
Abstract
The high numbers and diversity of protists in soil systems have long been presumed, but their true diversity and community composition have remained largely concealed. Traditional cultivation-based methods miss a majority of taxa, whereas molecular barcoding approaches employing PCR introduce significant biases in reported community composition of soil protists. Here, we applied a metatranscriptomic approach to assess the protist community in 12 mineral and organic soil samples from different vegetation types and climatic zones using small subunit ribosomal RNA transcripts as marker. We detected a broad diversity of soil protists spanning across all known eukaryotic supergroups and revealed a strikingly different community composition than shown before. Protist communities differed strongly between sites, with Rhizaria and Amoebozoa dominating in forest and grassland soils, while Alveolata were most abundant in peat soils. The Amoebozoa were comprised of Tubulinea, followed with decreasing abundance by Discosea, Variosea and Mycetozoa. Transcripts of Oomycetes, Apicomplexa and Ichthyosporea suggest soil as reservoir of parasitic protist taxa. Further, Foraminifera and Choanoflagellida were ubiquitously detected, showing that these typically marine and freshwater protists are autochthonous members of the soil microbiota. To the best of our knowledge, this metatranscriptomic study provides the most comprehensive picture of active protist communities in soils to date, which is essential to target the ecological roles of protists in the complex soil system.
Similar content being viewed by others

Introduction
Soils harbour a spectacular microbial diversity. Among the least-studied soil microorganisms are single-celled protists. They display a high diversity of fundamentally different taxa, based on both morphological features and phylogenetic relatedness (Adl et al., 2012; Pawlowski, 2013). Despite being microscopic, protist biomass in soils has been estimated to exceed that of most soil animal taxa (Schaefer and Schauermann, 1990; Zwart et al., 1994; Schröter et al., 2003). Protists play important ecological roles in controlling bacterial turnover and community composition, recycling of nutrients and plant growth promotion (Clarholm, 1985; de Ruiter et al., 1993; Bonkowski, 2004). However, we still lack basic knowledge of the protist communities in soils, and therefore a comprehensive understanding about their distribution and ecological functions in different soil systems has not yet been achieved.
The pervasive lack of knowledge on soil protist communities is mainly caused by the need to establish enrichment cultures, as the majority of protist taxa are difficult to extract and cultivate from soils, and the opaqueness of soil particles that prevents direct microscopic observation of the majority of taxa (Foissner, 1987; Clarholm et al., 2007). Expert knowledge is needed for the time-consuming microscopic identification (Foissner, 1987; Smirnov et al., 2008; Fenchel, 2010; De Jonckheere et al., 2012). Therefore, these traditional attempts to describe the full diversity of protist taxa in natural soils are rare and identification is usually only possible to a rather shallow taxonomic level (Finlay et al., 2000; Bamforth, 2007; Geisen et al., 2014a). Cultivation-based approaches also introduce bias, as different growth media select for different species, and only a subset of taxa is likely to be cultivable (Ekelund and Rønn, 1994; Foissner, 1999; Smirnov and Brown, 2004).
The advent of molecular techniques along with a revised species concept that usually includes molecular information such as the universal protist barcode, the small subunit ribosomal RNA (SSU rRNA) gene (Pawlowski et al., 2012), has fundamentally altered the view on the 'protist world'. Consequently, the classification of and relatedness between protist taxa are constantly being revised (Adl et al., 2012; Pawlowski, 2013). Further, environmental sequencing studies based on the SSU rRNA gene have revealed a huge diversity of previously unknown protists (Bass and Cavalier-Smith, 2004; Berney et al., 2004; Lara et al., 2007; Lejzerowicz et al., 2010; Bates et al., 2013). Despite molecular tools having diminished some of the problems associated to deciphering the community structure of soil protists, they introduce new biases that still obscure the true protist diversity in soils. Fundamental problems include (i) the lack of SSU rRNA reference sequences for a substantial number of protist species and genera, (ii) numerous mislabelled sequences in public databases and (iii) a large seed bank of dormant protist cysts that may survive for decades (Goodey, 1915; Moon van der Staay et al., 2006; Smirnov et al., 2008; Epstein and López-García, 2008; Adl et al., 2012; De Jonckheere et al., 2012). Further biases are introduced by the PCR step of SSU rRNA gene studies (for example, Bachy et al., 2013). The often applied 'general' eukaryotic primers to decipher the community structure of protists are in fact far from being truly universal (Adl et al., 2014). Thus, a strongly biased view of the protist community in soils is being depicted as only a subset of its diversity can be recovered (Jeon et al., 2008; Hong et al., 2009), whereas, on the other hand, some taxa within this subset will be overrepresented because of preferential PCR amplification (Berney et al., 2004; Medinger et al., 2010; Stoeck et al., 2014). Taxa of the common supergroup Amoebozoa as one of the dominant soil protists for instance, are notoriously underrepresented in molecular surveys because of long SSU rRNA sequences, frequent mismatches in primer regions and presence of introns (Berney et al., 2004; Fiore-Donno et al., 2010; Pawlowski et al., 2012). Ciliates on the contrary are highly overrepresented because of their shorter SSU rRNA sequences that ease amplification, and the presence of extremely high SSU rRNA gene copy numbers (Gong et al., 2013).
Most of these obstacles are avoided when directly targeting SSU rRNA transcripts instead of genes, especially by random hexamer-primed reverse transcription as in metatranscriptomic approaches. These rRNA transcripts are indicative of ribosomes and thus are likely derived from metabolically active cells and can be considered markers for living biomass (Urich and Schleper, 2011). The generated cDNA fragments originate from different regions of the SSU rRNA molecule unlike PCR-primed specific sites, and are therefore insensitive to the presence of introns or primer mismatches (Urich et al., 2008). These cDNA fragments can further be assembled into longer fragments, or even full-length SSU rRNA molecules for phylogenetic analyses (Urich et al., 2014). However, metatranscriptomics also has biases; the reported community composition is influenced, for example, by the different accessibility of SSU rRNA regions to primers and reverse transcriptase and also the physiological status of cells resulting in varying ribosomal content. Using this approach, Urich et al. (2008) generated cDNA from the total extracted RNA of soil communities. The cDNA was subjected directly to high throughput sequencing without any SSU rRNA gene PCR steps. Although the fraction of SSU rRNA originating from protists is comparably small in metatranscriptomes, recent studies showed that the sequencing depth even with 454 pyrosequencing yielded sizable datasets of protist SSU rRNAs (Urich et al., 2008; Turner et al., 2013; Tveit et al., 2013).
We used this PCR-free metatranscriptomic approach to reveal the diversity of the active soil protist communities within five different natural soil systems in Europe, including forest, grassland and peat soils as well as beech litter. We annotated all protist SSU rRNA sequences to a reference database consisting of manually curated, published protist SSU rRNA sequences and revealed high protist diversity, with communities strongly differing between sites. We show that Rhizaria and Amoebozoa are the most abundant protist groups and detect abundant potential plant and animal pathogens. Further, we perform for the first time an in-depth molecular analysis of the amoebozoan community structure. Finally, we report the widespread presence of protist clades that are usually associated with marine and freshwater environments, but not considered typical soil inhabitants.
Materials and methods
Soil sampling and processing
Arctic peat soils were sampled as described in the study by Tveit et al. (2013). The grassland site (Park Grass, untreated control plot 3d, Rothamsted) was sampled by coring, with intact cores being brought to the laboratory, topsoil (5–10 cm) sieved (5 mm mesh size) and subsequently flash-frozen in liquid nitrogen. Beech (Fagus sylvatica L) forest soils were sampled as described in the study by Kaiser et al. (2010), the top soil (5–10 cm) sieved (5 mm mesh size) and subsequently flash-frozen in liquid nitrogen. Beech litter from the same site was homogenized with a sterilised coffee grinder for 10 s and flash-frozen in liquid nitrogen.
Nucleic acid extraction, cDNA synthesis and sequencing
Nucleic acids were extracted from 5 g of mineral soil and 1.2 g of litter and peat and processed as previously described (Urich et al., 2008). cDNA synthesis was performed as described before (Radax et al., 2012; Tveit et al., 2013). 454-pyrosequencing was done either with FLX (forest soil and litter) or FLX Titanium (grassland, peat soil) chemistry. Sequencing was carried out at the CEES at the University of Oslo (Norway).
Sequence processing and analysis
Raw reads were processed as described in the study by Tveit et al. (2013). Sequences were first filtered using LUCY (Chou and Holmes, 2001), removing short (<150 bp) and low-quality sequences (>0.2% error probability). SSU ribosomal RNA sequences of eukaryotes were identified by MEGAN analysis of BLASTn files against a 3-domain SSU rRNA reference database (Lanzén et al., 2011; parameters: min. bit score 150, min. support 1, top percent 10; 50 best blast hits). All identified eukaryotic SSU rRNAs were reanalysed with CREST (Lanzén et al., 2012) using the Silvamod database and MEGAN with LCA parameters min bit score 250, top percent 2 (50 best blast hits) for classification of protist sequences. Correct taxonomic assignment of rRNA reads was verified by manual BLASTn searches against the NCBI GenBank nucleotide database. For the high-resolution taxonomic annotation of amoebozoan rRNA sequences, a custom-made database was constructed consisting of 1164 sequences from Silva (www.arb-silva.de) and sequences of in-house cultivated and newly described species (Geisen et al., 2014b, c, d). The taxonomy was set according to the most recent taxonomy of Amoebozoa (Smirnov et al., 2011; Adl et al., 2012; Lahr et al., 2013) to enable high-resolution taxonomic placement of sequences. Reference database and taxonomy were then generated with the CREST scripts (Lanzén et al., 2012), and amoebozoan sequences were classified using the same parameters in MEGAN as described above. The database can be obtained from TU or SG upon request.
SSU rRNA sequences of Choanoflagellida and Foraminifera were assembled into ribo-contigs using CAP3 (Huang and Madan, 1999), performing two subsequent rounds of assembly with (i) a minimum overlap of 150 bp with a minimum similarity threshold of 99% and mismatch and gap scores of −130 and 150, and (ii) minimum overlap of 150 bp and minimum 97% similarity threshold, respectively (Radax et al., 2012).
Phylogenetic analysis
Assembled Choanoflagellate contig sequences were subjected to BLASTn searches against the NCBI nucleotide database and manually aligned with their respective five best hits in Seaview 4 (Gouy et al., 2010). Additionally, SSU rRNA sequences of described choanoflagellates were added to this alignment. For phylogenetic analyses, 1438 unambiguously aligned positions of a total of 71 unique sequences were retained, excluding ambiguous positions and several positions in variable regions especially in V4 (helix E23). Maximum likelihood phylogenetic analyses were run using RAxML v. 7.2.6 (Stamatakis, 2006) using the GTR+γ+I model of evolution, as proposed by jModeltest v. 2.1.3 under the Akaike Information Criterion (Darriba et al., 2012), γ approximated by 25 categories. A total of 1000 non-parametric bootstrap pseudoreplicates were performed. Subsequently, Bayesian phylogenetic analyses were run in Mr Bayes v. 3.2.1 with GTR+γ+I model of evolution and eight categories (Huelsenbeck and Ronquist, 2001). Two runs of four simultaneous Markov chains were performed for 2 000 000 generations (with the default heating parameters) and sampled every 100 generations; convergence of the two runs (average deviation of split frequencies<0.01) was reached after 590 000 generations. Therefore, we discarded the first 5900 trees and built a consensus tree from the remaining 14 100 trees.
Unweighted Pair Group Method as a cluster analysis was applied to evaluate differences between the protist community composition in all samples (Sokal, 1961).
Data deposition
The sequence data generated in this study were deposited in the Sequence Read Archive of NCBI under accession number SRP014474 and SAMN03365922-24.
Results
Community composition of protist supergroups
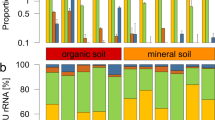
In total, 32 808 SSU rRNA transcripts of protists were obtained from 12 soil metatranscriptomes (Table 1). In all cases, the biological replicates of each site yielded a very similar community composition (Figure 1) and grouped together in a cluster analysis (Figure 2). All five protist supergroups according to Adl et al. (2012) were found at each site (Table 1; Figure 1). The SAR group, consisting of the formerly independent supergroups Stramenopiles, Alveolata and Rhizaria, dominated the active communities at all sites with sequences of Rhizaria being most numerous. We observed a clear dichotomy in protist community composition (Figure 2) with dominance of Alveolata in peat soils with their high moisture and high organic matter content, versus dominance of Rhizaria and Amoebozoa in grassland and forest soils, including forest litter (Figure 1).

Community composition of protist supergroups in the investigated soils. For detailed information on soil parameters, see Table 1.

Unweighted Pair Group Method (UPGMA) clustering analysis to evaluate differences between soil protist communities. For detailed information on soil characteristics and abbreviations, see Table 1.
The Rhizaria were almost exclusively comprised of Cercozoa (Figure 3a) with dominance of rRNAs of the small flagellates belonging to the class Filosa-Sarcomonadea (formerly combined into Cercomonadida) and both, flagellated and amoeboid Cercozoa in the class Filosa-Imbricatea (formerly Silicofilosea). Foraminiferan SSU rRNAs occurred in relatively constant, albeit low abundances in all samples (Figure 3a) except for the peatland site 'Knutsen', where they were more abundant and comprised 22% of all Cercozoa. Alveolate SSU rRNAs were quite variable among samples and dominated in the peat soils (Figure 1) with the phylum Ciliophora and its main orders Spirotrichea, Colpodea and Oligohymenophorea being most abundant, whereas the exclusively parasitic Apicomplexa still represented up to 11% of all Alveolata (Figure 3b). The third group in SAR, Stramenopiles, was less abundant, with Oomycetes representing the dominant stramenopiles in forest soils (Figure 3c), while abundant transcripts of the photosynthetic Bacillariophyta were characteristic in the waterlogged peatland soils. Chrysophyceaen SSU rRNAs were abundantly found in grassland soils and litter, as were transcripts of the Bicosoecida. The supergroup Amoebozoa represented up to 30% of SSU rRNAs, with highest abundances in the grassland and forest soil samples (Figure 1 and Table 1). The other protist supergroups, that is, Excavata, Archaeplastida and Opisthokonta (excluding fungi and animals) were generally less abundant (Figure 1 and Table 1; see Supplementary Table 1 for more information).

SSU rRNA transcripts of protist clades highly represented in cultivation-based approaches were analysed to evaluate whether these clades were also recovered in our metatranscriptomes. Among the clades targeted were flagellates of the supergroups SAR (Glissonomadida and Cercomonadida (Sarcomonadea; Rhizaria), Chrysophyceae and Bicosoecida (Stramenopiles) and Excavata (Bodonidae and Euglenida), as well as amoebae in the supergroup Excavata (Heterolobosea). All clades were found at each location, with cercomonads being highly abundant in grassland and forest habitats. Glissomonads were abundant especially in grasslands (5% of all protists). Bodonids were also common (1.9% of all protists) whereas euglenids, chrysophytes, bicosoecids and heteroloboseans comprised only about 1% of all protist transcripts (Supplementary Table 1). Exhaustive analyses of the most abundant clades of amoebae are presented in the next section.
Community composition of amoebozoa
The supergroup Amoebozoa was of particular interest, as no comprehensive molecular taxonomic analysis of this protist supergroup in soil exists to date because of PCR-primer biases (Baldwin et al., 2013; Bates et al., 2013). For this purpose, we constructed a high-resolution reference database and taxonomy of Amoebozoa (see Table 2 and Materials and Methods for details). Using this taxonomic assignment approach, the rather short SSU rRNA sequence reads could reliably be classified at least to the order, often even to the genus level. Amoebozoa were highly diverse at each sampling site, with rRNAs assigned to four classes, that is, Tubulinea, Discosea, Variosea and Mycetozoa with major orders Euamoebida, Leptomyxida, Arcellinida and Centramoebida (Smirnov et al., 2011) (Figure 4). The community composition within Amoebozoa differed significantly between sites. For instance, sequences assigned to the dominant class Tubulinea made up between 27.4% and 69.2% in Solvatn peat and forest soils, respectively, and were inversely related to Discosea with 41.6–12.7% in Solvatn peat and forest soils. Similar to the patterns observed at the class level, the proportional distribution within classes differed between sites. Among Tubulinea, the dominant order Euamoebida reached highest relative abundances in grasslands and forest mineral soils, while testate amoebae of the order Arcellinida became more abundant in organic rich substrates of litter and peat soils, and Leptomyxida were characteristic of forest habitats. The remaining tubulinean orders Echinamoebida and Nolandida were generally rare. Among the class Discosea, SSU rRNAs assigned to the subclass Longamoebia were almost entirely (96.1%) composed of the order Centramoebida. Sequences of the discosean subclass Flabellinia were generally less abundant (7.0% oas) and mostly derived from the order Vannellida (50.1% of flabellinian SSU rRNAs). Sequences assigned to the subphylum Conosa could only reliably be assigned to the class level, as taxonomy and the phylogenetic affiliations, especially of protists in the class Variosea, are still largely unresolved (Adl et al., 2012). Variosea was the dominant conosan class in grassland, forest and the Solvatn peat soil, while Mycetozoa were more abundant in forest litter and Knutsen peat soil (Figure 4).

Widespread were amoebozoan SSU rRNA sequences with high sequence identity to potential parasites. At all sites occurred diverse sequences related to facultative human pathogens of the genus Acanthamoeba (⩾97% sequence identity; 1.1–4.4% oas) while sequences most closely resembling Balamuthia were discovered in low relative abundance (0.1–0.5% oas) in all except the grassland soils. Transcripts related to groups of non-Amoebozoan parasitic taxa were also found, such as sequences most closely resembling the human pathogen Naegleria fowleri (Heterolobosea in the supergroup Excavata; ⩾97% sequence identity) in all four arctic peat samples. Further, opisthokonta Ichthyosporea (mainly animal parasites) were ubiquitously found (0.4–3.3% oas; Supplementary Table 1) as well as predominantly plant-parasitic plasmodiophorans (up to 0.8% oas).
Widespread presence of foraminifera and choanoflagellida in soils
SSU rRNA transcripts of the typically marine groups Foraminifera and Choanoflagellida revealed their general presence and activity in all samples (Figure 5), and for the first time, allowed to estimate the relative abundance of these taxa in soils. They comprised between 0.1% and 3.5% of all protist SSU rRNAs.

To enable better taxonomic assignment and phylogenetic placement of these enigmatic soil protist groups we, assembled larger SSU rRNA sequences from the short 454 reads. Three assembled SSU rRNA contigs of Foraminifera (742–890 bp length) were quite similar (⩾91%) to sequences obtained in a recent focused PCR-based molecular survey targeting soil Foraminifera (Lejzerowicz et al., 2010), but showed substantial sequence dissimilarity to described species (maximum SSU rRNA sequence identity of ≤76%). Several unassembled SSU rRNA sequences, however, closely matched sequences typically obtained from freshwater and marine environments, such as the genera Astrammina, Bathysiphon, Allogromia and diverse uncultured species (Supplementary Table 2).
Many Choanoflagellida-affiliated SSU rRNA sequences closely matched (with ⩾99% maximum identity) published sequences of uncultivated choanoflagellates (for example, with GenBank accession numbers HQ219439 (freshwater), EF024012 (soil), JF706236 and GQ330606 (peat soil)) while others reached similarities of >95% with uncultivated choanoflagellate sequences among typical freshwater and marine genera, such as Monosiga, Codonosiga, Salpingoeca, and more rarely with Lagenoeca, Stephanoeca, Didymoeca, Diaphanoeca, Desmarella and Acanthoeca. Five assembled long SSU rRNAs (763–1163 bp) had high sequence similarities of 96–99% to uncultivated freshwater choanoflagellates (Figure 6), but the closest hit to a formally described species was <92%.

Maximum likelihood tree of Choanoflagellida placing assembled long SSU rRNA contigs (red) with sequences of described and uncultivated choanoflagellates. Seventy-one sequences with 1232 unambiguously aligned positions were used; the tree is unrooted; values higher than 50 for maximum likelihood analyses (left) and 0.50 for Bayesian analyses (right) shown. Black circles indicate full support.
Discussion
Metatranscriptomics-enabled census of active soil protists
Cultivation-based studies have shown that soil protists are diverse, abundant and ecologically important (Ekelund and Rønn, 1994; de Ruiter et al., 1995; Bonkowski, 2004; Pawlowski et al., 2012), but fail to detect a majority of taxa (Epstein and López-García 2008). In contrast, cultivation-independent PCR-based SSU rRNA gene sequencing studies have revealed a much higher diversity of soil protists than previously anticipated (Lara et al., 2007; Lejzerowicz et al., 2010; Bates et al., 2013); however, they still fail to amplify the SSU rRNA genes of a wide range of protists and provide a highly skewed picture of protist communities (Epstein and López-García, 2008; Weber and Pawlowski, 2013; Stoeck et al., 2014). This is exemplified by the negative selection of amoebozoan sequences in PCR-primer-based approaches (Berney et al., 2004; Amaral-Zettler et al., 2009), explaining the virtual absence of nearly the entire supergroup in high throughput sequencing surveys (Baldwin et al., 2013; Bates et al., 2013), whereas the high rRNA gene copy numbers in ciliates (Gong et al., 2013) generally lead to their over-proportional representation.
Through the usage of metatranscriptomics, we avoided some of the above-mentioned issues to gain a more accurate picture of the protist diversity in soil systems. In fact, this study and earlier metatranscriptomes have revealed a different soil protist community structure than suggested before (Urich et al., 2008; Baldwin et al., 2013). The only similarity to primer-based high throughput sequencing surveys is merely the dominance of the supergroup SAR, which was because of high sequence abundances of Rhizaria (and Alveolata in arctic peat soils) (Baldwin et al., 2013; Bates et al., 2013). However, SSU rRNA sequences of Amoebozoa dominated over those of Alveolata in all non-peat soils. This is in line with cultivation-based studies, which show that small flagellates, especially cercomonads and glissomonads (Rhizaria) (Finlay et al., 2000; Ekelund et al., 2001; Howe et al., 2009) and amoebae represent the numerically dominant soil protists (Schaefer and Schauermann, 1990; Finlay et al., 2000; Robinson et al., 2002). Similarly, other taxonomically highly divergent heterotrophic protist taxa especially abundant in cultivation-based studies such as bodonid and euglenid euglenozoans (Finlay et al., 2000; Geisen et al., 2014a), chrysophytes (Finlay et al., 2000; Boenigk et al., 2005; Chatzinotas et al., 2013), bicosoecids (Ekelund and Patterson, 1997; Lentendu et al., 2014) and heteroloboseans (Geisen et al., 2014a) were identified, but contributed relatively little to the entire protist community.
It should be noted here that metatranscriptomics also has several biases and shortcomings. For example, the reported community composition can be biased by different accessibilities of SSU rRNA regions to primers and enzymes owing to RNA secondary structure. Further, the number of ribosomes can vary dependent of the physiological status of the cell. The higher costs related to cDNA synthesis and missing specificity as compared with PCR-amplicons make this approach rather low-throughput.
Diversity of Amoebozoa: the neglected supergroup in soil
In addition to the above-mentioned negative biases of Amoebozoa in high throughput sequencing approaches, a lack of molecular data and SSU rRNA reference sequences until now prevented thorough molecular diversity analyses of this supergroup. Therefore, the diversity of Amoebozoa in terrestrial samples is still largely unknown (Smirnov et al., 2005; Smirnov et al., 2008). Applying the most recent taxonomy and a newly built reference database allowed us to detect a large diversity within all amoebozoan classes. The general dominance of the classes Tubulinea and Discosea confirmed former cultivation-based studies (for example, Finlay et al., 2000; Bass and Bischoff, 2001), whereas the high abundance of Variosea is novel. This class was only constructed in 2004, hosting mostly large plasmodial, branching or reticulate amoebae (Cavalier-Smith et al., 2004; Smirnov et al., 2008; Smirnov et al., 2011). To date, only few variosean species have formally been described, and molecular information on those taxa is even more limited (Berney et al.). Recent cultivation studies specifically targeting Variosea confirm the results obtained here and revealed an unprecedented diversity of these large amoebae in soils (Geisen et al., 2014a). The addition of several of these recently published variosean SSU rRNA sequences (Geisen et al., 2014a) to our reference database resulted in improved sequence assignments of variosean-specific SSU rRNAs and confirmed that this group of amoebae has been entirely overlooked in earlier studies.
Protist parasites
Terrestrial Oomycetes, distinguished for containing diverse and devasting plant-pathogens (Latijnhouwers et al., 2003) such as Phytophtora infestans, the causative agent of potato blight (Martin et al., 2012), also include a variety of pathogens of other organisms (Benhamou et al., 1999; Phillips et al., 2008). Oomycetes in soil have been targeted with little success (Coince et al., 2013). Our metatranscriptome approach revealed that Oomycetes were ubiquitous, abundant and active members among soil protists, suggesting a significant role as structuring elements of natural plant communities. Unlike in the study of Urich et al. (2008), plant parasitic plasmodiophorids were little abundant in this study.
Apicomplexa are common parasites of soil invertebrates and may potentially play a comparable role in structuring soil food webs (Altizer et al., 2003; Field and Michiels, 2005). Similar to a recent DNA-based study (Bates et al., 2013), we found Apicomplexa in all samples. However, we found higher relative abundance in the dry grassland and forest habitats, in contrast to Bates et al. (2013), who detected apicomplexan SSU rRNA genes with higher relative abundance in the wet soils. As the detected sequences were derived from RNA, they probably did not originate from encysted apicomplexans (Ruiz et al., 1973), but most likely from parasitized soil invertebrates.
Most ichthyosporean taxa are animal parasites and have been described as inhabitants of aquatic organisms, but have recently been found as parasites in terrestrial animals (Glockling et al., 2013). We support these recent findings as we detected Ichthyosporea in significant abundances in all habitats studied.
Protist sequences closely resembling potential human pathogens were detected in all soils. Similar to cultivation-based studies (Page, 1988; Geisen et al., 2014b), Acanthamoeba spp. were common, some of which might be causative agents of amoebic keratitis and amoeboic encephalitis. However, as the SSU rRNA sequences never showed perfect sequence matches with described species and only functional studies on cultivated species allow drawing reliably conclusions on potential pathogenicity, we can only state that these could potentially act as pathogens. Amoebic encephalitis can also be caused by Balamuthia mandrillaris and Naegleria fowleri (Excavata, Heterolobosea) (Visvesvara et al., 1993; Schuster and Visvesvara, 2004; Visvesvara et al., 2007; Siddiqui and Ahmed Khan, 2012). The finding of related SSU rRNA sequences support recent studies showing the presence of both B. mandrillaris (Lares-Jiménez et al., 2014) and N. fowlerii in soil (Moussa et al., 2015).
Unexpected presence of typically marine and freshwater protists
The surprising finding of several typically aquatic protist groups corroborates the notion that a plethora of soil protist taxa have formerly been missed in both, cultivation- and PCR-primer-based studies. For example, we detected Choanoflagellida in all samples; these protists are typically marine and only a few taxa are known from freshwater systems (Tong et al., 1997; Arndt et al., 2000; Stoupin et al., 2012). Mostly, anecdotic evidence from few cultivation-based studies exists on the existence of the choanoflagellate genera Monosiga, Codosiga and Salpingoeca in soils (Ekelund and Patterson, 1997; Finlay et al., 2000; Ekelund et al., 2001; Tikhonenkov et al., 2012), and only a few molecular soil surveys have reported choanoflagellate SSU rRNA sequences (Lesaulnier et al., 2008; Lara et al., 2011), several of which closely resemble SSU rRNA sequences obtained in this study. However, other sequences, among them our assembled rRNA contigs (Figure 5), more closely resembled sequences obtained in freshwater surveys (Chen et al., 2008; Monchy et al., 2011; Stoupin et al., 2012). As most transcripts showed highest similarity with SSU rRNAs of uncultivated species, the future linkage of sequence information with morphological and functional information on the respective protist species in soil will be essential (Bachy et al., 2013).
Foraminifera are another group of typically marine protists commonly not associated with the soil environment, with Edaphoallogromia australica representing the only foraminiferan species reported from soil (Meisterfeld et al., 2001). Our study is the first to assess their relative abundance among active soil protists. Although Foraminifera appeared to comprise a small fraction of the active protist community, they were diverse and present at all sites. This is supported by a recent targeted DNA survey that detected diverse foraminiferan SSU rRNA genes in 17 out of 20 soils samples (Lejzerowicz et al., 2010). Most sequences and SSU rRNA contigs closely matched the sequences obtained by Lejzerowicz et al. (2010), but several non-assembled transcripts more closely resembled the typically marine genera Astrammina, Bathysiphon and Allogromia.
The presence of rRNA transcripts of Choanoflagellida and Foraminifera in soil has important implications. First, these taxa are genuine autochthonous, active inhabitants of soils and not simply dormant states accidentally dispersed to soil by wind or water. Second, there is a need to cultivate these protists, because this will provide information about their ecological roles, morphology and adaptations to the terrestrial habitat, as well as enable comparative studies with their marine relatives that may provide insights in the evolutionary origin of soil protists.
Conclusions
Our study demonstrates the power of metatranscriptomics for obtaining a census of soil protist communities by circumventing major biases commonly associated with cultivation and molecular studies. Still, significant gaps in the taxonomic information prevail in all soil protist supergroups, especially in Amoebozoa where reliable assignment of several SSU rRNA sequences beyond order or even class level was still impeded. Cultivation-based approaches (for example, Geisen et al., 2014c, 2014d) are necessary to fill these gaps. Furthermore, taxonomic expertise remains a crucial prerequisite to interpret protist SSU rRNA data and is as indispensible as a reliable reference database for the correct assignment of taxa. Taking the high sequence coverage into account, we are confident that our study provides the most detailed and potentially closest picture of the true composition of active protist communities in soils so far.
Accession codes
References
Adl SM, Simpson AGB, Lane CE, Lukeš J, Bass D, Bowser SS et al. (2012). The revised classification of eukaryotes. J Eukaryot Microbiol 59: 429–514.
Adl SM, Habura A, Eglit Y . (2014). Amplification primers of SSU rDNA for soil protists. Soil Biol Biochem 69: 328–342.
Altizer S, Harvell D, Friedle E . (2003). Rapid evolutionary dynamics and disease threats to biodiversity. Trends Ecol Evol 18: 589–596.
Amaral-Zettler LA, McCliment EA, Ducklow HW, Huse SM . (2009). A method for studying protistan diversity using massively parallel sequencing of V9 hypervariable regions of small-subunit ribosomal RNA genes. PloS One 4: e6372.
Arndt H, Dietrich D, Auer B, Cleven E-J, Gräfenhan T, Weitere M et al. (2000). Functional diversity of heterotrophic flagellates in aquatic ecosystems. In: Leadbeater BSC, Green JC et al. The Flagellates: Unity, Diversity and Evolution. Taylor & Francis: London, pp 240–268.
Bachy C, Dolan JR, Lopez-Garcia P, Deschamps P, Moreira D . (2013). Accuracy of protist diversity assessments: morphology compared with cloning and direct pyrosequencing of 18 S rRNA genes and ITS regions using the conspicuous tintinnid ciliates as a case study. ISME J 7: 244–255.
Baldwin DS, Colloff MJ, Rees GN, Chariton AA, Watson GO, Court LN et al. (2013). Impacts of inundation and drought on eukaryote biodiversity in semi-arid floodplain soils. Mol Ecol 22: 1746–1758.
Bamforth SS . (2007). Protozoa from aboveground and ground soils of a tropical rain forest in Puerto Rico. Pedobiologia 50: 515–525.
Bass P, Bischoff PJ . (2001). Seasonal variability in abundance and diversity of soil gymnamoebae along a short transect in southeastern USA. J Eukaryot Microbiol 48: 475–479.
Bass D, Cavalier-Smith T . (2004). Phylum-specific environmental DNA analysis reveals remarkably high global biodiversity of Cercozoa (Protozoa). Int J Syst Evol Micr 54: 2393–2404.
Bates ST, Clemente JC, Flores GE, Walters WA, Parfrey LW, Knight R et al. (2013). Global biogeography of highly diverse protistan communities in soil. ISME J 7: 652–659.
Benhamou N, Rey P, Picard K, Tirilly Y . (1999). Ultrastructural and cytochemical aspects of the interaction between the mycoparasite Pythium oligandrum and soilborne plant pathogens. Phytopathol 89: 506–517.
Berney C, Fahrni J, Pawlowski J . (2004). How many novel eukaryotic 'kingdoms'? Pitfalls and limitations of environmental DNA surveys. BMC Biol 2: 13.
Berney C, Geisen S, Van Wichelen J, Nitsche F, Vanormelingen P, Bonkowski M et al. Expansion of the 'Reticulosphere': diversity of novel branching and network-forming amoebae helps to define Variosea (Amoebozoa). Protist.
Boenigk J, Pfandl K, Stadler P, Chatzinotas A . (2005). High diversity of the ‘Spumella-like’ flagellates: an investigation based on the SSU rRNA gene sequences of isolates from habitats located in six different geographic regions. Environ Microbiol 7: 685–697.
Bonkowski M . (2004). Protozoa and plant growth: the microbial loop in soil revisited. New Phytol 162: 617–631.
Cavalier-Smith T, Chao EEY, Oates B . (2004). Molecular phylogeny of Amoebozoa and the evolutionary significance of the unikont Phalansterium. Eur J Protistol 40: 21–48.
Chatzinotas A, Schellenberger S, Glaser K, Kolb S . (2013). Assimilation of cellulose-derived carbon by microeukaryotes in oxic and anoxic slurries of an aerated soil. Appl Environ Microbiol 79: 5777–5781.
Chen M, Chen F, Yu Y, Ji J, Kong F . (2008). Genetic diversity of eukaryotic microorganisms in Lake Taihu, a large shallow subtropical lake in China. Microb Ecol 56: 572–583.
Chou H-H, Holmes MH . (2001). DNA sequence quality trimming and vector removal. Bioinformatics 17: 1093–1104.
Clarholm M . (1985). Interactions of bacteria, protozoa and plants leading to mineralization of soil nitrogen. Soil Biol Biochem 17: 181–187.
Clarholm M, Bonkowski M, Griffiths B . (2007). Protozoa and other protista in soil. In: Van Elsas J, Jansson J, Trevors J (eds), Modern Soil Microbiology 2nd edn CRC, Boca Raton London New York. CRC Press: Boca Raton, FL, USA, pp 148–175.
Coince A, Caël O, Bach C, Lengellé J, Cruaud C, Gavory F et al. (2013). Below-ground fine-scale distribution and soil versus fine root detection of fungal and soil oomycete communities in a French beech forest. Fungal Ecol 6: 223–235.
Darriba D, Taboada GL, Doallo R, Posada D . (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9: 772–772.
De Jonckheere JF, Gryseels S, Eddyani M . (2012). Knowledge of morphology is still required when identifying new amoeba isolates by molecular techniques. Eur J Protistol 48: 178–184.
de Ruiter PC, Moore JC, Zwart KB, Bouwman LA, Hassink J, Bloem J et al. (1993). Simulation of nitrogen mineralization in the below-ground food webs of two winter wheat fields. J Appl Ecol 30: 95–106.
de Ruiter PC, Neutel AM, Moore JC . (1995). Energetics, patterns of interaction strengths, and stability in real ecosystems. Science 269: 1257–1260.
Ekelund F, Rønn R . (1994). Notes on protozoa in agricultural soil with emphasis on heterotrophic flagellates and naked amoebae and their ecology. FEMS Microbiol Rev 15: 321–353.
Ekelund F, Patterson DJ . (1997). Some heterotrophic flagellates from a cultivated garden soil in Australia. Arch Protistenkd 148: 461–478.
Ekelund F, Rønn R, Griffiths BS . (2001). Quantitative estimation of flagellate community structure and diversity in soil samples. Protist 152: 301–314.
Epstein S, López-García P . (2008). “Missing” protists: a molecular prospective. Biodivers Conserv 17: 261–276.
Fenchel T . (2010). The life history of Flabellula baltica Smirnov (Gymnamoebae, Rhizopoda): Adaptations to a spatially and temporally heterogeneous environment. Protist 161: 279–287.
Field SG, Michiels NK . (2005). Parasitism and growth in the earthworm Lumbricus terrestris: fitness costs of the gregarine parasite Monocystis sp. Parasitology 130: 397–403.
Finlay BJ, Black HIJ, Brown S, Clarke KJ, Esteban GF, Hindle RM et al. (2000). Estimating the growth potential of the soil protozoan community. Protist 151: 69–80.
Fiore-Donno AM, Kamono A, Chao EE, Fukui M, Cavalier-Smith T . (2010). Invalidation of Hyperamoeba by transferring its species to other genera of Myxogastria. J Eukaryot Microbiol 57: 189–196.
Foissner W . (1987). Soil protozoa: fundamental problems, ecological significance, adaptations in ciliates and testaceans, bioindicators, and guide to the literature. Prog Protistol 2: 69–212.
Foissner W . (1999). Soil protozoa as bioindicators: pros and cons, methods, diversity, representative examples. Agr Ecosyst Environ 74: 95–112.
Geisen S, Bandow C, Römbke J, Bonkowski M . (2014a). Soil water availability strongly alters the community composition of soil protists. Pedobiologia 57: 205–213.
Geisen S, Fiore-Donno A, Walochnik J, Bonkowski M . (2014b). Acanthamoeba everywhere: high diversity of Acanthamoeba in soils. Parasitol Res 113: 3151–3158.
Geisen S, Kudryavtsev A, Bonkowski M, Smirnov A . (2014c). Discrepancy between species borders at morphological and molecular levels in the genus Cochliopodium (Amoebozoa, Himatismenida), with the description of Cochliopodium plurinucleolum n. sp. Protist 165: 364–383.
Geisen S, Weinert J, Kudryavtsev A, Glotova A, Bonkowski M, Smirnov A . (2014d). Two new species of the genus Stenamoeba (Discosea, Longamoebia): cytoplasmic MTOC is present in one more amoebae lineage. J Eukaryot Microbiol 50: 153–165.
Glockling SL, Marshall WL, Gleason FH . (2013). Phylogenetic interpretations and ecological potentials of the Mesomycetozoea (Ichthyosporea). Fungal Ecol 6: 237–247.
Gong J, Dong J, Liu X, Massana R . (2013). Extremely high copy numbers and polymorphisms of the rDNA operon estimated from single cell analysis of oligotrich and peritrich ciliates. Protist 164: 369–379.
Goodey T . (1915). Note on the remarkable retention of vitality by protozoa from old stored soils. Ann Appl Biol 1: 395–399.
Gouy M, Guindon S, Gascuel O . (2010). SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 27: 221–224.
Hong S, Bunge J, Leslin C, Jeon S, Epstein SS . (2009). Polymerase chain reaction primers miss half of rRNA microbial diversity. ISME J 3: 1365–1373.
Howe AT, Bass D, Vickerman K, Chao EE, Cavalier-Smith T . (2009). Phylogeny, taxonomy, and astounding genetic diversity of Glissomonadida ord. nov., the dominant gliding zooflagellates in soil (Protozoa: Cercozoa). Protist 160: 159–189.
Huang X, Madan A . (1999). CAP3: A DNA sequence assembly program. Genome Res 9: 868–877.
Huelsenbeck JP, Ronquist F . (2001). MRBAYES: bayesian inference of phylogenetic trees. Bioinformatics 17: 754–755.
Jeon S, Bunge J, Leslin C, Stoeck T, Hong S, Epstein S . (2008). Environmental rRNA inventories miss over half of protistan diversity. BMC Microbiol Biol 8: 222.
Kaiser C, Koranda M, Kitzler B, Fuchslueger L, Schnecker J, Schweiger P et al. (2010). Belowground carbon allocation by trees drives seasonal patterns of extracellular enzyme activities by altering microbial community composition in a beech forest soil. New Phytol 187: 843–858.
Lahr DJG, Grant J, Katz LA . (2013). Multigene phylogenetic reconstruction of the Tubulinea (Amoebozoa) corroborates four of the six major lineages, while additionally revealing that shell composition does not predict phylogeny in the Arcellinida. Protist 164: 323–339.
Lanzén A, Jørgensen SL, Bengtsson MM, Jonassen I, Øvreås L, Urich T . (2011). Exploring the composition and diversity of microbial communities at the Jan Mayen hydrothermal vent field using RNA and DNA. FEMS Microbiol Ecol 77: 577–589.
Lanzén A, Jørgensen SL, Huson DH, Gorfer M, Grindhaug SH, Jonassen I et al. (2012). CREST – Classification Resources for Environmental Sequence Tags. PloS one 7: e49334.
Lara E, Berney C, Ekelund F, Harms H, Chatzinotas A . (2007). Molecular comparison of cultivable protozoa from a pristine and a polycyclic aromatic hydrocarbon polluted site. Soil Biol Biochem 39: 139–148.
Lara E, Mitchell EA, Moreira D, López García P . (2011). Highly diverse and seasonally dynamic protist community in a pristine peat bog. Protist 162: 14–32.
Lares-Jiménez LF, Booton GC, Lares-Villa F, Velázquez-Contreras CA, Fuerst PA . (2014). Genetic analysis among environmental strains of Balamuthia mandrillaris recovered from an artificial lagoon and from soil in Sonora, Mexico. Exp Parasitol 145 (Supplement): S57–S61.
Latijnhouwers M, de Wit PJGM, Govers F . (2003). Oomycetes and fungi: similar weaponry to attack plants. Trends Microbiol 11: 462–469.
Lejzerowicz F, Pawlowski J, Fraissinet-Tachet L, Marmeisse R . (2010). Molecular evidence for widespread occurrence of Foraminifera in soils. Environ Microbiol 12: 2518–2526.
Lentendu G, Wubet T, Chatzinotas A, Wilhelm C, Buscot F, Schlegel M . (2014). Effects of long-term differential fertilization on eukaryotic microbial communities in an arable soil: a multiple barcoding approach. Mol Ecol 23: 3341–3355.
Lesaulnier C, Papamichail D, McCorkle S, Ollivier B, Skiena S, Taghavi S et al. (2008). Elevated atmospheric CO2 affects soil microbial diversity associated with trembling aspen. Environ Microbiol 10: 926–941.
Martin FN, Abad ZG, Balci Y, Ivors K . (2012). Identification and detection of Phytophthora: reviewing our progress, identifying our needs. Plant Dis 96: 1080–1103.
Medinger R, Nolte V, Pandey RV, Jost S, Ottenwälder B, Schlötterer C et al. (2010). Diversity in a hidden world: potential and limitation of next generation sequencing for surveys of molecular diversity of eukaryotic microorganisms. Mol Ecol 19: 32–40.
Meisterfeld R, Holzmann M, Pawlowski J . (2001). Morphological and molecular characterization of a new terrestrial allogromiid species: Edaphoallogromia australica gen. et spec. nov. (Foraminifera) from northern Queensland (Australia). Protist 152: 185–192.
Monchy S, Sanciu G, Jobard M, Rasconi S, Gerphagnon M, Chabé M et al. (2011). Exploring and quantifying fungal diversity in freshwater lake ecosystems using rDNA cloning/sequencing and SSU tag pyrosequencing. Environ Microbiol 13: 1433–1453.
Moon van der Staay SY, Tzeneva VA, Van Der Staay GW, De Vos WM, Smidt H, Hackstein JH . (2006). Eukaryotic diversity in historical soil samples. FEMS Microbiol Ecol 57: 420–428.
Moussa M, Tissot O, Guerlotté J, De Jonckheere J, Talarmin A et al. (2015). Soil is the origin for the presence of Naegleria fowleri in the thermal recreational waters. Parasitol Res 114: 311–315.
Page FC . (1988) A new key to freshwater and soil gymnamoebae with instructions for culture. Freshwater Biological Association: Ambleside.
Pawlowski J, Audic S, Adl S, Bass D, Belbahri L, Berney C et al. (2012). CBOL protist working group: barcoding eukaryotic richness beyond the animal, plant, and fungal kingdoms. PLoS Biol 10: e1001419.
Pawlowski J . (2013). The new micro-kingdoms of eukaryotes. BMC Biol 11: 40.
Phillips AJ, Anderson VL, Robertson EJ, Secombes CJ, van West P . (2008). New insights into animal pathogenic oomycetes. Trends Microbiol 16: 13–19.
Radax R, Rattei T, Lanzen A, Bayer C, Rapp HT, Urich T et al. (2012). Metatranscriptomics of the marine sponge Geodia barretti: tackling phylogeny and function of its microbial community. Environ Microbiol 14: 1308–1324.
Robinson BS, Bamforth SS, Dobson PJ . (2002). Density and diversity of protozoa in some arid Australian soils. J Eukaryot Microbiol 49: 449–453.
Ruiz A, Frenkel J, Cerdas L . (1973). Isolation of Toxoplasma from soil. J Parasitol 59: 204–206.
Schaefer M, Schauermann J . (1990). The soil fauna of beech forests: comparison between a mull and a moder soil. Pedobiologia 34: 299–314.
Schröter D, Wolters V, de Ruiter PC . (2003). C and N mineralisation in the decomposer food webs of a European forest transect. Oikos 102: 294–308.
Schuster FL, Visvesvara GS . (2004). Free-living amoebae as opportunistic and non-opportunistic pathogens of humans and animals. Int J Parasitol 34: 1001–1027.
Siddiqui R, Ahmed Khan N . (2012). Biology and pathogenesis of Acanthamoeba. Parasit Vectors 5: 1–13.
Smirnov AV, Brown S . (2004). Guide to the methods of study and identification of soil gymnamoebae. Protistology 3: 148–190.
Smirnov AV, Nassonova E, Berney C, Fahrni J, Bolivar I, Pawlowski J . (2005). Molecular phylogeny and classification of the lobose amoebae. Protist 156: 129.
Smirnov AV, Nassonova ES, Cavalier-Smith T . (2008). Correct identification of species makes the amoebozoan rRNA tree congruent with morphology for the order Leptomyxida Page 1987; with description of Acramoeba dendroida n. g., n. sp., originally misidentified as ‘Gephyramoeba sp. Eur J Protistol 44: 35–44.
Smirnov AV, Chao E, Nassonova ES, Cavalier-Smith T . (2011). A revised classification of naked lobose amoebae (Amoebozoa: Lobosa). Protist 162: 545–570.
Sokal RR . (1961). Distance as a measure of taxonomic similarity. Syst Zool 10: 70–79.
Stamatakis A . (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22: 2688–2690.
Stoeck T, Breiner H-W, Filker S, Ostermaier V, Kammerlander B, Sonntag B . (2014). A morphogenetic survey on ciliate plankton from a mountain lake pinpoints the necessity of lineage-specific barcode markers in microbial ecology. Environ Microbiol 16: 430–444.
Stoupin D, Kiss AK, Arndt H, Shatilovich AV, Gilichinsky DA, Nitsche F . (2012). Cryptic diversity within the choanoflagellate morphospecies complex Codosiga botrytis - phylogeny and morphology of ancient and modern isolates. Eur J Protistol 48: 263–273.
Tikhonenkov DV, Mylnikov AP, Gong YC, Feng WS, Mazei Y . (2012). Heterotrophic flagellates from freshwater and soil habitats in subtropical China (Wuhan Area, Hubei province). Acta Protozool 51: 65.
Tong S, Vørs N, Patterson D . (1997). Heterotrophic flagellates, centrohelid heliozoa and filose amoebae from marine and freshwater sites in the Antarctic. Polar Biol 18: 91–106.
Turner TR, Ramakrishnan K, Walshaw J, Heavens D, Alston M, Swarbreck D et al. (2013). Comparative metatranscriptomics reveals kingdom level changes in the rhizosphere microbiome of plants. ISME J 7: 2248–2258.
Tveit A, Schwacke R, Svenning MM, Urich T . (2013). Organic carbon transformations in high-Arctic peat soils: key functions and microorganisms. ISME J 7: 299–311.
Urich T, Lanzén A, Qi J, Huson DH, Schleper C, Schuster SC . (2008). Simultaneous assessment of soil microbial community structure and function through analysis of the meta-transcriptome. PloS One 3: e2527.
Urich T, Schleper C . (2011). The “Double-RNA” approach to simultaneously assess the structure and function of a soil microbial community. de Brujin FJ. Handbook of Molecular Microbial Ecology I. John Wiley & Sons, Inc.: Hoboken, NJ, USA, pp 587–596.
Urich T, Lanzén A, Stokke R, Pedersen RB, Bayer C, Thorseth IH et al. (2014). Microbial community structure and functioning in marine sediments associated with diffuse hydrothermal venting assessed by integrated meta-omics. Environ Microbiol 16: 2699–2710.
Visvesvara GS, Schuster FL, Martinez AJ . (1993). Balamuthia mandrillaris, N. G., N. Sp., agent of amebic meningoencephalitis in humans and other animals. J Eukaryot Microbiol 40: 504–514.
Visvesvara GS, Moura H, Schuster FL . (2007). Pathogenic and opportunistic free living amoebae: Acanthamoeba spp., Balamuthia mandrillaris, Naegleria fowleri, and Sappinia diploidea. FEMS Immunol Med Mic 50: 1–26.
Weber AAT, Pawlowski J . (2013). Can abundance of protists be inferred from sequence data: a case study of Foraminifera. PloS one 8: e56739.
Zwart KB, Burgers SLGE, Bloem J, Bouwman LA, Brussaard L, Lebbink G et al. (1994). Population dynamics in the belowground food webs in two different agricultural systems. Agr Ecosyst Environ 51: 187–198.
Acknowledgements
We thank Ave Tooming-Klunderud and colleagues at the Centre for Ecological and Evolutionary Synthesis (CEES, University of Oslo, Norway) for 454-sequencing. We thank Sarah Jennings (NIOO, Netherlands) for help in cDNA generation from Rothamsted soil and thank Christoph Bayer (Vienna) for help in sequence processing. Christa Schleper (Vienna) is thanked for her generous and continuous support. Special thanks to Fionn Clissman for his helpful suggestions on the wording. Part of this work was supported by a research grant from the EU-project ‘EcoFINDERS’ No. 264465 and by a research council of norway grant 191696/V49.
Author Contributions
TU, SG and MB designed research. TU, AT, MS and IC collected the soil samples. Metatranscriptomes were generated by TU and AT. TU processed metatranscriptome data, analysed by SG and TU. SG and TU generated Amoebozoa-CREST. SG, TU and MB wrote the manuscript, aided by all authors.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Geisen, S., Tveit, A., Clark, I. et al. Metatranscriptomic census of active protists in soils. ISME J 9, 2178–2190 (2015). https://doi.org/10.1038/ismej.2015.30
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2015.30
This article is cited by
-
Soil microbial ecology through the lens of metatranscriptomics
Soil Ecology Letters (2024)
-
Community dynamics of microbial eukaryotes in intertidal mudflats in the hypertidal Bay of Fundy
ISME Communications (2023)
-
Metagenomics and metatranscriptomics as potential driving forces for the exploration of diversity and functions of micro-eukaryotes in soil
3 Biotech (2023)
-
Protists: the hidden ecosystem players in a wetland rice field soil
Biology and Fertility of Soils (2023)
-
Microeukaryotic gut parasites in wastewater treatment plants: diversity, activity, and removal
Microbiome (2022)
