamic target. Antimicrob. Agents Chemother. 56, 2795–2805 (2012).
Velkov, T ., Bergen, P. J ., Lora-Tamayo, J ., Landersdorfer, C. B . & Li, J . PK/PD models in antibacterial development. Curr. Opin. Microbiol. 16, 573–579 (2013).
Li, J . et al. Simple method for assaying colistin methanesulfonate in plasma and urine using high-performance liquid chromatography. Antimicrob. Agents Chemother. 46, 3304–3307 (2002).
Wan, E. C ., Ho, C ., Sin, D. W . & Wong, Y. C . Detection of residual bacitracin A, colistin A, and colistin B in milk and animal tissues by liquid chromatography tandem mass spectrometry. Anal. Bioanal. Chem. 385, 181–188 (2006).
Ma, Z ., Wang, J ., Gerber, J. P . & Milne, R. W . Determination of colistin in human plasma, urine and other biological samples using LC-MS/MS. J. Chromatogr. B 862, 205–212 (2008).
Gobin, P ., Lemaitre, F ., Marchand, S ., Couet, W . & Olivier, J. C . Assay of colistin and colistin methanesulfonate in plasma and urine by liquid chromatography-tandem mass spectrometry. Antimicrob. Agents Chemother. 54, 1941–1948 (2010).
Dotsikas, Y ., Markopoulou, C. K ., Koundourellis, J. E . & Loukas, Y. L . Validation of a novel LC-MS/MS method for the quantitation of colistin A and B in human plasma. J. Sep. Sci. 34, 37–45 (2011).
Xu, Y . et al. Analysis of colistin A and B in fishery products by ultra performance liquid chromatography with positive electrospray ionization tandem mass spectrometry. J. Chromatogr. B 899, 14–20 (2012).
Li, J . et al. A simple method for the assay of colistin in human plasma, using pre-column derivatization with 9-fluorenylmethyl chloroformate in solid-phase extraction cartridges and reversed-phase high-performance liquid chromatography. J. Chromatogr. B 761, 167–175 (2001).
Jansson, B ., Karvanen, M ., Cars, O ., Plachouras, D . & Friberg, L. E . Quantitative analysis of colistin A and colistin B in plasma and culture medium using a simple precipitation step followed by LC/MS/MS. J. Pharm. Biomed. Anal. 49, 760–767 (2009).
Bergen, P. J . et al. Pharmacokinetic/pharmacodynamic investigation of colistin against Pseudomonas aeruginosa using an in vitro model. Antimicrob. Agents Chemother. 54, 3783–3789 (2010).
Plachouras, D . et al. Population pharmacokinetic analysis of colistin methanesulfonate and colistin after intravenous administration in critically ill patients with infections caused by Gram-negative bacteria. Antimicrob. Agents Chemother. 53, 3430–3436 (2009).
US Department of Health and Human Services. Food and Drug Administration, Center for Drug Evaluation and Research, Center for Veterinary Medicine. Guidance for Industry: Bioanalytical Method Validation, US Department of Health and Human Services, (2013).
Liang, W . et al. Pharmacokinetics and pharmacodynamics of nemonoxacin against Streptococcus pneumoniae in an in vitro infection model. Antimicrob. Agents Chemother. 57, 2942–2947 (2013).
Acknowledgements
This study was supported by the New Drug Creation and Manufacturing Program of the Ministry of Science and Technology of China (2012ZX09303004-001) and the National Natural Science Foundation of China (81373493).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article

Cite this article
Zhao, M., Cao, YR., Guo, BN. et al. LC-MS/MS determination of colistin in Mueller–Hinton broth for in vitro pharmacodynamic studies. J Antibiot 67, 825–829 (2014). https://doi.org/10.1038/ja.2014.73
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2014.73
This article is cited by
-
A physicochemical assessment of the thermal stability of dextrin–colistin conjugates
Scientific Reports (2021)
-
Quantification of Colistin in Plasma by Liquid Chromatography-Tandem Mass Spectrometry: Application to a Pharmacokinetic Study
Scientific Reports (2020)
