
Abstract
Bacteria and fungi drive the decomposition of dead plant biomass (litter), an important step in the terrestrial carbon cycle. Here we investigate the sensitivity of litter microbial communities to simulated global change (drought and nitrogen addition) in a California annual grassland. Using 16S and 28S rDNA amplicon pyrosequencing, we quantify the response of the bacterial and fungal communities to the treatments and compare these results to background, temporal (seasonal and interannual) variability of the communities. We found that the drought and nitrogen treatments both had significant effects on microbial community composition, explaining 2–6% of total compositional variation. However, microbial composition was even more strongly influenced by seasonal and annual variation (explaining 14–39%). The response of microbial composition to drought varied by season, while the effect of the nitrogen addition treatment was constant through time. These compositional responses were similar in magnitude to those seen in microbial enzyme activities and the surrounding plant community, but did not correspond to a consistent effect on leaf litter decomposition rate. Overall, these patterns indicate that, in this ecosystem, temporal variability in the composition of leaf litter microorganisms largely surpasses that expected in a short-term global change experiment. Thus, as for plant communities, future microbial communities will likely be determined by the interplay between rapid, local background variability and slower, global changes.
Similar content being viewed by others

Introduction
Leaf litter microorganisms have an important role in terrestrial ecosystems through their effects on decomposition rates and carbon cycling (Swift et al., 1979; Schimel and Schaeffer, 2012). Microbial communities in general (Allison and Martiny, 2008; Singh et al., 2010), and leaf litter communities in particular (Allison et al., 2013), are sensitive to global changes in climate and nutrient availability. The variety of pathways by which such changes can affect leaf litter communities, however, complicates predictions of future responses. Changes in the abiotic environment can directly alter the growth and survival of particular microbial taxa. For example, moisture determines the physical connectivity of the terrestrial matrix, and desiccation stress can result in population extinction and decreased community diversity (Seifert, 1961; Treves et al., 2003; Castro et al., 2010; Sheik et al., 2011). In addition, environmental change might affect the leaf litter community indirectly via changes in the plant community and the quality or quantity of leaf litter (Kominoski et al., 2009; Cleveland et al., 2014). To complicate matters further, shifts in microbial composition are just one route by which environmental change impacts leaf litter decomposition. Decomposition rates are also influenced directly by abiotic conditions and changes in litter quality (for example, Mcclaugherty et al., 1985; Cotrufo et al., 1994; Cleveland et al., 2014).
To understand the importance of these different response pathways, we investigated leaf litter microbes and their functioning in a well-replicated, global change experiment (Potts et al., 2012; Kimball et al., 2014). Southern California grasslands are currently experiencing a range of environmental changes. Projections for California over the next 50 years suggest that annual temperature will increase by 3 °C and that precipitation will decrease by 10–25% (CEC, 2003). These changes are expected to drastically affect Mediterranean-type ecosystems where water availability is a key environmental constraint (Specht et al., 1983; Larcher, 2000). Indeed, most of the mean annual precipitation falls almost exclusively from November through April, with a predictable summer drought from May through October (Arguez et al., 2012). Further, nitrogen deposition rates in southern California are among the highest in the United States, with deposition ranging from 2.5 to 4.0 g N m−2 year−1 in many regions (Fenn et al., 2003). If these rates continue, nitrogen loading will remain a significant driver of southern California’s ecosystems (Padgett and Allen, 1999; Fenn et al., 2003; Zavaleta et al., 2003).
In temperate ecosystems, microbial responses to global change must be considered in light of temporal variation and, particularly, seasonal variation. Although previous studies have examined the response of microbial composition to environmental change over multiple years (for example, Sheik et al., 2011; Gutknecht et al., 2012), only a handful have considered the effect of seasonal variation on these responses (for example, Lage et al., 2010; Bell et al., 2014). However, the fast generation times of microbes mean that composition can turnover quickly, even across seasons (Bardgett et al., 1999; Kennedy et al., 2006; Habekost et al., 2008; Cregger et al., 2012; Gutknecht et al., 2012). We hypothesize that this high background variability will have several, important consequences for global change responses. First, the response of microbial communities to an incremental, mean change in the abiotic environment will be relatively difficult to detect compared with slower growing organisms, such as plants. Specifically, seasonal variation will greatly exceed the variation owing to global change (or global change treatments). Second, the response of microbial communities to environmental change will depend on the time of year. Seasonal variation in the traits of the microbial community could alter its response to environmental change (Bardgett et al., 1999; Niklaus et al., 2003; Cregger et al., 2012). Third, season-dependent responses will be particularly strong when the environmental parameter also varies seasonally. For instance, a drought treatment might shift the phenology of the microbial community (for example, promote an early appearance of the dry season community during the wet season). To test these hypotheses, we compared the compositional and functional responses of leaf litter microorganisms over 2 years (years 4 and 5 of the ecosystem manipulation). We characterized bacterial and fungal composition by amplicon pyrosequencing of the 16S and 28S rRNA genes and assayed functioning by potential enzyme activities and litter decomposition rates.
Materials and methods
Experimental design
The study area is a California grassland ecosystem 5 km north of Irvine, CA, USA (33°44’ N, 117°42’ W, 365 m elevation) that is dominated by exotic annual grasses and forbs. For this experiment, we used a subset of plots from an existing field manipulation of precipitation and nitrogen inputs that began in February 2007 (Potts et al., 2012; Allison et al., 2013). We utilized two levels of precipitation (ambient and ~50% reduction) applied at the plot scale (6.1 m × 12.2 m) and two levels of nitrogen (ambient or 60 kg N ha−1 year−1 added) applied to subplots within precipitation treatments. This design is replicated in eight experimental blocks. Within each block, we used subplots with (1) ambient precipitation and ambient nitrogen (‘control’ plots), (2) reduced precipitation+ambient nitrogen (‘drought’ plots), and (3) ambient precipitation+added nitrogen (‘nitrogen addition’ plots). Drought was imposed by covering the plots (using polyethylene sheeting over steel arch frames) only when rainfall was forecast, and the covers were removed soon after rainfall. A previous study at this site reported that litter derived from the nitrogen addition treatment contained significantly more nitrogen, cellulose and hemicellulose but lower concentrations of lignin (Allison et al., 2013). Similarly, litter decaying in the drought plots contained significantly more nitrogen and protein and significantly lower concentrations of cellulose, hemicellulose and lignin (Allison et al., 2013). Surface soil moisture (0 cm depth) was also significantly lower in drought plots than plots in the ambient treatment (two sample t-test, P>0.001; unpublished data).
Litter sample collection
Surface litter was collected from eight replicate plots for each environmental treatment (ambient, reduced precipitation and nitrogen addition) four times a year for 2 years (3 treatments × 8 times × 8 replicates=192 samples). The collection dates roughly corresponded to seasonal sampling and occurred on 14 April 2010, 20 August 2010, 17 December 2010, 28 February 2011, 10 June 2011, 21 September 2011, 14 December 2011 and 12 March 2012. We define the first four dates as year 1 and the last four dates as year 2. For each collection, litter was randomly sampled three times from a roughly 4 m2 section of a plot, combined and stored on ice for approximately 2 h. Litter samples were ground using a blade coffee grinder (KitchenAid model BCG111OB, Benton Harbor, MI, USA) for approximately 1 min, and 0.05 g of ground litter was flash frozen using liquid nitrogen and stored at −80 °C for DNA extraction.
DNA extraction
Microbial DNA from all litter samples was extractedfollowing a previously published procedue (DeAngelis et al., 2009), with a few modifications. Briefly, samples were extracted in lysing matrix E tubes (MP Biomedicals, Santa Ana, CA, USA) using a hexadecyl-trimethylammonium bromide (CTAB) extraction buffer, which is 10% CTAB, 500 mM phosphate buffer, and 1m sodium chloride; a final concentration of 100 mm aluminum ammonium sulfate was also added (Braid et al., 2003), along with phenol–chloroform–isoamylalcohol (25:24:1). Following bead-beating in a FastPrep FP120 (Bio101, Vista, CA, USA) at 5.5 m s−1 for 45 s, extracts were purified with chloroform. Nucleic acids were precipitated in 30% polyethylene glycol (PEG–NaCl) 6000, washed once with 70% ethanol and reconstituted in water. These extracted nucleic acids were then cleaned using an AllPrep Kit (Qiagen, Valencia, CA, USA) and then used for PCR amplification.
Microbial community sequencing
For the bacteria, 2 μl of a 1:10 dilution of DNA (median of 7.4 ng DNA; lower and upper quartiles of 4.8 and 10.1 ng DNA, respectively) from each extract was added to a PCR cocktail containing 1.2 units of HotStarTaq polymerase (Qiagen), 1 × PCR buffer supplied by the manufacturer, 200 μM of each dNTP, 0.7 μM of each primer and H2O to a final volume of 25 μl. 16S rRNA gene amplification used concatemers containing the universal primer 907 R (Lane, 1991), an 8- bp multiplex tag and the 454 ‘B’ adaptor CCTATCCCCTGTGTGCCTTGGCAGTCTCAG (in the reverse direction) and the complimentary primer 515F (Turner et al., 1999) and the 454 ‘A’ adaptor CCATCTCATCCCTGCGTGTCTCCGACTCAG (in the forward direction). Following an initial denaturation step at 95 °C for 5 min, PCR was cycled 35 times at 95 °C for 30 s, 63 °C for 45 s, 72 °C for 30 s, with a final extension at 72 °C for 10 min. We acknowledge that 35 cycles is relatively high and may exacerbate amplification bias, but we wanted to follow the Earth Microbiome protocol (Caporaso et al., 2012) for comparison to their extensive database.
For the fungi, the same PCR cocktail was used as above except the concentration of each primer was reduced to a final concentration of 0.4 μM. 28S rRNA gene amplification used concatemers containing the fungal-specific LROR (Tedersoo et al., 2008), an 8-bp multiplex tag, and the 454 ‘B’ adaptor (in the forward direction) and the complimentary primer LR5F (Tedersoo et al., 2008) with the 454 ‘A’ adaptor (in the reverse direction). Following an initial denaturation step at 95 °C for 3 min, PCR was cycled 35 times at 95 °C for 30 s, 54 °C for 45 s, 72 °C for 50 s, with a final extension at 72 °C for 10 min. For both the bacteria and fungi, each aliquot was amplified twice and subsequently pooled to reduce the effect of random PCR amplification.
PCR products were cleaned using the Agencourt AMPure XP PCR Purification Kit (Beckman Coulter Inc., Indianapolis IN, USA) following the manufacturer’s instructions, quantified using a Quant-iT (Life Technologies, Grand Island, NY, USA) Assay Kit and a Synergy 4 microplate reader (BioTek, Winooski, VT, USA), pooled into equimolar concentrations and pyrosequenced at the Duke University ISGP Sequencing Facility on a 454 Life Sciences FLX sequencer using Titanium chemistry (454 Life Sciences, Branford, CT, USA). 16S and 28S rRNA amplicons were run in separate regions of a gasketed plate. Additionally, the two sample years were prepared and sequenced separately, which could contribute additional, artificial variation across the sample years.
Characterization of the microbial community using metagenomic sequencing was obtained from Berlemont et al., 2014. Briefly, DNA extracts were pooled such that each collection date and environmental treatment combination contained two replicates (8 dates × 3 treatments × 2 replicates=48 DNA libraries in total). Metagenomic libraries were prepared using a Truseq Library Kit (Illumina, San Diego, CA, USA) and sequenced with an Illumina HiSeq2000 (100-bp paired ends). Taxonomic origin of the sequences down to the genus level was obtained using the M5NR database.
Plant community
Plant community composition data used in this study are from Kimball et al. (2014). Species composition and fractional cover was determined in all plots by point intercept during early to mid-April of 2010 and 2011, coinciding with late flowering and maximum seed set. Briefly, two 160 × 60 cm2 PVC frames with 10-cm interval grids were positioned within each plot. A stiff wire was dropped from each grid point, and the first-intercepted species was recorded. The point was recorded as plant litter or bare soil if live plant material was not encountered. The number of interceptions for each species was summed within a plot to calculate fractional cover. Fractional cover data of all species observed (22 in total) were used to generate a Jaccard and Bray–Curtis distance matrix. Only plant data collected from plots used for microbial sampling were used in the analyses.
Extracellular enzyme activity
The potential activities of nine extracellular enzymes involved in carbon and nutrient cycling were assayed as described in Alster et al. (2013) (Table 1). Briefly, litter samples were collected seasonally from September 2011 to March 2013 (seven sample dates in total) and frozen at −80 °C until analysis. Sample homogenates were prepared by mechanically homogenizing 0.1 g of litter in 60 ml of 25 mM maleate buffer (pH 6.0). The homogenates were continuously stirred while dispensing 200 μl per well into 96-well microplates with eight replicate wells per sample per assay. Fluorimetric enzyme assays were performed according to the methods described in German et al. (2011) and Alster et al. (2013), and oxidative enzymes were measured using a colorimetric assay described in Allison and Jastrow (2006) and Alster et al. (2013). The potential activities (in μmol g−1 h−1) of all nine enzymes were used to generate a Euclidian distance matrix of all samples.
Leaf litter decomposition
To assess how the environmental treatments influenced leaf litter decomposition rates, we analyzed a subset of the data from a litterbag study previously conducted at this site (Allison et al., 2013). Briefly, litterbags containing 2 g (dry weight) of litter from the drought, nitrogen addition or ambient plots were placed into nylon membrane bags with 0.45-μm pores and sterilized with at least 22 kGy gamma irradiation. Sterile litter bags were then reinoculated with 50 mg of air-dried, ground (Wiley mill, 1-mm mesh, Swedesboro, NJ, USA) non-sterile litter collected from drought, nitrogen addition or ambient plots. Litterbags were placed in the field on 15 December 2010, and subsets were retrieved on 3 March 2011, 14 June 2011 and 14 November 2011 for analysis of percentage of mass loss. Fresh litter in each bag was weighed, and a subsample was dried to a constant mass at 65 °C to obtain dry weight; all mass losses are reported as percentage of initial dry mass. For this study, we only analyzed ‘control’ litterbags whose litter contents, microbial inoculum and deployment environment were all from the same environmental treatment (8 litterbags × 3 treatments × 3 dates=72 litterbags in total).
Sequence analysis
Pyrosequencing data were processed using the QIIME (version 1.6.0) toolkit (Caporaso et al., 2010) with the following parameters: quality score >50, sequence length >300 and <700 for fungi and >200 and <550 for bacteria, maximum homopolymer of 6, 6 maximum ambiguous bases, and 0 mismatched bases in the primer. Sequences were denoised using Denoiser (Reeder and Knight, 2010), and operational taxonomic units (OTUs) were picked at the 97% identity level using UCLUST (Edgar, 2010) in QIIME. Fifteen of the 192 samples were excluded from the analyses for which quality data were not obtained. Because the number of reads in the remaining samples varied widely (623–42 560, median=6410), we generated median Bray–Curtis distance matrices based on 100 random sub-sampling of the full fungal and bacterial OTU-by-sample matrices. Briefly, 100 OTU-by-sample matrices were created in QIIME such that each matrix contained samples with the same number of bacterial (623) or fungal (2269) sequences (the number of sequences in the smallest sample). For each of these matrices, a Bray–Curtis distance matrix was created. Then, for each pairwise sample-to-sample comparison, the median value across the 100 distance matrices was chosen to make up the final median Bray–Curtis matrix used in the data analyses.
The taxonomic identity of OTUs was assigned to bacteria by reference to the Greengenes database (DeSantis et al., 2006) using the naive RDP classifier within QIIME (Wang et al., 2007) and to fungi by reference to the 28S LSU RDP database using BLAST. QIIME was also used to determine the Shannon diversity (α diversity metric) of all libraries on the rarefied OTU matrix (Caporaso et al., 2010). Unprocessed sequences are available through NCBI’s Sequence Read Archive (accession number SRP041807).
Data analysis
We analyzed microbial diversity and enzyme activity data using a factorial mixed-model analysis of variance (ANOVA; hereafter ‘overall ANOVA’). The model included plot treatment (ambient, drought or nitrogen addition) and collection date (hereafter referred to as season) nested within year as fixed, categorical effects and year and block as random effects, along with the season and plot treatment interaction. Plant data were analyzed using the same model but without the season and season and plot treatment interaction variables. Decomposition data were analyzed using a mixed-model ANOVA with plot treatment (ambient, drought or nitrogen addition) and time (3, 6 and 11 months) as fixed factors and block as a repeated-measure, random effect. All analyses were conducted in the R software environment.
Permutational multivariate analysis of variance (PERMANOVA) was used to test the effects of experimental factors on the distribution of microbial enzyme activity and bacterial, fungal and plant community composition. The microbial and enzyme models included plot treatment (ambient, drought or nitrogen addition) and season nested within year as fixed effects and year and block as random effects, along with the season and treatment interaction. The plant model included plot treatment as a fixed effect and year and block as random effects. PERMANOVA analyses were conducted using partial sums of squares on 999 permutations of residuals under a reduced model. When the model returned nonsignificant variables, these terms were removed and model was run again until all terms in model had a P-value of <0.05. Terms with lowest mean square were removed first. Multivariate analysis was conducted using PRIMER6 and PERMANOVA+ (Primer-E Ltd, Ivybridge, UK). Statistical routines are described in (Clarke and Warwick, 2001) and (Anderson et al., 2008).
To test whether the compositional differences were correlated between the microbial and plant communities, we performed a Mantel test based on 999 permutations using both Jaccard and Bray–Curtis matrices. We also determined whether the overall profile of enzymatic activity was correlated with bacterial or fungal composition. For litter samples where we had both microbial composition and potential enzyme activity data (72 samples), we first used a Mantel test to compare a Euclidean distance matrix of all enzyme activities and the microbial Bray–Curtis matrix. To further investigate whether overall enzyme activity could be explained by particular microbial taxa, we performed a distanced-based linear model test in PRIMER6 using the relative abundance of the top 20 bacterial and fungal OTUs as predictor variables. Analyses were conducted using a step-wise selection procedure to maximize the adjusted R2 based on 999 permutations.
Results
Leaf litter microbial composition
We characterized 2199 bacterial and 830 fungal OTUs using a 97% sequence similarity cutoff from roughly 1.2 and 1.3 million high-quality sequences, respectively (Table 2). Most of the bacterial OTUs were classified as Proteobacteria (28%), Bacteriodetes (16%) or Actinobacteria (15%), with Planctomycetes (11%) and Firmicutes (10%) comprising notable fractions as well. The vast majority of the fungal OTUs were classified as Ascomycota (92%) or Basidiomycota (8%), but OTUs classified as Chytridiomycota or Blastocladiomycota were also detected. The majority of bacterial and fungal OTUs were detected more than once (only 30% and 35% were singletons, respectively) and in multiple samples. However, much of this diversity was not evenly distributed across samples; 873 bacterial OTUs (40%) and 281 fungal OTUs (34%) were unique to one sample.
The most diverse bacterial phyla were also the most abundant; Proteobacteria, Bacteriodetes, and Actinobacteria made up 23, 26 and 49% of the sequences, respectively. Notably, four OTUs (from the genera Duganella, Curtobacterium, Frigoribacterium and Kineococcus) were observed in all samples and together represented approximately 40% of all sequences (Supplementary Table S1). Indeed, despite high total richness, a small number of bacterial taxa dominated the samples; the two most abundant OTUs, Curtobacterium and Frigoribacterium (both in the Microbacteriaceae family), represented approximately 34% of the sequences and 20 OTUs made up 73% of all the sequences (Table 1 and Supplementary Table S1). This trend was even more striking in the fungal community; six OTUs (Capnodiales, Davidiellaceae OTU 1, Phaeosphaeriaceae OTU 1, Pleosporaceae OTU 1, Agaricostilbaceae and Tremellaceae) were observed in all samples and together represented 83% of the sequences (Supplementary Table S1). The top 20 most abundant fungal OTUs made up 94% of the sequences (Table 2, Supplementary Table S1).
Although amplicon sequencing is known to suffer from a variety of methodological biases, the broad compositional results observed in our data sets were also observed using metagenomic sequencing of the same samples (Berlemont et al., 2014; Supplementary Figure S1). The most abundant bacterial and fungal phyla were detected in both the data sets. Although families (the finest taxonomic resolution that can generally be assigned to the metagenomic sequences) were distributed more evenly in the metagenomes, their relative abundances were positively correlated across ribosomal DNA and metagenomic data sets (Supplementary Figure S2). Additionally, the families only detected in the metagenomes represented just 14% and 11% of the bacterial and fungal metagenomic sequences, respectively.
Treatment and temporal effects on microbial composition
The drought and nitrogen addition treatments had small but significant effects on microbial community composition analysis (white and gray bars in Figure 1; PERMANOVA: P<0.002 in all instances). The main effect of the drought treatment explained approximately 3% of the bacterial and 6% of the fungal community composition. Additionally, the main effect of the nitrogen treatment was consistent for both the bacterial and fungal communities, explaining roughly 2% of the changes in community composition. Bacterial and fungal community evenness (mean±s.e.m.) was not significantly affected by either of the environmental treatments (Table 3).

PERMANOVA multivariate components of variation for the bacterial (white), fungal (gray), microbial enzymes (black) and plant (checkered) communities for each the drought (a) and nitrogen addition (b) treatments (average based on 999 unique permutations). Data include samples from all collection dates (8 in total for microbial communities, 7 for enzyme concentrations and 2 for plant community). The values plotted are the proportional sizes of the estimates of the variance components. All P-values <0.05.
Although microbial composition was altered by the environmental treatments, it was even more strongly influenced by annual and seasonal variation (Figures 1 and 2, Supplementary Figure S3). The interannual variation can even be seen at the phylum level. For example, 16S rRNA sequences identified as Actinobacteria, Bacteriodetes and Proteobacteria represented 43, 28 and 28% of all sequences in year 1 but shifted to 60, 25 and 22% in year 2. Microbial evenness also varied by season (Table 3) and was lower during the summer months than the rest of the year (Figure 3c).

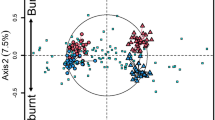
Non-metric multidimensional scaling ordination based on Bray–Curtis similarities depicting (a) fungal and (b) bacterial community composition in the second sampling year. The samples collected during the same collection date are displayed in the same symbol. The color of the symbols represent the plot treatment: ambient conditions (blue), drought (yellow), or nitrogen addition (green).

Average (±s.e.) maximum daytime temperature (a), total monthly precipitation (b) and shannon index values (c) for bacterial (white) and fungal (gray) communities across all collection dates.
Drought, but not nitrogen addition, interacted with season to affect bacterial and fungal composition, explaining 3% of the variation (Figure 1). Drought was more likely to affect composition in samples collected during the rainy season (Table 4). Further, the drought × season interaction was apparent when we repeated the analyses with just the abundant (top 20 OTUs) or rare taxa (not in the top 20) (Supplementary Figure S5). Finally, the effect of drought on fungal evenness also varied by collection date (Table 3), although a seasonal trend was less apparent (Table 4).
Given that the microbial response varied by season, we next asked whether taxa that responded to the drought manipulation responded in a similar way to the dry, summer season. In general, the majority of fungal taxa that decreased in relative abundance in the drought treatment tended to have reduced relative abundances during the summer, although this trend was not statistically significant (χ2=0.98, P=0.32; Supplementary Figure S4b). The same trend was not apparent for the bacteria; taxa that decreased in relative abundance during the dry season were equally likely to show increased or decreased relative abundances in the drought manipulation (χ2=0.25, P=0.62; Supplementary Figure S4a).
Although the degree to which microbial composition responded to the two environmental treatments was similar (comparing panels a and b in Figure 1), the direction of responses did not appear to be linked (Figure 4). Some taxa, such as Methylobacterium OTU 2, had a higher relative abundance in the nitrogen addition plots but decreased in relative abundance in the drought plots. Other taxa, such as Kineococcus and Pleosporaceae OTU 1, showed a positive response to the drought treatment but no response to the nitrogen addition treatment. Three of the most abundant fungal taxa (belonging to the Dothioraceae, Phaeosphaeriaceae and Tricholomataceae families) also had significantly lower relative abundances in both the nitrogen addition and drought treatments (Figure 4b).

Percentage of change (mean±s.e.) in relative abundance of the 20 most common (a) bacterial and (b) fungal OTUs in nitrogen addition (diagonal) or drought (white) plots relative to ambient plots across all collection dates. Asterisks (*) denote P-value of <0.05 in two sample t-test between the ambient and drought or nitrogen treatments.
Extracellular enzyme activities
\To investigate potential functioning of the microbial decomposers, we assayed the potential activity of extracellular enzymes in the leaf litter. Similar to the bacterial and fungal communities, variation in enzyme composition was not only most strongly influenced by temporal variation (black bars in Figure 1) but was also affected significantly by the nitrogen treatment. When each enzyme was analyzed individually, we also observed strong seasonal effects, and in general, activities were lower in the dry summer months (Supplementary Tables S2 and S3). Some enzyme activities varied significantly across environmental treatments. For example, acid phosphatase activity was significantly higher in the nitrogen treatment than in the ambient treatment, and polyphenol oxidase activity was significantly lower in the drought treatment (Supplementary Table S2). The potential activities of the three enzymes (polyphenol oxidase, β-xylosidase and leucine aminopeptidase) also had a significant interaction between drought and season and were generally lower in the dry, summer months (Supplementary Table S2). Although microbial composition and enzyme activities varied similarly through time and in response to the environmental treatments, microbial composition was not correlated with the overall profile of potential enzyme activities (Mantel tests: P>0.05). However, a selection of the most abundant microbial taxa did correlate with enzyme activities. A model of eight bacterial OTUs or five fungal OTUs explained 13% and 23% of total variation in the overall profile of enzymatic activity, respectively (Supplementary Table S4).
Comparison to plant community
To put the variation in microbial composition into perspective, we analyzed plant composition from the same plots. Although the plant and microbial data sets are not entirely comparable (plant composition was not measured across seasons), we could examine interannual versus treatment variation. As with the microbial communities, plant composition varied largely through time (checkered bars in Figure 1). Drought also altered plant composition (Figure 1a), owing to a significant reduction of native grasses in drought plots (Supplementary Table S5). Unlike the microbial communities, however, nitrogen addition treatment did not affect plant community composition (Figure 1b), although it did decrease the relative abundance of native grasses and increase the relative abundance of non-native grasses (Supplementary Table S5). When we compared the plant and microbial communities directly, we found a significant positive correlation between plant community composition (binary Jaccard distance) at the end of each growing season and the microbial communities (Bray–Curtis distance) the following winter (rM=0.31, P=0.001 and rM=0.22, P=0.001 for bacterial and fungal communities, respectively). Thus plant and microbial communities appeared to be similarly sensitive to the treatments and temporal variability in this system.
Litter decomposition rates
Leaf litter mass generally declined through time, with an average of 78–83% of initial litter mass remaining after 11 months (Figure 5). Across all time points, the drought and nitrogen addition treatments did not significantly affect the fraction of leaf litter decomposed (mixed-model ANOVA: χ2=0.14, P=0.71 (drought) and χ2=0.13, P=0.72 (nitrogen)). The response of decomposition to drought varied by season, however. Decomposition was significantly lower in the drought litterbags after 6 months (mixed-model ANOVA: χ2=22.2, P<0.001; Figure 5). By the end of the dry season (month 11), mass loss was again similar across treatments.

Mean (±s.e.) percentage of mass remaining over time in each of the three environmental treatments. N=8 for each treatment × sample date combination. Asterisks (*) represent significant differences between treatments, based on a Tukey's post-hoc test.
Discussion
Climate and plant production are highly seasonal in California grasslands. These factors are also known to affect microbial composition either directly or indirectly (Zak et al., 2003; Fierer et al., 2009; de Vries et al., 2012). In light of this background temporal variation, we quantified the compositional and functional responses of grassland litter microbes to two simulated global changes: drought and nitrogen addition. The responses of microbial composition to these treatments were consistent with our three hypotheses. Both bacterial and fungal composition varied strongly over seasons and years, and these effects were larger than either environmental treatment (Hypothesis 1). The response of both microbial groups to drought depended on season, however (Hypothesis 2). In contrast, their responses to nitrogen were consistent among seasons, supporting the idea that seasonal dependence is stronger for highly seasonal factors, such as precipitation (Hypothesis 3).
The response of microbial composition to the treatments and temporal factors was mostly concordant with the response of the functional potential of the litter communities, as assayed by the potential activities of extracellular enzymes. Of the variation that could be explained, most of the variation in enzymatic activity could be attributed to seasonal and interannual variation and less so to the nitrogen treatment. Unlike microbial composition, however, drought did not significantly affect overall enzyme activity, although the activity of many individual enzymes (AG, AP, BX, NAG and PPO) tended to be lower in the drought plots than the ambient plots. Further, the enzyme response to drought seemed to be less season dependent than the compositional response. Although three of the nine enzymes responded differently to drought over time, the overall enzyme profile did not (that is, there was no drought × time interaction). These results indicate some degree of functional redundancy among the extracellular enzyme-producing microbes among the wet and dry season communities. In contrast, the nitrogen treatment had a small but similar impact on the variation in microbial composition and its functional potential, suggesting that added nitrogen selects for taxa that differ in their enzymatic activities. Indeed, a selection of some of the dominant bacterial and fungal taxa explained a small (13% or 23%, respectively) but significant amount of variation in the profiles of enzyme activities across our samples.
Similar to microbial composition and enzyme activity, plant community composition also varied primarily over time and was altered by drought. Nitrogen addition did not affect overall plant species composition but did alter the relative abundance of native and non-native grasses. In addition, plant community composition was positively correlated with microbial composition on litter collected later in the year. The parallel response patterns of microbes and plants and the direct correlation between their composition both indicate that the plant community might be driving some of the changes in microbial composition and its potential functioning (as seen in Zak et al., 2003; Loranger-Merciris et al., 2006). Indeed, litter chemistry was altered in both treatments (Allison et al., 2013). For instance, drought increased lignin and decreased cellulose and hemicellulose concentrations, whereas added nitrogen had the opposite effects.
Together, however, the plant and microbial responses to the global change treatments did not translate into discernible impacts on litter decomposition. Drought slowed decomposition during the spring; however, neither drought nor nitrogen addition altered total mass loss over a full year. This result is surprising considering the mounting evidence for the influence of microbial composition (Bell et al., 2005; Tiunov and Scheu, 2005; Strickland et al., 2009; Matulich and Martiny, 2015) as well as that of the plants (Hobbie, 1996; Hattenschwiler et al., 2005). In this system, in particular, we used a reciprocal transplant experiment to tease apart the microbial effect on decomposition (Allison et al., 2013). Microbial communities from the drought environment were slower to decompose litter than communities from the ambient environment. Thus it appears that, when combined, the various pathways that the treatments alter may work in different directions and buffer any changes on litter decomposition. Given the importance of temporal variability, however, we recognize that additional decomposition measurements are needed to confirm this result over multiple years.
Despite its high temporal variability, the microbial community was dominated by only a handful of highly abundant bacterial and fungal taxa throughout the 2-year study. Such an uneven abundance distribution was also observed in oak leaf litter, where approximately 80% of all fungal sequences belonged to only 30 OTUs (Voriskova and Baldrian, 2013). Nevertheless, little is known about the taxa that dominate leaf litter. Broadly speaking, the taxonomic composition observed in this study is similar to that found in other terrestrial litter and soil systems. The majority of fungal sequences in this study were Ascomycota and Basidiomycota (Barnard et al., 2013; Voriskova and Baldrian, 2013; Weber et al., 2013), and Proteobacteria, Bacteriodetes and Actinobacteria made up most of the bacterial diversity (Castro et al., 2010; Sheik et al., 2011; Barnard et al., 2013; Kim et al., 2014). In contrast, many soils contain a large fraction of Acidobacteria (Castro et al., 2010; Sheik et al., 2011; Barnard et al., 2013; Kim et al., 2014), but they represented <1% of our litter communities.
Previous studies in California ecosystems have also observed general effects of drought and nitrogen on microbial composition. For example, Yuste et al. (2011) found that soil bacterial communities were significantly affected by a simulated drought treatment in both a holm-oak forest and a scrubland. Additionally, microbial composition in a grassland ecosystem varied in response to added nitrogen (Gutknecht et al., 2012). The specific taxonomic responses were not always consistent with previous studies, however. For instance, drought reduced the relative abundance of Proteobacteria in our study, as previously reported in a constructed old-field ecosystem (Castro et al., 2010); two abundant Proteobacteria OTUs (Methylobacterium 2 and Mitsuaria) had a significantly lower relative abundance in the drought treatment. In contrast, Weber et al. (2013) reported a positive response of Ascomycetes to nitrogen addition, whereas many of the abundant Ascomycete OTUs decreased in relative abundance in our nitrogen treatment. Also, in contrast to prior studies, added nitrogen or drought did not increase the relative abundance of Actinobacteria (Ramirez et al., 2012; Barnard et al., 2013). Further work is needed to understand whether these taxonomic groups are not consistent in their responses (Philippot et al., 2010), or whether such responses depend on the community and system context.
In summary, the responses of leaf litter microbes to the global change manipulations might have been easily overlooked without consideration of background seasonal variability. This pattern is consistent with other recent experiments (Cruz-Martinez et al., 2009; Yuste et al., 2011; Cregger et al., 2012) and suggests that future changes in seasonality may be even more important than changes in annual averages. We predict in this system, for instance, that additional summer rain will have a greater impact on litter decomposition than a slight increase in mean annual precipitation. Consequently, information about the seasonal phenology of microbial taxa might be useful to predict longer-term responses to climate variables, as it seems to be for plants (Menzel et al., 2006; Khanduri et al., 2008; Wolkovich et al., 2012). Indeed, the responses of litter fungi to the dry season and drought were correlated in this study. Thus, as with larger organisms, the future of microbial communities will likely depend on the interplay between rapid, local background variability and slower, global change.
Accession codes
References
Allison SD, Jastrow JD . (2006). Activities of extracellular enzymes in physically isolated fractions of restored grassland soils. Soil Biol Biochem 38: 3245–3256.
Allison SD, Lu Y, Weihe C, Goulden ML, Martiny AC, Treseder KK et al. (2013). Microbial abundance and composition influence litter decomposition response to environmental change. Ecology 94: 714–725.
Allison SD, Martiny JBH . (2008). Resistance, resilience, and redundancy in microbial communities. Proc Natl Acad Sci USA 105: 11512–11519.
Alster CJ, German DP, Lu Y, Allison SD . (2013). Microbial enzymatic responses to drought and to nitrogen addition in a southern California grassland. Soil Biol Biochem 64: 68–79.
Anderson MJ, Gorley RN, Clarke KR . (2008) PERMANOVA+ for PRIMER: Guide to Software and Statistical Methods PRIMER-E Ltd Plymouth, UK.
Arguez A, Durre I, Applequist S, Vose RS, Squires MF, Yin XG et al. (2012). NOAA's 1981-2010 U.S. climate normals: an overview. B Am Meteorol Soc 93: 1687–1697.
Bardgett RD, Lovell RD, Hobbs PJ, Jarvis SC . (1999). Seasonal changes in soil microbial communities along a fertility gradient of temperate grasslands. Soil Biol Biochem 31: 1021–1030.
Barnard RL, Osborne CA, Firestone MK . (2013). Responses of soil bacterial and fungal communities to extreme desiccation and rewetting. ISME J 7: 2229–2241.
Bell CW, Tissue DT, Loik ME, Wallenstein MD, Acosta-Martinez V, Erickson RA et al. (2014). Soil microbial and nutrient responses to 7 years of seasonally altered precipitation in a Chihuahuan Desert grassland. Global Change Biol 20: 1657–1673.
Bell T, Newman JA, Silverman BW, Turner SL, Lilley AK . (2005). The contribution of species richness and composition to bacterial services. Nature 436: 1157–1160.
Berlemont R, Allison SD, Weihe C, Lu Y, Brodie EL, Martiny JBH et al. (2014). Cellulolytic potential under environmental changes in microbial communities from grassland litter. Front Microbiol 5: 639.
Braid MD, Daniels LM, Kitts CL . (2003). Removal of PCR inhibitors from soil DNA by chemical flocculation. J Microbiol Meth 52: 389–393.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336.
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6: 1621–1624.
Castro HF, Classen AT, Austin EE, Norby RJ, Schadt CW . (2010). Soil microbial community responses to multiple experimental climate change drivers. Appl Environ Microbiol 76: 999–1007.
CEC. (2003) Global Climate Change and California: Potential Implications for Ecosystems, Health, and the Economy—Consultant Report. Publication# 500-03-058CF. California Energy Commission: USA.
Clarke KR, Warwick RM . (2001) Change in Marine Communities: An Approach to Statistical Analysis and Interpretation, 2nd edn. PRIMER-E Ltd Plymouth, UK.
Cleveland CC, Reed SC, Keller AB, Nemergut DR, O'Neill SP, Ostertag R et al. (2014). Litter quality versus soil microbial community controls over decomposition: a quantitative analysis. Oecologia 174: 283–294.
Cotrufo MF, Ineson P, Rowland AP . (1994). Decomposition of tree leaf litters grown under elevated CO2 - effect of litter quality. Plant Soil 163: 121–130.
Cregger MA, Schadt CW, McDowell NG, Pockman WT, Classen AT . (2012). Response of the soil microbial community to changes in precipitation in a semiarid ecosystem. Appl Environ Microbiol 78: 8587–8594.
Cruz-Martinez K, Suttle KB, Brodie EL, Power ME, Andersen GL, Banfield JF . (2009). Despite strong seasonal responses, soil microbial consortia are more resilient to long-term changes in rainfall than overlying grassland. ISME J 3: 738–744.
de Vries FT, Manning P, Tallowin JR, Mortimer SR, Pilgrim ES, Harrison KA et al. (2012). Abiotic drivers and plant traits explain landscape-scale patterns in soil microbial communities. Ecol Lett 15: 1230–1239.
DeAngelis KM, Brodie EL, DeSantis TZ, Andersen GL, Lindow SE, Firestone MK . (2009). Selective progressive response of soil microbial community to wild oat roots. ISME J 3: 168–178.
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72: 5069–5072.
Edgar RC . (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460–2461.
Fenn ME, Poth MA, Bytnerowitcz A, Sickman JO, Takemoto BK . (2003). Effects of ozone, nitrogen deposition, and other stressors on montane ecosystems in the Sierra Nevada. Dev Environ Sci 2: 111–155.
Fierer N, Strickland MS, Liptzin D, Bradford MA, Cleveland CC . (2009). Global patterns in belowground communities. Ecol Lett 12: 1238–1249.
German DP, Weintraub MN, Grandy AS, Lauber CL, Rinkes ZL, Allison SD . (2011). Optimization of hydrolytic and oxidative enzyme methods for ecosystem studies. Soil Biol Biochem 43: 1387–1397.
Gutknecht JLM, Field CB, Balser TC . (2012). Microbial communities and their responses to simulated global change fluctuate greatly over multiple years. Global Change Biol 18: 2256–2269.
Habekost M, Eisenhauer N, Scheu S, Steinbeiss S, Weigelt A, Gleixner G . (2008). Seasonal changes in the soil microbial community in a grassland plant diversity gradient four years after establishment. Soil Biol Biochem 40: 2588–2595.
Hattenschwiler S, Tiunov AV, Scheu S . (2005). Biodiversity and litter decomposition interrestrial ecosystems. Annu Rev Ecol Evol Syst 36: 191–218.
Hobbie SE . (1996). Temperature and plant species control over litter decomposition in Alaskan tundra. Ecol Monogr 66: 503–522.
Kennedy N, Brodie E, Connolly J, Clipson N . (2006). Seasonal influences on fungal community structure in unimproved and improved upland grassland soils. Can J Microbiol 52: 689–694.
Khanduri VP, Sharma CM, Singh SP . (2008). The effects of climate change on plant phenology. Environmentalist 28: 143–147.
Kim M, Kim WS, Tripathi BM, Adams J . (2014). Distinct bacterial communities dominate tropical and temperate zone leaf litter. Microb Ecol 67: 837–848.
Kimball S, Goulden ML, Suding KN, Parker S . (2014). Altered water and nitrogen input shifts succession in a Southern California coastal sage community. Ecol Appl 24: 1390–1404.
Kominoski JS, Hoellein TJ, Kelly JJ, Pringle CM . (2009). Does mixing litter of different qualities alter stream microbial diversity and functioning on individual litter species? Oikos 118: 457–463.
Lage MD, Reed HE, Weihe C, Crain CM, Martiny JB . (2010). Nitrogen and phosphorus enrichment alter the composition of ammonia-oxidizing bacteria in salt marsh sediments. ISME J 4: 933–944.
Lane DJ . (1991) 16S/23S rRNA sequencing. In: Stackebrandt E, Goodfellow M (eds). Nucleic Acid Techniques in Bacterial Systematics. John Wiley and Sons: New York, NY, USA, pp 115–175.
Larcher W . (2000). Temperature stress and survival ability of Mediterranean sclerophyllous plants. Plant Biosyst 134: 279–295.
Loranger-Merciris G, Barthes L, Gastine A, Leadley P . (2006). Rapid effects of plant species diversity and identity on soil microbial communities in experimental grassland ecosystems. Soil Biol Biochem 38: 2336–2343.
Matulich KL, Martiny JBH . (2015). Microbial composition alters the response of litter decomposition to environmental change. Ecology 96: 154–163.
Mcclaugherty CA, Pastor J, Aber JD, Melillo JM . (1985). Forest litter decomposition in relation to soil-nitrogen dynamics and litter quality. Ecology 66: 266–275.
Menzel A, Sparks TH, Estrella N, Koch E, Aasa A, Ahas R et al. (2006). European phenological response to climate change matches the warming pattern. Global Change Biol 12: 1969–1976.
Niklaus PA, Alphei D, Ebersberger D, Kampichler C, Kandeler E, Tscherko D . (2003). Six years of in situ CO(2) enrichment evoke changes in soil structure and soil biota of nutrient-poor grassland. Global Change Biol 9: 585–600.
Padgett PE, Allen EB . (1999). Differential responses to nitrogen fertilization in native shrubs and exotic annuals common to Mediterranean coastal sage scrub of California. Plant Ecol 144: 93–101.
Philippot L, Andersson SG, Battin TJ, Prosser JI, Schimel JP, Whitman WB et al. (2010). The ecological coherence of high bacterial taxonomic ranks. Nat Rev Microbiol 8: 523–529.
Potts DL, Suding KN, Winston GC, Rocha AV, Goulden ML . (2012). Ecological effects of experimental drought and prescribed fire in a southern California coastal grassland. J Arid Environ 81: 59–66.
Ramirez KS, Craine JM, Fierer N . (2012). Consistent effects of nitrogen amendments on soil microbial communities and processes across biomes. Global Change Biol 18: 1918–1927.
Reeder J, Knight R . (2010). Rapidly denoising pyrosequencing amplicon reads by exploiting rank-abundance distributions. Nat Methods 7: 668–669.
Schimel JP, Schaeffer SM . (2012). Microbial control over carbon cycling in soil. Front Microbiol 3: 348.
Seifert J . (1961). The influence of moisture and temperature on the number of bacteria in the soil. Folia Microbiol 6: 268–272.
Sheik CS, Beasley WH, Elshahed MS, Zhou XH, Luo YQ, Krumholz LR . (2011). Effect of warming and drought on grassland microbial communities. ISME J 5: 1692–1700.
Singh BK, Bardgett RD, Smith P, Reay DS . (2010). Microorganisms and climate change: terrestrial feedbacks and mitigation options. Nat Rev Microbiol 8: 779–790.
Specht RL, Moll EJ, Pressinger F, Sommerville J . (1983) . Moisture regime and nutrient control of seasonal growth in Mediterranean ecosystems. In: Kruger FJ, Mitchell DT, Jarvis JUM (eds). Mediterranean-Type Ecosystems: The Role of Nutrients. Springer: Berlin, Germany, pp 120–132.
Strickland MS, Lauber C, Fierer N, Bradford MA . (2009). Testing the functional significance of microbial community composition. Ecology 90: 441–451.
Swift MJ, Heal OW, Anderson JM . (1979) Decomposition in Terrestrial Ecosystems. Blackwell Scientific Publications: Oxford, UK.
Tedersoo L, Jairus T, Horton BM, Abarenkov K, Suvi T, Saar I et al. (2008). Strong host preference of ectomycorrhizal fungi in a Tasmanian wet sclerophyll forest as revealed by DNA barcoding and taxon-specific primers. New Phytol 180: 479–490.
Tiunov AV, Scheu S . (2005). Facilitative interactions rather than resource partitioning drive diversity-functioning relationships in laboratory fungal communities. Ecol Lett 8: 618–625.
Treves DS, Xia B, Zhou J, Tiedje JM . (2003). A two-species test of the hypothesis that spatial isolation influences microbial diversity in soil. Microb Ecol 45: 20–28.
Turner S, Pryer KM, Miao VPW, Palmer JD . (1999). Investigating deep phylogenetic relationships among cyanobacteria and plastids by small submit rRNA sequence ansalysis. J Eukaryot Microbiol 46: 327–338.
Voriskova J, Baldrian P . (2013). Fungal community on decomposing leaf litter undergoes rapid successional changes. ISME J 7: 477–486.
Wang Q, Garrity GM, Tiedje JM, Cole JR . (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73: 5261–5267.
Weber CF, Vilgalys R, Kuske CR . (2013). Changes in fungal community composition in response to elevated atmospheric CO2 and nitrogen fertilization varies with soil horizon. Front Microbiol 4: 78.
Wolkovich EM, Cook BI, Allen JM, Crimmins TM, Betancourt JL, Travers SE et al. (2012). Warming experiments underpredict plant phenological responses to climate change. Nature 485: 494–497.
Yuste JC, Penuelas J, Estiarte M, Garcia-Mas J, Mattana S, Ogaya R et al. (2011). Drought-resistant fungi control soil organic matter decomposition and its response to temperature. Global Change Biol 17: 1475–1486.
Zak DR, Holmes WE, White DC, Peacock AD, Tilman D . (2003). Plant diversity, soil microbial communities, and ecosystem function: are there any links? Ecology 84: 2042–2050.
Zavaleta ES, Shaw MR, Chiariello NR, Thomas BD, Cleland EE, Field CB et al. (2003). Grassland responses to three years of elevated temperature, CO(2), precipitation, and N deposition. Ecol Monogr 73: 585–604.
Acknowledgements
This work was funded by the US Department of Energy Program in Ecosystem Research; the Office of Science (BER), US Department of Energy (program in Microbial Communities and Carbon Cycling), the NSF Major Research Instrumentation program and the US Department of Education Graduate Assistance in Areas of National Need (GAANN) fellowship. We thank Orange County Parks and the Irvine Ranch Conservancy for site access and Michaeline Nelson, Eoin Brodie and Kathleen Treseder for comments on previous versions of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Matulich, K., Weihe, C., Allison, S. et al. Temporal variation overshadows the response of leaf litter microbial communities to simulated global change. ISME J 9, 2477–2489 (2015). https://doi.org/10.1038/ismej.2015.58
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2015.58
This article is cited by
-
Wavelet clustering analysis as a tool for characterizing community structure in the human microbiome
Scientific Reports (2023)
-
Curtobacterium allii sp. nov., the actinobacterial pathogen causing onion bulb rot
Antonie van Leeuwenhoek (2023)
-
Sodium as a subsidy in the spring: evidence for a phenology of sodium limitation
Oecologia (2023)
-
Response of fungal and bacterial communities in fine root litter with different diameters to nitrogen application
Journal of Soils and Sediments (2023)
-
Routes and rates of bacterial dispersal impact surface soil microbiome composition and functioning
The ISME Journal (2022)
