
Abstract
Symbioses represent a frequent and successful lifestyle on earth and lichens are one of their classic examples. Recently, bacterial communities were identified as stable, specific and structurally integrated partners of the lichen symbiosis, but their role has remained largely elusive in comparison to the well-known functions of the fungal and algal partners. We have explored the metabolic potentials of the microbiome using the lung lichen Lobaria pulmonaria as the model. Metagenomic and proteomic data were comparatively assessed and visualized by Voronoi treemaps. The study was complemented with molecular, microscopic and physiological assays. We have found that more than 800 bacterial species have the ability to contribute multiple aspects to the symbiotic system, including essential functions such as (i) nutrient supply, especially nitrogen, phosphorous and sulfur, (ii) resistance against biotic stress factors (that is, pathogen defense), (iii) resistance against abiotic factors, (iv) support of photosynthesis by provision of vitamin B12, (v) fungal and algal growth support by provision of hormones, (vi) detoxification of metabolites, and (vii) degradation of older parts of the lichen thallus. Our findings showed the potential of lichen-associated bacteria to interact with the fungal as well as algal partner to support health, growth and fitness of their hosts. We developed a model of the symbiosis depicting the functional multi-player network of the participants, and argue that the strategy of functional diversification in lichens supports the longevity and persistence of lichens under extreme and changing ecological conditions.
Similar content being viewed by others

Introduction
Symbiosis, one of the most common lifestyles on earth, is a long-term interaction, which acts as source of evolutionary innovation (Margulis and Fester, 1991; Martin and Schwab, 2013). The term symbiosis was introduced by Frank (1877) in a study of lichens, which are today considered a classic example for self-sustaining partnerships of species belonging to different kingdoms of life (Nash, 2008). Lichens represent one of most diversified and oldest symbiotic lifestyles of fungi, with more than 18 000 recognized fungal species and a typical stratified morphology that evolved at least 415 million years ago (Honegger et al., 2013). The light-exposed lichen thalli are shaped by outer layers of fungal hyphae, which shelter internalized phototrophic partners. Lichen thalli develop only with proper combinations of fungal and algal species. Once the symbiotic phenotype is established, lichens may reach indeterminate ages and may even survive even the harshest conditions on earth (Øvstedal and Lewis-Smith, 2001). One reason for the ecological success of this fungal-algal partnership is the mutually enhanced ability to survive oxidative stress by suspended animation and rapid resumption of metabolism under permissive conditions (Kranner et al., 2005). However, additional and hitherto unidentified forces might have helped lichens to adapt to nutrient-poor and hostile habitats with strong fluctuation of abiotic parameters.
Most biology textbooks characterize lichens as an association solely between a fungal (mycobiont) and an algal (photobiont) partner. Our recent studies, however, revealed a high diversity of bacteria that are also associated with lichens. We have visualized biofilm-like communities, dominated by Alphaproteobacteria on the surfaces of thalli (Cardinale et al., 2008, 2012a, 2012b). Their structure suggests some degree of host specificity of the bacterial communities (Grube et al., 2009; Hodkinson et al., 2011; Bates et al., 2011). The ubiquity and abundance of lichen-associated bacteria challenge the classic view of lichens as a two-tier partnership and support an extended concept that also includes the universally present bacterial communities. With this new perspective in mind, we have now explored the potential functions of the bacterial communities with the goal of understanding their symbiotic context in a more holistic way. Functional analysis of the culturable fraction of the lichen-associated bacteria suggested their involvement in several roles, including iron and phosphate mobilization, hormone production, nitrogen fixation as well as several lytic activities (Liba et al., 2006; Grube et al., 2009). The culturable fraction usually represents only a minor part of the total lichen-associated microbiome and therefore most likely does not cover the host-dependent majority of the bacterial species (Cardinale et al., 2008). Recently developed omics approaches and subsequent bioinformatics tools are therefore required for a more comprehensive understanding of the role of the microbiome.
The objective of our study was therefore to investigate the function and metabolic potential of the bacterial lichen microbiome using a combined omics approach, together with a comprehensive spectrum of molecular, microscopic and physiological assays. The results were comparatively assessed and visualized using bioinformatics tools. For our study, we chose the lung lichen Lobaria pulmonaria (L.) Hoffm., a lichen which is considered to be endangered in many areas. L. pulmonaria serves as an indicator of primeval forest ecological continuity (Scheidegger and Werth, 2009). The mycobiont of L. pulmonaria engulfs a green-algal photobiont (Dictyochloropsis reticulata; found in 90% of the lichen surface) and a minor cyanobacterial partner, Nostoc sp. (Cornejo and Scheidegger, 2013). Schneider et al. (2011) provided an initial insight into the L. pulmonaria proteome and suggested Lobaria as an ideal model to study symbiotic processes. In our study, we extended this work and now provide results from the comparison of metagenomic and metaproteomic data.
Material and methods
Sampling strategy and preparation
L. pulmonaria was sampled from a rich population on maple tress (Acer spp.) in the Alps (Johnsbach, Austria; N 47°32′35′′, E 14°37′38′′) after visual inspection to avoid contamination by lichenicolous fungi and other organisms (Supplementary Figure S1). Using integrated sampling, a total amount of 176.3 g lichen was shock frozen with liquid nitrogen and immediately ground with mortar and pestle. The sample was homogenized in 360 ml 0.85% NaCl and filtered using a 63 μm mesh sieve; larger lichen parts were retained and colonizing bacteria were enriched in the filtrate. The filtrate was centrifuged at 8,000 r.p.m. at 4 °C for 20 min and the pellet was resuspended in 16 × 1.5 ml 0.85% NaCl aliquots. After a subsequent centrifugation step at 13 000 r.p.m. at 4 °C for 20 min, the supernatant was discarded and the pellets were used for DNA isolation (PowerSoil DNA Isolation Kit, MO BIO Laboratories Inc., Carlsbad, CA, USA). Following the DNA isolation, an aliquot containing 22 μg of metagenomic DNA was sent to GATC Biotech (Konstanz, Germany) for Illumina sequencing (HiSeq 2000 paired-end runs, Illumina Inc., San Diego, CA, USA).
Quality control and assembly of Illumina reads
Illumina HiSeq 2000 paired-end metagenomic DNA sequencing reads (GATC Biotech) were initially quality-checked using the FastQC program. The adapter sequence (5′-GATC GGAA GAGC ACAC GTCT GAAC TCCAG TCAC GTCC GCAC ATCT CGTAT-3′: identified as part of the Illumina TruSeq Indexed Adapter) was found in over 1% of the reads in set 1, and diminishing quality scores were observed towards the end of reads. Based on this observation, quality trimming and filtering was performed on each raw read set, using a custom-developed Perl script (written in-house by JS). The adapter sequences were removed and the sequence ends were trimmed, when the base quality score was smaller than 20. After the trimming step, reads were filtered out (i) if the length was shorter than 75 bp; (ii) or the read contained one or more ambiguous (N) bases; or (iii) the average quality score overall bases of the read was <25. After quality control, we did not observe overrepresented sequences, significant degradation of base qualities, or any other major quality issues. The sequence quality was confirmed again using the FastQC program. After quality control, some reads from set 1 did not have matching reads from set 2 due to read filtering, and vice versa. Therefore, to use paired-end information during assembly, only those reads that formed a matching pair were retained for further analysis.
Assembly of the reads into contiguous sequences (contigs) was performed using the Velvet de novo assembly software (Zerbino and Velvet, 2008; http://www.ebi.ac.uk/~zerbino/velvet/). Multiple trial assemblies with different k-mer lengths (57, 61, 65, 67, 69, 71 and 73) were performed and assembly statistics were compared with find the best k-mer length. All assemblies were conducted with an insert length of 350. The final k-mer length chosen was 71, which resulted in the maximum N50 value (2411 nts).
Taxonomic and functional analysis of assembled Illumina reads
The number of actual contigs used for this analysis was 368 424 (out of 503 528 overall contigs), based on the default Velvet minimum contig-length threshold of two times the k-mer length and the default coverage cutoff of half the median coverage. The Tera-BLASTN program (www.timelogic.com/documents/TeraBLAST_2009.pdf) was run on the 368 424 contigs, using TimeLogic (Active Motif, Carlsbad, CA, USA) DeCypher boards against the ‘nt’ database from NCBI (ftp://ftp.ncbi.nlm.nih.gov/blast/db). The blastn results were imported into MEGAN (Metagenome Analyzer, v4.70.4; Huson et al., 2011) to produce several taxonomy profiles. For assembly-based functional analysis, we used a similar approach as above but using BLASTX (www.timelogic.com/documents/TeraBLAST_2009.pdf), which was run against the ‘non-redundant protein sequence’ database from NCBI (ftp://ftp.ncbi.nlm.nih.gov/blast/db). The blastx results were imported into MEGAN (v4.70.4) for functional analysis. Both SEED and KEGG functional analyses were conducted with MEGAN.
MG-RAST analysis of Illumina reads
Unprocessed Illumina HiSeq 2000 reads were uploaded on the MG-RAST v3 public server (Meyer et al., 2008) to undergo paired-end reads joining and quality filtering (with default settings). 67 731 962 (88.8%) out of 76 310 051 sequences passed quality control; therein 60 015 088 (78,7%) sequences contained predicted proteins of either known or unknown function, while 1 788 100 (2.3%) sequences contained ribosomal RNA genes. SEED Subsystems Annotation was conducted with a maximum e-value cutoff of 1e−5 and a minimum 60% identity cutoff. Rarefaction analysis based on identified ribosomal RNA genes was done using best-hit classification and the Greengenes database (http://greengenes.lbl.gov) as the annotation source (with a minimum e-value cutoff of 1e−5; Supplementary Figure S2). The Lobaria metagenome was compared within a Principal Coordinates Analysis (annotation source: subsystems) with 20 publicly available datasets on MG-RAST. The functional abundance of eight particular habitats was compared using a minimum e-value cutoff of 1e−5. A table with all compared habitats together with their MG-RAST accession numbers is provided in the Supplementary Material (Supplementary Table S1). The metagenomic dataset is available under MG-RAST ID 4530091.3.
Quantitative real-time PCR
Quantification of nifH genes in the lichen DNA extract was conducted with primer pair nifH-F/nifH-R, as described by Hai et al. (2009). Standards containing the nifH fragments were prepared according to Bragina et al. (2013). Briefly, the gene fragments from Erwinia carotovora subsp. atroseptica SCRI1043 were cloned into the pGEM-T Easy Vector (Promega, Madison, WI, USA) and later re-amplified with vector-specific primers. Amplification-grade DNase I (Sigma-Aldrich, St Louis, MI, USA) treated total DNA extract was used to determine inhibitory effects of co-extracted substances. Based on this experiment, the total community DNA was diluted to 1:25 and target genes were amplified using KAPA SYBR FAST qPCR Kit (Kapa Biosystems, Woburn, MA, USA). Two independent runs, with three replicates for each sample, were performed on the Rotor Gene 6000 (Corbett Research, Mortlake, VIC, Australia), according to Bragina et al. (2013). The specificity of the amplicons was confirmed with both melting-curve analysis and gel electrophoresis of the qPCR products, respectively. Gene copy numbers for nifH were calculated per gram of lichen fresh weight.
Sample preparation for protein extraction
The collected thalli (Johnsbach, Austria; N 47°32′35′′, E 14°37′38′′) were cleaned with sterile tweezers from moss, bark and other visible contaminations. Samples from different thalli were pooled to a total amount of 2 g. Liquid nitrogen was added to the pooled thalli, which were subsequently ground to a fine powder using mortar and pestle. Proteins were extracted as described by Wang et al. (2006).
Gel electrophoresis with extracted proteins
One-dimensional SDS-polyacrylamide gel electrophoresis was performed as described earlier (Laemmli, 1970), and by loading 50 μg of extracted lichen protein mixture per lane. The sample was analyzed in three technical replicates (three lanes). Electrophoresis was carried out at 150 V and 250 mA for 45 min, afterwards proteins were fixed by shaking the gel in an aqueous solution containing 40% ethanol and 10% acetic acid for 30 min. Finally, proteins were stained with 25 ml of colloidal Coomassie Brilliant Blue G (Sigma-Aldrich, Steinheim, Germany), as described earlier (Neuhoff et al., 1988). The gel was scanned on a standard light scanner (Microtek, Hsinchu, Taiwan) for documentation. Afterwards each of the three lanes was cut into 20 pieces, as shown in the Supplementary Material (Supplementary Figure S3).
In-gel digestion after SDS-polyacrylamide gel electrophoresis
Each of the 20 gel pieces from the three technical replicates was cut into small cubes (1 mm3), which were destained by adding 700 μl of 30% acetonitrile containing 0.2 M NH4HCO3, and shaking for 15 min at 37 °C and 1500 r.p.m. This step was repeated twice. After destaining, the pieces were dried in a vacuum concentrator (Eppendorf, Hamburg, Germany). In-gel digestion was performed with 50-μl-modified sequence-grade trypsin (Promega), with an enzyme concentration of 2 μg ml−1 overnight at 37 °C. Subsequently, peptides were eluted by sonication for 15 min (Ultra sonic cleaner, VWR, Darmstadt, Germany). The eluted peptides were transferred into vials, dried in a vacuum concentrator (Eppendorf) again, and finally dissolved in 10 μl of ultrapure water.
Mass spectrometry analysis
Peptide mixtures resulting from in-gel tryptic cleavage were subjected to liquid chromatography-tandem mass spectroscopy measurements, using an EASYnLC 1000 (Thermo Scientific, Odense, Denmark), with self-packed columns (Luna 3 μ C18(2) 100 A, Phenomenex, Aschaffenburg, Germany) in a one-column setup online, coupled to an Orbitrap Elite mass spectrometer (Thermo Fisher, Bremen, Germany). Samples were loaded and desalted in 0.1% acetic acid, with a flow rate of 700 nl min−1, followed by peptide separation achieved by a binary non-linear 170 min gradient from 5–50% acetonitrile in 0.1% acetic acid at a flow rate of 300 nl min−1. Mass spectrometric measurement was performed in the Orbitrap Elite (Thermo Fisher Scientific) at a spray voltage of 2.4 kV applied to the emitter. After a survey scan in the Orbitrap (R=30 000) tandem mass spectroscopy data were recorded for the 20 most intensive precursor ions in the linear ion trap. Singly charged ions were not taken into account for tandem mass spectroscopy analysis. The lock mass option was enabled throughout all analyses.
Database construction for metaproteome analysis, database search and metaproteome data analysis
Due to the lack of translated metagenomic sequences of L. pulmonaria, a synthetic metagenomic-based database was created. This database contained all protein sequences available in the NCBI NR protein database (state of 13.09.26) which have been predicted to be present in the sample based on similarity searches of the metagenomic reads by BLAST (Altschul et al., 1990). Because neither the fungal nor the algal symbiont genomes of L. pulmonaria have been sequenced yet, all available NCBI protein sequences from fungi and algae were added to the database, together with a set of typical contaminations (for example, porcine trypsin, human keratin). Entries with the same protein sequence, but different headers were combined into one entry by an in-house php script (written by SF). The final database contained 2 473 550 protein sequences, pointing to 2 581 850 GenBank identifiers (bacteria: 1 564 300; algae: 226 723; fungi: 513 152; archaea: 26 952; other unclassified: 250 723).
The raw files were converted to mgf-files by MSconvert (www.Proteowizard.org), and searched with the Mascot search engine (version 2.2.04, Matrix Science Inc., Boston, MA, USA) with the following parameters: parent mass tolerance 10 ppm, fragment mass tolerance 0.5 Da, maximum missed cleavages 2; charge state 1+; variable modifications and oxidation of methionine. The Mascot search was followed by an X-tandem analysis in Scaffold (version 4.0.7, Proteome Software Inc., Portland, OR, USA). This analysis was performed as a ‘MudPit experiment’ to merge the individual mascot result files into a single file. The results were filtered as follows: 99% peptide probability, 1 peptide, 99% protein probability. Only proteins detected in two out of three technical replicates were considered for further analyses.
For functional classification and taxonomical distribution the in-house developed metaproteome analyses pipeline ‘Prophane 2.0’ was used (http://www.prophane.de; Schneider et al., 2011). Briefly, peptide to protein matches were clustered in groups by the Scaffold software. To standardize functional annotation all peptide to protein matches were functionally characterized based on TIGRFAMs (release 12; Haft et al., 2013) using HMMER3 (e-value⩽1E−10; Haft et al., 2013; Eddy, 2011). Functional data were transferred to the protein group if the members share the same predicted function. If these proteins share multiple predicted functions the function with the lowest overall e-value were assigned to the respective protein group. Groups of proteins sharing no functional prediction were named heterogeneous on functional level.
Moreover, protein groups were taxonomically classified based on the annotation of the respective protein members. Protein quantification was based on normalized spectral abundance factor values (Zybailov et al., 2006), however, only exclusive spectral counts were considered.
Voronoi treemaps
Voronoi treemaps were generated using Paver (Decodon, Greifswald, Germany, http://www.decodon.com/).
Fluorescence In Situ Hybridization and Confocal Laser Scanning Microscopy samples were collected from the same Lobaria population used for the metagenomics and metaproteomics analysis. Lichen thalli were fixed with 4% paraformaldehyde/phosphate-buffered saline (PBS) (v/v, 3:1) at 4 °C for at least 4 h, followed by three washing steps with ice-cold PBS. The samples were stored at −20 °C in ethanol absolute/PBS (v/v, 1:1). Before the hybridization step of the thallus, 30-μm thick cryosections were made. Fluorescence In Situ Hybridization was carried out in tubes, as outlined in Cardinale et al., (2008) and samples were analyzed using a Leica TCS SPE microscope (Leica Microsystems, Heidelberg, Germany), as well as image analysis and three-dimensional modeling, using the software Image Surfer (http://imagesurfer.cs.unc.edu/) and Imaris 7.0 (Bitplane, Zurich, Switzerland), respectively.
Isolation of lichen-associated bacteria
Lichen thalli of L. pulmonaria were sampled from three different locations in Austria (Tamischbachgraben, N 47°32′40′′, E 14°37′35′′, Johnsbach, N 47°32′35′′, E 14°37′38′′, and St. Oswald, N 46°44′50′′, E 15°04′26′′). After grinding lichen samples with mortar and pestle, a homogenate was prepared using sterile 0.85% NaCl in a 1:10 (w/v) ratio, together with a lab stomacher (BagMixer; Interscience, St Nom, France). Diluted fractions were plated on R2A agar (Carl Roth, Karlsruhe, Germany), R2A agar with 25 μg ml−1 cycloheximide, starch casein agar (Kuester and Williams, 1964) and ISP2 agar (Shirling and Gottlieb, 1966). Bacterial colonies were randomly picked within 5 days of incubation at room temperature.
Screening of isolates for in vitro antagonistic activity towards particular bacteria and fungi
Dual-culture experiments were carried out as confrontation assays, using different media and target organisms according to Berg et al. (2002) and Opelt et al. (2007). Lichen-associated isolates were spotted on solid media pre-inoculated with E. coli XL1 and S. aureus ATCC 25923 and assessed for inhibition zones after 4 days of incubation at 30 °C. Antagonistic activity against the fungus Botrytis cinerea Pers. (TU Graz culture collection, Graz, Austria) was tested by dual culture on Waksman agar, according to Berg et al. (2002) and assessed after 5–7 days of incubation at 20 °C. Cultures of the lichen-colonizing fungus Rhinocladiella sp. (TU Graz culture collection) were homogenized and resuspended in sterile 0.85% NaCl. Aliquots from one batch (50 μl) were used to inoculate each well of 24-well plates, containing solid potato dextrose agar (Carl Roth, Karlsruhe, Germany). Subsequently, 100-μl culture filtrate obtained from each lichen-associated isolate was added to particular wells. After 3 weeks of incubation, the wells were checked for growth reduction. All experiments were conducted with replicates and carried out twice.
Functional assays with Lobaria-associated bacteria
Altogether, 388 randomly selected bacterial cultures were subjected to functional assays based on different growth media. Protease, β-glucanase and chitinase activity were analyzed according to Berg et al. (2002), and phosphate solubilization of strains as described by Nautiyal (1999). Chromobacterium violaceum CV026 was used to detect C4–C6 AHL-mediated quorum sensing by visualizing purple pigmentation of the reporter strain (McClean et al., 1997). Pseudomonas putida F117 pAS-C8 and P. putida F117 pAS-C12 (Steidle et al., 2001) were used to detect C8 AHLs and C12 AHLs, respectively. Visualization of the green fluorescent protein-based AHL sensor was achieved through epifluorescence imaging, using an Universal Hood III (Bio-Rad, Hercules, CA, USA). All strains were incubated at 30 °C for 48 h.
Results
Taxonomic structure of the bacterial lichen microbiome
The analysis of 368 424 contigs revealed the taxonomic profile represented in Figure 1 that shows the overall composition of the metagenome and a more detailed structure of the dominant bacterial taxa within. Among the Proteobacteria, Alphaproteobacteria was the prominent taxon, with Rhizobiales and Sphingomonadales as the most frequently called orders. Within Rhizobiales, Methylobacteriaceae and Bradyrhizobiaceae are prominent, with Rhizobiaceae, Beijerinckiaceae, Xanthobacteriaceae and Phylobacteriaceae in minor quantities. Almost all of the Sphingomonadales belonged to the Sphingomonadaceae, and we estimate that a total of more than 800 bacterial species represent the diversity of the associated bacterial community, according to the rarefaction analysis (Supplementary Figure S2). Results derived from the present metaproteomic analysis presented an outmost similar community structure to the metagenome analysis. Proteobacteria were the predominant phylum and accounted for 361 distinct database hits, followed by Cyanobacteria with 47 hits and Acidobacteria with 28 hits. A complementary Fluorescence In Situ Hybridization visualization with Alphaproteobacteria and Betaproteobacteria specific probes, together with unspecific eubacteria probes illustrated and generally confirmed the taxonomic distribution observed by the metagenome analysis. Alphaproteobacteria were predominant and widespread on both, the upper and lower surface of the leaf-like lichen thallus, respectively, while Betaproteobacteria were less abundant and locally restricted (Figure 2a).

Taxonomic spectrum visualized with Krona (www.krona.sourceforge.net/) of contigs in the metagenome for all domains of life. Circles represent taxonomic classifications in ascending order up to the family level (outermost circle). Less-abundant taxa are listed outside the charts together with their relative abundance.

(a) Leaf-like thallus visualization of bacteria on a cross-section by 3D reconstruction of FISH image stacks. Eubacteria (red) and Alphaproteobacteria (yellow) were found widespread on both, the upper and the lower cortex, while Betaproteobacteria (pink) were less abundant and locally contained. Fungal hyphae (blue) and algae located under the upper cortex (green) were visualized without specific FISH probes, due to the naturally occurring fluorescence of the internal structures. (b) Model of the lichen symbiosis depicting the functional network of the participants. The model includes relevant functions of the colonizing bacteria, which are derived from metagenomic/metaproteomic analysis, as well as cultivation-dependent experiments.
Metagenome and proteome of the bacterial lichen microbiome
Functional analyses of the lichen metagenome focused on bacterial contigs, as the sampling procedure was designed to enrich the bacterial metagenome, using both SEED and KEGG functional analyses. SEED functional analysis was used for finding a set with functions of interest (that is, carbohydrate, virulence, cofactors and so on), many of which are standard SEED functional terms. Out of the 368 424 contigs, 69 823 were assigned to a functional term and the breakdown of the assignments at the top level is shown as a bar graph (Supplementary Figure S4). A significant number of contigs represented primary metabolic functions of bacteria (amino acids and derivatives: 6440; central carbohydrate metabolism: 2770; fatty acids, lipids and isoprenoids: 2721). Taxonomic separation of functional assignments revealed that up to 50% of different functional terms were covered by Alphaproteobacteria, while less-abundant bacteria account for the remaining contigs (Figure 3). Moreover MG-RAST visualization of functional abundance with SEED subsytems annotation showed presence of all level 4 functional terms. Out of the 368 424 contigs, 66 739 were assigned to KEGG pathways. Although the KEGG pathway assignment is primarily developed using mammalian genome information, it covers central capabilities of universal primary metabolism (carbohydrate metabolism: 12 823, energy metabolism: 7616, lipid metabolism: 5301, nucleotide metabolism: 5909, amino acid metabolism: 12 784, metabolism of other amino acids: 4068, glycan biosynthesis and metabolism: 2181 and genetic information processing: 12 258). Principal Coordinates Analysis carried out with MG-RAST revealed a unique functional distribution most similar to the one found on the plant phyllosphere (Figure 4; Supplementary Table S1).

Distribution of particular functions prior and after exclusion of less-abundant taxa. The number of contigs assigned to all organisms within the sample (blue bars) is visualized in contrast to assigned contigs of the eight most-abundant taxa (Alphaproteobacteria, Betaproteobacteria, Gammaproteobacteria, Deltaproteobacteria, Actinobacteria, Acidobacteria, Bacteroidetes and Firmicutes; red bars) and Alphaproteobacteria (green bars). The full colour version of this figure is available at ISME Journal online.

Principal Coordinates Analysis (PCoA) including 20 publicly available metagenomic datasets from MG-RAST and the Lobaria metagenome (red dot). All datasets were compared with subsytems and calculated using normalized values and the Bray-Curtis distance matrix. Single metagenomes from different biomes are labeled with their MG-RAST accession numbers and grouped in colored ellipses. The full colour version of this figure is available at ISME Journal online.
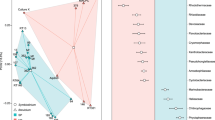
Lichen samples used for metagenome analyses were investigated in parallel on metaproteomics level. For this a in-gel tryptic digestion followed by liquid chromatography-tandem mass spectrometry based metaproteomics approach was used as recently described (Schneider et al., 2011) combined with a database containing all BLAST protein hits of the metagenomic reads (best hit per read). In total, our metaproteome analyses revealed 4405 different proteins, which were covered by at least one peptide-to-spectrum match (99% peptide probability, 99% protein probability) in at least two of three replicates. All proteins were divided into 3226 groups, based on shared peptide-to-spectrum matches by the Scaffold software (Koskinen et al., 2011). Out of these, 2676 and 541 groups could be unambiguously assigned to a eukaryotic and eubacterial origin, respectively, employing the Prophane workflow (Schneider et al., 2011). As our study aims for a better understanding of structure and functionality of the bacterial lichen microbiome, further functional analyses based on TIGRFAMs (e-value⩽1E−10) focused exclusively on protein groups of bacterial origin. Functional data were only transferred to bacterial protein groups whose members share the same functional prediction (424 out of 541, see material and methods for details). To gain a view on the role of bacterial proteins, the data were visualized with Voronoi treemaps, which also show the participation of different eubacterial groups in functions (Figure 5). In addition, we integrated the metaproteomic data with metagenomic data using the Voronoi treemap (Figure 6). Generally, most of the functional classes predicted by the metagenome analysis are also represented by at least one protein. As expected, numerous proteins assigned to functions involved in protein synthesis, DNA replication, recombination and repair were found in the metaproteome. Moreover, a significant number of proteins involved in protein fate and central energy metabolism, for example, tricarbonic acid cycle, were recovered from the bacterial lichen microbiome.

Voronoi Treemap visualization of the prokaryotic metaproteome on the taxonomic (a) and functional (b) level. The taxonomic and functional distributions were carried out by Prophane 2.0. Taxonomic groups are indicated by different colors (a and b) and the functional classes are separated by black lines (based on TIGRRoles). Each cell represents at least one protein (or protein group) assigned to the respective phylum.

Functional recovery of the bacterial lichen microbiome metagenome on the metaproteomic level using Voronoi treemaps. The protein sequences were compared with metagenomic contigs by BLAST and then functionally characterized using HMMER3 and TIGRFAMs. Upper panel shows all functions covered by the metagenome (third level) and the respective subroles (second level) and main roles (first level). The mainrole labels are shown. On the lower panel, metaproteomic coverage is shown (blue cells: functions present only in the metagenome; grey cells: functions present in both metagenome and metaproteome).
Selected functions of the bacterial lichen microbiome
Nutrient supply
About 2793 contigs suggested the presence of Ton and Tol transport systems, some of which are also involved in iron uptake. The TonB-dependent receptor, a family of beta-barrel proteins from the outer membrane of Gram-negative bacteria and responsible for siderophore transport into the periplasm, was present in 2094 contigs. Metaproteome analysis indicated at least four different types of TonB-dependent receptors, which were derived from two different bacterial phyla (Proteobacteria and Bacteroidetes, respectively). Phosphate metabolism is represented in 885 contigs, as well as in two PFAM classifications within the metaproteome and also includes proteins involved in solubilization of phosphates. Corresponding to this finding, 19.6% of all bacterial isolates from Lobaria formed clearing zones on NBRIP agar and thus underscored the potential role of lichen-associated bacteria in the solubilization of phosphates. Moreover, we utilized a quantitative real-time PCR approach, based on total community DNA, to evaluate the bacterial potential for nitrogen fixation. Therewith log10 5.0±0.1 nifH copies were identified per gram of lichen fresh weight.
Resistance against biotic stress factors (pathogen defense)
Virulence functions are common among the lichen-associated bacteria. About 1152 contigs were assigned to multidrug resistance efflux pumps, and multiple genes that code for resistance against antibiotics were also found (fluoroquinolone, vancomycin, methicillin, penicillin and cephalosporine) in addition to the 955 contigs that contribute to Type III, Type IV, Type VI and ESAT secretion systems. Type VI secretion systems were represented by three different variants in the metaproteome data, as well as one specific hit for an acriflavin-resistance protein. A moderate number of genes were also found which contribute to secondary metabolism, according to SEED analysis (548 contigs); some of the secondary metabolites are known as antibacterials or antifungals, but are represented by a fairly low number of contigs (phenazine biosynthesis protein PhzF: 26 contigs, clavulanic acid biosynthesis: 11 contigs). Furthermore, results from cultivation-dependent studies involving Lobaria-associated bacteria combined with taxonomic data from the present metagenomic approach suggest that around 7% of the taxa present have an antagonistic potential. The majority of these showed antagonism against fungi (data not shown).
Resistance against abiotic stress factors
Stress-related functions are distinct in the bacterial microbiome (2769 contigs; oxidative stress: 1238). About 891 contigs indicated genes conferring resistance to metals (copper, cobalt-zinc-cadmium, silver, mercury and arsenic). Interestingly, a similar proportion of oxidative-stress protectants to heavy metal efflux pumps was observed in the metaproteome (3 and 1, respectively). In addition, we looked for genes involved in dormancy and sporulation, as we expected this to be an important aspect of bacteria adapted to the poikilohydric life on lichens. However, only 13 contigs were related to this function.
Photosynthesis support by vitamin B12
Numerous genes are involved in the metabolism of cofactors, vitamins and prostethic groups (3799 contigs), according to SEED functional analysis, and a relatively high number of 1203 contigs was annotated to tetrapyrrole biosynthesis. Among these, for example, 365 contigs coded for coenzyme-B12 biosynthesis, 312 contigs for thiamine and 174 contigs indicated biotin biosynthesis. In the KEGG analysis, 5479 contigs were linked with the metabolism of cofactors and vitamins. Metaproteome analysis resulted in 2 hits that support the presence of enzymes involved in cobalamin biosynthesis, while folic acid and thiamine biosynthesis were represented by 1 hit each, respectively.
Hormone production
For the potential production of hormones, we found that auxin biosynthesis was represented by 156 contigs.
Detoxification of metabolites
In the KEGG analysis, many of the contigs that indicate xenobiotics biodegradation and metabolism (5482 contigs) seem to also be involved in the degradation of phenolic compounds. The presence of enzymes involved in degradation of phenolic compounds was reinforced by three hits in the metaproteome data.
Lytic activities
Degradation of older thallus parts could provide nutrients for the young and growing thallus parts. Such function is enhanced by bacterial enzymes involved in specific degradation procedures. 341 contigs within the metagenome are connected to chitin and N-acetylglucosamine utilization. Additionally, 1000 contigs were found for protein degradation of which 957 contigs were derived from bacteria. The cultivation-based functional screening underlines that Lobaria-associated bacteria are key-players in the recycling of old lichen parts: 33% of the isolates demonstrated protease activity, while 6.7 and 12.9% have shown chitinase and glucanase activity, respectively.
Other functions
In the KEGG analysis, the biosynthesis of polyketides and terpenoids was suggested in 2975 contigs, and the biosynthesis of other secondary metabolites is suggested by a further 2012 contigs. This set also includes known antibiotics such as betalain, penicillin, streptomycin, butirosin, neomycin and novobiocin, as well as phenylpropanoids, stilbenoids and alkaloids. A substantial number of contigs in the KEGG analysis was linked with pathways of environmental information processing (12 053 contigs), and of these, membrane transport forms a fraction of 7629 contigs. Some bacteria also undergo CO2-fixation, of which about 757 bacterial contigs were assigned. Notably, polyol utilization is represented in 301 contigs (ribitol-, xylitol-, arabitol-, mannitol- and sorbitol-utilization).
Quorum sensing was rather poorly represented in our dataset with only 25 contigs. Metaproteome analysis supports the under-represented role of quorum sensing within the Lobaria microbiome, no relevant sequences were identified. It was therefore not surprising, that cultivation-dependent experiments involving lichen-associated bacteria revealed that <1% of tested isolates produced detectable N-acyl homoserine–lactone-derived quorum sensing molecules. In addition, phages, prophages and transposable elements were not particularly common among the lichen-associated microbiome (275 contigs).
Comparison with an algal partner
To confirm that the selected functions noted above are truly dominant in the lichen-associated bacteria, we have compared the SEED function analysis of the Lobaria metagenome with that of a transcriptome of D. reticulata, the green-algal photobiont of L. pulmonaria. The transcriptome data were obtained from the Joint Genome Institute repository (sequencing of three cultured partners of L. pulmonaria, http://genome.jgi.doe.gov/Lobpulcupartners/Lobpulcupartners.info.html). There are a total of 102 102 contigs in the assembly (about 27.71% of the 368 424 contigs of the lichen metagenome). The SEED analysis for the D. reticulata data was performed in exactly the same way as was done for the lichen metagenome. Supplementary Table S2 shows a comparison of the selected microbiome function assignments between these two datasets, whereas Supplementary Table S3 shows the same kind of comparison of top-level SEED functions. As Supplementary Table S2 clearly shows, for every selected function discussed until now, the lichen data has far more contigs assigned than the D. reticulata data has. Although a genomic/transcriptomic comparison with both fungal and algal partners is currently not feasible, some bacterial functions, which contribute greatly to the overall symbiosis, have been confirmed.
Discussion
The analysis of our data has revealed metabolic capacities and potential roles of the lichen-associated bacteria, especially in the areas of stability and survival of the overall symbiosis. Using our multiphasic approach, combining omics technologies and physiological assays, we found diverse and previously unknown potential functions of the microbiome, such as nutrient supply, resistance against biotic and abiotic stress, support for the photosynthesis and for the growth of the two eukaryotic partners, as well as detoxification and thallus degradation abilities. Supportive roles of associated microbiomes are well-known from humans, animals and plants (Berg, 2009; Bragina et al., 2013; Cho and Blaser, 2012). With our present results, we have found new hints that a similar helper effect can be present in lichen symbioses. This supports our concept of lichens as complex microbial ecosystem (Grube et al., 2009). Combining metagenomic analysis of a bacteria-enriched Lobaria sample with a metaproteomic approach provided an additional insight into the functional and structural diversity of bacterial inhabitants. The identified potential functions of the lichen microbiome suggest interactions with the algal as well as with the fungal partner. Metagenomic and proteomic experiments have provided evidence for the capability of production of vitamin B12 and other cofactors supporting the beneficial algae–bacteria interaction. Many algae are auxotroph for vitamin B12, which is often synthesized by prokaryotes in symbiotic interactions (Croft et al., 2005). Other functions such as nutrient supply and resistance against biotic and abiotic stress factors indicate interactions with the fungal partner. Lichens are exposed to abiotic stress and a well-known target for parasitic fungi (Grube et al., 2012). In addition, hormones such as auxin produced by bacteria can support the growth of algae as well as fungi (Gutjahr, 2014). Although results confirmed by both metagenomics and proteomics data corroborated functionalities, a more detailed view is often limited by the availability of annotated data and the lack of completely sequenced genomes, for comparison. Some differences between the two approaches might also be explained by variation of the actual metabolic activity of the involved microorganisms at the time of sample preparation. However, the present results agree with a previous metaproteomic approach of an independent sample of the same lichen, covering the entire lichen holobiome. Schneider et al. (2011) showed that algal proteins are involved in energy production and a diverse set of functions of the fungal proteins relates to the role of the mycobiont in energy consumption and symbiotic control. Similar findings resulted from the analysis of the eukaryotic metatranscriptome in another lichen symbiosis (Juntilla and Rudd, 2012; Juntilla et al., 2013). The present study extends the previous studies in resolution of bacterial functionality. Focusing on the bacterial fraction, the comparison of the new proteomic and metagenomic datasets revealed an overall similarity in the taxonomic representation of bacterial organisms. In both datasets, Proteobacteria are the most prominent phylum, with Alphaproteobacteria as the most prevalent class. In contrast, cyanobacterial proteins were much more abundant as expected from the contig counts; they might be more active than other phylogenetic groups. As potential carbon and nitrogen fixing organisms, they are known for a long time as substantial part of lichen symbioses (Honegger et al., 2013).
Bacterial communities on long-living lichen thallus surface remain largely constant over seasons (Grube et al., 2009), despite they are exposed to substantial periodicity of abiotic factors in the habitat. Our data show that bacteria living on the surfaces of lichens are well-adapted to abiotic stress, in particular osmotic and oxidative stress. These properties match with the general property of lichens to tolerate periodic drought in their natural habitats. The periodic desiccation and rehydration cycles lead to repeated oxidative bursts at the surfaces of the fungal textures. Release of free radicals under these circumstances has been demonstrated and was interpreted as pathogen defense mechanism of lichens (Minibayeva and Beckett, 2001; Beckett et al., 2013). Thus, thallus-colonizing thalli bacteria without pronounced tolerance to oxidative stress and other selective conditions barely survive. By oxidative degradation of these non-adapted bacteria, a broad spectrum of additional nutrients is accessible. This source might be more important for oligotrophic lichens than for soil-provisioned plants (Paunfoo-Lonhienne et al., 2010; White et al., 2012). Consequently we hypothesize that periodic hydration acts as selective pressure for enrichment of specific and stress-tolerant bacterial communities, which can contribute to longevity and persistence of lichens under extreme and changing ecological conditions.
As we found little evidence of quorum sensing, we hypothesize that bacterial colonization of the thallus is mostly regulated by the fungal partner in the symbiotic community. It is well-established that the secondary metabolites of diverse lichen species have broad antibacterial properties (Boustie and Grube, 2005). The surprising abundance of bacteria on the surfaces and between crystals of secondary metabolites in lichens (for example, Lecanora polytropa; Grube et al., 2009) can only be explained by differences in the susceptibility to antibiotic compounds, which may be considered another factor of bacterial selection in lichens. Because we found significant numbers of multidrug resistance efflux pumps, the phylogenetically old lichen symbiosis could represent a natural reservoir of bacterial resistance mechanisms. Moreover, some of the adapted bacteria are potentially involved in the degradation of fungal secondary metabolites, as indicated by contigs of genes whose products may process complex and cyclic carbohydrates. These genes might also be interesting for biotechnological approaches, aiming at the degradation of xenobiotics. The presence of genes for the metabolism of typical bacterial antibiotics in our dataset suggests potential competition among bacterial strains on the lichen surfaces or a defense against other strains entering the microbial surface community. The ecological significance of these functions is pending further experimentation.
The morphological design of lichen structures could have a profound effect on the organization of the symbiotic networking. Bacterial communities primarily colonize the (hydrophilic) lichen surfaces, yet this pattern is strikingly different from the internalized symbionts in lichens, such as the green algae (D. reticulata in L. pulmonaria), which primarily contribute to the provision of photosynthetically produced carbohydrates. The green-algal strain is massively enriched within the fungal structures, while cyanobacterial Nostoc strains are acquired from the surfaces of L. pulmonaria recurrently during the life-time of the thallus to form internal organs devoted to nitrogen fixation in lichens (Hyvärinen et al., 2002, Cornejo and Scheidegger, 2013). Conversely, the external presence of other bacteria in the lichen symbiosis recalls the helper bacteria of mycorrhizal symbioses, which provide multiple functions to mutually support and stabilize the root symbioses, including exchange of carbohydrates and vitamin provision (Frey-Klett et al., 2007; Deveau et al., 2010). The long-living lichen thallus is formed by tightly packed fungal hyphae, which are conglutinated by their cell walls. Lichen-adapted bacteria benefit from the persistent cell walls for nutrition, and in return provide multiple helper functions for the longevity of lichen thalli (Figure 2b) to enhance fitness of the holobiome (symbiome). Although we have found a lot of supportive facts for this symbiosis model within our datasets, the evidence for fulfilled contribution of bacterial communities to the lichen symbiosis can only be found by additional experiments, for example, using isotope-labeled compounds and/or comparative physiological analysis between lichens with and without the bacterial microbiota. Both are currently difficult to establish due to slow metabolism and high diversity of bacteria. Despite these facts, we consider lichens as an interesting model for multi-biont symbioses, with different distributions of functions among the symbionts. It appears that partners with narrowly specified functions, such as carbohydrate or nitrogen fixation, are preferentially internalized by fungal structures and massively enriched in the lichen thallus. Internalization of a partner may help to provide a more uniform and stable environment for these partners. This symbiotic design has evolved as a convergence in unrelated fungal lineages (Grube and Hawksworth, 2007; Honegger, 2012). It has been optimized not only for the association with carbon-providing algal photobionts (Kranner et al., 2005), but also for the enrichment of bacterial supporters. Genome sequences of the eukaryotic partners, which are now becoming available (for example, Wang et al., 2014), will further help to address the intricacies of one of the oldest known symbiosis and its interactions with their bacterial helpers.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ . (1990). Basic local alignment search tool. J Mol Biol 215: 403–410.
Bates ST, Cropsey GWG, Caporaso JG, Knight R, Fierer N . (2011). Bacterial communities associated with the lichen symbiosis. Appl Environ Microbiol 77: 1309–1314.
Beckett RP, Zavarzina AG, Liers C . (2013). Oxidoreductases and cellulases in lichens: possible roles in lichen biology and soil organic matter turnover. Fungal Biol 117: 431–438.
Berg G . (2009). Plant-microbe interactions promoting plant growth and health: perspectives for controlled use of microorganisms in agriculture. Appl Microbiol Biotechnol 84: 11–18.
Berg G, Roskot N, Steidle A, Eberl L, Zock A, Smalla K . (2002). Plant-dependent genotypic and phenotypic diversity of antagonistic rhizobacteria isolated from different Verticillium host plants. Appl Environ Microbiol 68: 3328–3338.
Boustie J, Grube M . (2005). Lichens—a promising source of bioactive secondary metabolites. Plant Gen Res 3: 273–287.
Bragina A, Berg C, Müller H, Moser D, Berg G . (2013). Insights into functional bacterial diversity and its effects on Alpine bog ecosystem functioning. Sci Rep 3: 1995.
Cardinale M, Müller H, Berg G, de Castro JV Jr, Grube M . (2008). In situ analysis of the bacteria community associated with the reindeer lichen Cladonia arbuscula reveals predominance of Alphaproteobacteria. FEMS Microbiol Ecol 66: 63–71.
Cardinale M, Steinova J, Rabensteiner J, Berg G, Grube M . (2012a). Age, sun, and substrate: triggers of bacterial communities in lichens. Environ Microbiol Rep 4: 23–28.
Cardinale M, Grube M, de Castro JV Jr, Müller H, Berg G . (2012b). Bacterial taxa associated with the lung lichen Lobaria pulmonaria are differentially shaped by geography and habitat. FEMS Microb Lett 329: 111–115.
Cho I, Blaser MJ . (2012). The human microbiome: at the interface of health and disease. Nat Rev Gen 13: 260–270.
Cornejo C, Scheidegger C . (2013). New morphological aspects of cephalodium formation in the lichen Lobaria pulmonaria (Lecanorales, Ascomycota). Lichenologist 45: 77–87.
Croft MT, Lawrence AD, Raux-Deery E, Warren MJ, Smith AG . (2005). Algae acquire vitamin B12 through a symbiotic relationship with bacteria. Nature 438: 90–93.
Deveau A, Brulé C, Palin B, Champmartin D, Rubini P, Garbaye J et al. (2010). Role of fungal trehalose and bacterial thiamine in the improved survival and growth of the ectomycorrhizal fungus Laccaria bicolor S238N and the helper bacterium Pseudomonas fluorescens BBc6R8. Environ Microbiol Rep 4: 560–568.
Eddy SR . (2011). Accelerated profile HMM searches. PLoS Comput Biol 7: e1002195.
Frank AB . (1877). Über die biologischen Verhältnisse des Thallus einiger Krustenflechten. Beitr Biol Pflanzen 2: 123–200.
Frey-Klett P, Garbaye J, Tarkka M . (2007). The mycorrhiza helper bacteria revisited. New Phytol 176: 22–36.
Grube M, Hawksworth DL . (2007). Trouble with lichen: the re-evaluation and re-interpretation of thallus form and fruit body types in the molecular era. Mycol Res 111: 1–1132.
Grube M, Cardinale M, Vieira de Castro J, Müller H, Berg G . (2009). Species-specific structural and functional diversity of bacterial communities in lichen symbioses. ISME J 3: 1105–1115.
Grube M, Köberl M, Lackner S, Berg C, Berg G . (2012). Host-parasite interaction and microbiome response: effects of fungal infections on the bacterial community of the Alpine lichen Solorina crocea. FEMS Microbiol Ecol 82: 472–481.
Gutjahr C . (2014). Phytohormone signaling in arbuscular mycorhiza development. Curr Opin Plant Biol 21: 26–34.
Hai B, Diallo NH, Sall S, Haesler F, Schauss K, Bonzi M et al. (2009). Quantification of key genes steering the microbial nitrogen cycle in the rhizosphere of sorghum cultivars in tropical agroecosystems. Appl Environ Microbiol 75: 4993–5000.
Haft DH, Selengut JD, Richter RA, Harkins D, Basu MK, Beck E . (2013). TIGRFAMs and genome properties in 2013. Nucleic Acids Res 41 (Database issue): D387–D395.
Hodkinson BP, Gottel NR, Schadt CW, Lutzoni F . (2011). Photoautotrophic symbiont and geography are major factors affecting highly structured and diverse bacterial communities in the lichen microbiome. Environ Microbiol 14: 147–161.
Honegger R . (2012). The symbiotic phenotype of lichen-forming Ascomycetes and their endo- and epibionts. In: Hock B (ed.) The Mycota IX. Springer: Berlin, pp 287–339.
Honegger R, Edwards D, Axe L . (2013). The earliest records of internally stratified cyanobacterial and algal lichens from the Lower Devonian of the Welsh Borderland. New Phytol 197: 264–275.
Huson DH, Mitra S, Weber N, Ruscheweyh H, Schuster SC . (2011). Integrative analysis of environmental sequences using MEGAN4. Genome Res 21: 1552–1560.
Hyvärinen M, Härdling R, Tuomi J . (2002). Cyanobacterial lichen symbiosis: the fungal partner as an optimal harvester. Oikos 98: 498–502.
Juntilla S, Rudd S . (2012). Characterization of a transcriptome from a non-model organism, Cladonia rangiferina, the grey reindeer lichen, using high-throughput next generation sequencing and EST sequence data. BMC Genomics 13: 575.
Junttila S, Laiho A, Gyenesei A, Rudd S . (2013). Whole transcriptome characterization of the effects of dehydration and rehydration on Cladonia rangiferina, the grey reindeer lichen. BMC Genomics 14: 870.
Kranner I, Cram WJ, Zorn M, Wornik S, Yoshimura I, Stabentheiner E, Pfeifhofer HW . (2005). Antioxidants and photoprotection in a lichen as compared with its isolated symbiotic partners. PNAS 102: 3142–3146.
Koskinen VR, Emery PA, Creasy DM, Cottrell JS . (2011). Hierarchical clustering of shotgun proteomics data. Mol Cell Proteomics 10: M110.003822.
Kuester E, Williams ST . (1964). Selection of media for isolation of Streptomycetes. Nature 202: 928–929.
Laemmli UK . (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685.
Liba CM, Ferrara FIS, Mangio GP, Fantinatti-Garboggini F, Albuquerque RC, Pavan C et al. (2006). Nitrogen-fixing chemo–organotrophic bacteria isolated from cyanobacteria-deprived lichens and their ability to solubilize phosphate and to release amino acids and phytohormones. J Appl Microbiol 101: 1076–1086.
Margulis L, Fester R . (1991) Symbiosis as a source of evolutionary innovation: speciation and morphogenesis. MIT Press: Cambridge, pp 454.
Martin BD, Schwab E . (2013). Current usage of symbiosis and associated terminology. Int J Biol 5: 32–45.
McClean KH, Winson MK, Fish L, Taylor A, Chhabra SR, Camara M et al. (1997). Quorum sensing and Chromobacterium violaceum: exploitation of violacein production and inhibition for the detection of N-acylhomoserine lactones. Microbiology 143: 3703–3711.
Meyer F, Paarmann D, D’Souza M, Olson R, Glass EM, Kubal M et al. (2008). The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9: 386.
Minibayeva F, Beckett RP . (2001). High rates of extracellular superoxide production in bryophytes and lichens, and an oxidative burst in response to rehydration following desiccation. New Phytol 152: 333–343.
Mushegian AA, Peterson CN, Baker CCM, Pringle A . (2011). Bacterial diversity across individual lichens. Appl Environ Microbiol 77: 4249–4252.
Nash TH III . (2008) Lichen Biology 2nd edn Cambridge University Press: Cambridge, UK.
Nautiyal CS . (1999). An efficient microbiological growth medium for screening phosphate solubilizing microorganisms. FEMS Microbiol Lett 170: 265–270.
Neuhoff V, Arold N, Taube D, Ehrhardt W . (1988). Improved staining of proteins in polyacrylamide gels including isoelectric focusing gels with clear background at nanogram sensitivity using Coomassie Brilliant Blue G-250 and R-250. Electrophoresis 9: 255–262.
Opelt K, Chobot V, Hadacek F, Schönmann S, Eberl L, Berg G . (2007). Investigations of the structure and function of bacterial communities associated with Sphagnum mosses. Environ Microbiol 9: 2795–2809.
Øvstedal D, Lewis-Smith R . (2001) Lichens of Antarctica and South Georgia, A guide to their identification and ecology. Cambridge University Press: Cambridge, UK.
Paungfoo-Lonhienne C, Rentsch D, Robatzek S, Webb RI, Sagulenko E, Näsholm T et al. (2010). Turning the table: plants consume microbes as a source of nutrients. PLoS One 5: e11915.
Scheidegger C, Werth S . (2009). Conservation strategies for lichens: insights from population biology. Fungal Biol Rev 23: 55–66.
Schneider T, Schmid E, de Castro JV Jr, Cardinale M, Eberl L, Grube M et al. (2011). Structure and function of the symbiosis partners of the lung lichen (Lobaria pulmonaria L. Hoffm.) analyzed by metaproteomics. Proteomics 11: 2752–2756.
Shirling EB, Gottlieb D . (1966). Methods for characterization of Streptomyces species. Int J Syst Bacteriol 16: 313–340.
Steidle A, Sigl K, Schuhegger R, Ihring A, Schmid M, Gantner S et al. (2001). Visualization of N-acylhomoserine lactone-mediated cell-cell communication between bacteria colonizing the tomato rhizosphere. Appl Environ Microbiol 67: 5761–5770.
Wang W, Vignani R, Scali M, Cresti M . (2006). A universal and rapid protocol for protein extraction from recalcitrant plant tissues for proteomic analysis. Electrophoresis 27: 2782–2786.
Wang YY, Liu B, Zhang XY, Zhou QM, Zhang T, Li H et al. (2014). Genome characteristics reveal the impact of lichenization on lichen-forming fungus Endocarpon pusillum Hedwig (Verrucariales, Ascomycota). BMC Genomics 15: 34.
White JF Jr, Crawford H, Torres MS, Mattera R, Irizarry I, Bergen M . (2012). A proposed mechanism for nitrogen acquisition by grass seedlings through oxidation of symbiotic bacteria. Symbiosis 57: 161–171.
Zerbino DR, Velvet BE . (2008). Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18: 821–829.
Zybailov B, Mosley AL, Sardiu ME, Coleman MK, Florens L, Washburn MP . (2006). Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J Proteome Res 5: 2339–2347.
Acknowledgements
This work was supported by a joint grant of the Austrian and German Science Foundation (FWF, DFG) to GB, MG and KR (FWF-DACH Project I882).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Grube, M., Cernava, T., Soh, J. et al. Exploring functional contexts of symbiotic sustain within lichen-associated bacteria by comparative omics. ISME J 9, 412–424 (2015). https://doi.org/10.1038/ismej.2014.138
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2014.138
This article is cited by
-
Cotton microbiome profiling and Cotton Leaf Curl Disease (CLCuD) suppression through microbial consortia associated with Gossypium arboreum
npj Biofilms and Microbiomes (2023)
-
Bacterial microbiome in tropical lichens and the effect of the isolation method on culturable lichen-derived actinobacteria
Scientific Reports (2023)
-
Tackling fungal diversity in lichen symbioses: molecular and morphological data recognize new lineages in Chaetothyriales (Eurotiomycetes, Ascomycota)
Mycological Progress (2023)
-
Core taxa and photobiont-microbial interaction within the lichen Heterodermia obscurata (Physcsiaceae, Heterodermia)
Symbiosis (2022)
-
Metagenomic analysis of lichen-associated bacterial community profiling in Roccella montagnei
Archives of Microbiology (2022)
