
Abstract
Antibiotic resistance is a dire clinical problem with important ecological dimensions. While antibiotic resistance in human pathogens continues to rise at alarming rates, the impact of environmental resistance on human health is still unclear. To investigate the relationship between human-associated and environmental resistomes, we analyzed functional metagenomic selections for resistance against 18 clinically relevant antibiotics from soil and human gut microbiota as well as a set of multidrug-resistant cultured soil isolates. These analyses were enabled by Resfams, a new curated database of protein families and associated highly precise and accurate profile hidden Markov models, confirmed for antibiotic resistance function and organized by ontology. We demonstrate that the antibiotic resistance functions that give rise to the resistance profiles observed in environmental and human-associated microbial communities significantly differ between ecologies. Antibiotic resistance functions that most discriminate between ecologies provide resistance to β-lactams and tetracyclines, two of the most widely used classes of antibiotics in the clinic and agriculture. We also analyzed the antibiotic resistance gene composition of over 6000 sequenced microbial genomes, revealing significant enrichment of resistance functions by both ecology and phylogeny. Together, our results indicate that environmental and human-associated microbial communities harbor distinct resistance genes, suggesting that antibiotic resistance functions are largely constrained by ecology.
Similar content being viewed by others

Introduction
Multidrug resistance in clinical pathogens continues to rise, whereas the pipeline for new antibiotic development and approval continues to dwindle (Spellberg et al., 2004). Consequently, we face the prospect of returning to a preantibiotic era, where an increasing number of infections can no longer be treated effectively with our current arsenal of drugs. Although it is clear that dissemination of antibiotic resistance (AR) extends beyond the clinic (Wright, 2010; Forsberg et al., 2012) and includes many routes through agricultural and environmental microbial communities, the full impact of environmental AR on human health is still unknown (Finley et al., 2013). A deep quantitative understanding of the ecological relationship between environmental and human-associated microbial resistomes is necessary to evaluate the relative importance of these diverse ecologies in AR gene acquisition by human pathogens.
Hindering our ability to study the ecology and transmission of AR genes between environmental and human-associated reservoirs is the difficulty in accurately identifying AR functions from sequence alone. This is emphasized by continual identification of sequence-novel AR genes in almost every microbial community, including soil (Riesenfeld et al., 2004; Allen et al., 2008; Torres-Cortes et al., 2011; Forsberg et al., 2012), activated sludge (Mori et al., 2008; Parsley et al., 2010), human gut and oral microbiomes (Diaz-Torres et al., 2003, 2006; Sommer et al., 2009; Cheng et al., 2012) and animal gut microbiomes (Kazimierczak et al., 2009). This enforces the need for functional validation in studies of AR transmission (Pehrsson et al., 2013) and for improved methods to identify sequence-divergent AR determinants in silico.
In order to quantitatively analyze the relationship between environmental and human-associated resistomes, we developed Resfams, a curated database of protein families and associated profile hidden Markov models (HMMs), organized by ontology and confirmed for AR function. Profile HMMs have been widely adopted for improved annotation of general functions in microbial genomes and metagenomes (Meyer et al., 2008; Markowitz et al., 2012). However, they have not yet been specifically applied to AR functions in microbial communities or genomes. Once developed and validated, Resfams profile HMMs were applied to quantitatively understand the relationship between human-associated and environmental resistomes using both functional selections to 18 antibiotics from the soil and human microbiota and analysis of over 6000 microbial genomes representing diverse phylogenies and habitats.
Materials and methods
Building of profile hidden Markov models (HMMs)
Resfams AR family profile HMMs were built by (1) generating a multiple sequence alignment for each AR family (see Supplementary Methods) using MUSCLE (Edgar, 2004) v3.8.31 with default parameters and (2) training profile HMMs using the hmmbuild function of the HMMER3 (Finn et al., 2011) software package using default parameters. Gathering thresholds were added to the profile HMMs by using a test set of known AR proteins and optimizing precision and recall metrics (see Supplementary Methods). Resfams profile HMMs were first trained using 2097 unique AR protein sequences from the Comprehensive Antibiotic Resistance Database (CARD) database (McArthur et al., 2013), the Lactamase Engineering Database (LacED) (Thai et al., 2009) and Jacoby and Bush’s collection of curated β-lactamase proteins (http://www.lahey.org/Studies/).
This core database of 119 profile HMMs was supplemented with an additional 47 profile HMMs from the Pfam (Bateman et al., 2000) and TIGRFam (Haft et al., 2003) databases to generate the full Resfams profile HMM database, resulting in a total of 166 AR-specific profile HMMs (Supplementary Table S1). The 47 additional profile HMMs included in this version of Resfams have been curated and demonstrated to identify protein families that commonly contribute to AR, such as acetyltransferases, AraC transcriptional regulators, Major Facilitor Superfamily (MFS) transporters, ATP-binding cassette (ABC) efflux pumps and so on (see Supplementary Table S1 for HMMs with ‘HMM Source’ of Pfam or TIGRFam). These supplementary profile HMMs were verified using functional assays, including functional metagenomic selections of the soil microbiota and the human gut microbiota. The full version of the Resfams database is only utilized when there is previous functional evidence of AR activity, such as functional metagenomic selections. All versions of the Resfams database and supporting datafiles are available at http://dantaslab.wustl.edu/resfams.
Protein annotation using Resfams profile HMMs or BLAST to AR-specific databases
Proteins were aligned to the core Resfams database of profile HMMs (Resfams.hmm) for microbial genome annotation or the full Resfams database of profile HMMs (Resfams-full.hmm) for functional metagenomic selections using the hmmscan function of the HMMER3 (Finn et al., 2011) software package using the following parameters: —cut_ga and —tblout. Antibiotic mechanism and β-lactamase class classification used in this analysis can be found in Supplementary Table S1.
The Antibiotic Resistance Database (ARDB; Liu and Pop, 2009) and CARD (McArthur et al., 2013) were used for comparison of Resfams profile HMMs to pairwise sequence alignment against known AR proteins. A protein was called an AR protein if it had an amino acid identity greater than or equal to the class-specific identity threshold defined by ARDB documentation over >85% of the length of the target sequence. If no class-specific identity threshold was defined or the top hit was to a protein from the CARD (McArthur et al., 2013) database, a protein was called an AR protein if it had ⩾80% amino acid identity over >85% of the length of the target sequence. To classify AR proteins by resistance mechanism or β-lactamase Ambler class, we used the mapping table in Supplementary Table S6.
Functional metagenomic selections
All functional metagenomic selections in this analysis were selected, sequenced and assembled into contigs as previously described (Forsberg et al., 2012, 2014; Moore et al., 2013). Briefly, metagenomic plasmid libraries prepared in Escherichia coli DH10B host were selected for resistant inserts on Luria-Bertani or Mueller-Hinton agar plates containing kanamycin (50 μg ml−1; plasmid resistance marker) plus the antibiotic of interest at a concentration toxic to wild-type E. coli host. Resistant inserts were then amplified and sequenced using the Illumina (San Diego, CA, USA) Hi-Seq Pair-End (PE) 76 or 101 bp sequencing protocol. Sequencing reads were then assembled into contigs using the PARFuMS (Parallel Annotation and Re-assembly of Functional Metagenomic Selections) pipeline (Forsberg et al., 2012). Open reading frames were predicted in assembled contigs using the gene-finding algorithm MetaGeneMark (Zhu et al., 2010) using default parameters. Functional metagenomic selections used for this study were prepared from (1) multidrug resistant (MDR) cultured soil isolates, (2) pediatric human fecal samples and (3) Cedar Creek and Kellogg Biological Station soils, as outlined in Supplementary Table S2. Resistome comparisons were performed using a combination of R, Cytoscape and the QIIME software package (Shannon et al., 2003; Caporaso et al., 2010) (see Supplementary Methods).
Microbial genomes
A total of 6179 microbial proteomes were downloaded from the Integrated Microbial Genomes database (IMG) on 18 August 2013. Open reading frames were called using the IMG pipelines as previously described (Markowitz et al., 2012) and protein sequences downloaded from IMG were used in all downstream annotation and analysis. A complete list of the microbial genomes analyzed is available in Supplementary Table S4, including bacterial phyla, habitat and potential pathogen status. Habitat and potential pathogen status was curated using the metadata available from the IMG database, using the ‘Habitat’ and ‘Diseases’ fields. A mapping of values listed in the IMG metadata ‘Habitat’ and ‘Disease’ fields to the ‘Habitat’ and ‘Pathogen Status’ used in this study is available in Supplementary Table S7.
Results and discussion
Optimization of antibiotic resistance profile HMMs (Resfams)
Following extensive curation and functional validation (see Supplementary Methods), the full Resfams HMM database contains 166 profile HMMs representing all major AR gene classes, including AR genes against β-lactams, aminoglycosides, fluoroquinolones, glycopeptides, macrolides, tetracyclines as well as efflux pumps and transcription factors modulating AR. To improve Resfams prediction accuracy, we optimized profile-specific gathering thresholds (see Supplementary Methods) that set an inclusion bit score cutoff for a protein sequence alignment on a profile-by-profile basis (Punta et al., 2012). We achieved perfect precision and high recall (99±0.02%) of all AR proteins used to train the Resfams profile HMMs (Supplementary Figure S1B). We tested the prediction accuracy of these optimized Resfams families on AR proteins from the ARDB (Liu and Pop, 2009) not used in training of the original profile HMMs. We predicted the AR function of 2454 unique sequences, representing 54 Resfams protein families, and achieved perfect precision and high recall (98±0.03%) for all protein families (Supplementary Figure S1C). These recruited protein sequences were subsequently incorporated into the corresponding Resfams protein families, resulting in the final database of AR-specific profile HMMs used for all further analyses in this study.
Resfams accurately predicts novel resistance functions from sequence alone
The prediction sensitivity and specificity of Resfams was evaluated using recent functional metagenomic studies investigating the resistomes of cultured soil microbiota and human gut microbiota (see Supplementary Methods). We compared Resfams HMMs with pairwise sequence alignment (BLAST) against the ARDB (Liu and Pop, 2009) and CARD (McArthur et al., 2013) databases for their ability to predict AR function. Resfams demonstrated improved sensitivity: 64% of AR proteins identified using Resfams in both the soil and the human gut microbiota were not identified by BLAST (Figure 1). Focusing on the hand-curated, gold standard set of AR proteins identified in the set of 95 MDR cultured soil isolates, we show that Resfams was able to predict over 95% of the full-length AR proteins (>90 amino acids) (Figure 1a). In contrast, pairwise sequence alignment to AR-specific databases using widely accepted identity thresholds for AR proteins (Liu and Pop, 2009) annotated <34% of full-length AR proteins. Importantly, Resfams did not identify any false positive AR proteins (that is, not predicted by intensive hand-curation), ensuring that AR potential of microbial communities is not overestimated. Generating the gold standard set of AR genes involved extensive hand-curation, including functional characterization, phylogenetic analysis and primary literature validation to identify causative AR genes. This time-intensive process is prohibitive for large-scale studies of AR potential of microbial communities. In comparison, Resfams enables automated, rapid, accurate and high-resolution predictions of AR proteins.

Annotation of functional metagenomic selections using Resfams. Unique full-length open reading frames (ORFs; >90 amino acids (a.a.)) annotated as AR proteins from functional metagenomic selections using Resfams HMM database (blue) compared with hand curation (black) and BLAST to AR databases (red) from (a) MDR cultured soil bacteria and (b) human gut microbiota. The Resfams HMM database identified no false positive annotations as measured by the hand-curated gold standard for cultured soil bacteria. For the amoxicillin selection, MetaGeneMark predicted one ORF in the hand-curated set as two independent ORFs and both were correctly identified by Resfams.
As a demonstration of the high-resolution and high-specificity AR functional predictions of Resfams, we focused on β-lactamases, one of the most widely disseminated and clinically relevant class of AR genes (Davies and Davies, 2010). In the previously discussed soil and human gut functional metagenomic data sets, 113 unique, full-length β-lactamase proteins were predicted by Resfams that were not predicted as an AR gene by pairwise sequence alignment to AR databases (representing 45% of all identified β-lactamases). β-lactamases are commonly classified into four molecular classes (Ambler classes A, B, C and D) based on primary structure (Ambler, 1980), with over 1000 unique AR-related β-lactamases identified to date (Davies and Davies, 2010). We accurately predict the molecular class or subclass for over 60% of the novel β-lactamases from the soil (Figure 2a) and over 80% of the novel β-lactamases from the human gut (Figure 2b). Importantly, all of these predicted class or subclass β-lactamases clustered with previously reported β-lactamases of the same molecular class on a phylogenetic tree (Figure 2c), confirming that Resfams accurately categorized these β-lactamases by sequence class where conventional computational methods were unable to even predict general AR function.

Resfams improves the resolution of annotation of antibiotic resistance proteins. The majority of β-lactamases identified from (a) MDR cultured soil bacteria and (b) human gut microbiota in functional metagenomic selections that are highly sequence divergent from any protein in the ARDB or CARD databases (<80% amino acid (a.a.) identity over 85% of target sequence) are annotated according to Ambler class (red) or subclass (cyan) level by Resfams. (c) All highly divergent β-lactamases from (a) and (b) that are annotated at the Ambler class and subclass level (colored branches) by Resfams accurately cluster on a phylogenetic tree with previously verified β-lactamases from all four Ambler classes (black branches). Pie charts in (c) depict the fraction of Resfams identified β-lactamases and previously verified β-lactamases represented on the phylogenetic tree in each Ambler class clade.
Antibiotic resistomes cluster by ecology
Environmental, nonpathogenic and commensal organisms have long been shown to harbor functional AR genes (Benvenis and Davies, 1973; Marshall et al., 1998; Riesenfeld et al., 2004). However, it was only recently established that exchange of multiple classes of AR genes has occurred between nonpathogenic environmental bacteria and human pathogens (Forsberg et al., 2012). These findings indicate that environmental bacteria serve as potential reservoirs of AR genes primed for exchange with pathogenic bacteria. This motivates a high-resolution characterization of the AR genes distinct to and shared between environmental and human-associated microbial communities. Analyses that employ Resfams protein families are capable of addressing this goal by reducing bias in comparisons across ecological barriers. As a demonstration, we re-annotated functional metagenomic selections against 18 antibiotics from uncultured human gut, soil microbiota and MDR soil-dwelling cultured isolates using Resfams (summarized in Supplementary Table S2).
To compare the diversity of underlying AR functions that give rise to observed AR profiles across samples and ecologies (Supplementary Figure S2), a count matrix of unique protein sequences per Resfams AR family identified in each resistome was generated by summing across a subset of five antibiotic selections included in all three data sets (selections encompass four major classes of antibiotics: β-lactams, tetracyclines, amphenicols and glycopeptides). Although previous studies have predicted an enrichment of AR genes in the human gut microbiota (Hu et al., 2013), we found no significant differences in the number of distinct AR genes between the human gut and soil microbiota when screened for functional AR. Resistomes cluster by ecology using both Bray-Curtis and Jaccard distance metrics (P<0.001, ANISOM; Figure 3, Supplementary Figure S3), suggesting that the soil and human gut microbiota consist of functionally distinct AR gene architectures.

Resistomes differ by ecology. (a) Principal coordinate analysis (PCoA) plot depicting Bray-Curtis distances between resistomes of the soil (green), human gut (magenta) and MDR soil isolates (cyan), calculated using unique AR protein counts. Resistomes of different ecologies cluster separately (P<0.001, ANOSIM). Function biplot coordinates (squares) represent the weighted average of the top six most discriminating AR functions between ecologies across all samples. The size of the biplot squares represent the aggregate abundance of the unique AR family members. Separation of resistomes is heavily influenced by β-lactamase (orange squares) and tetracycline resistance functions (yellow squares). (b) Bipartite network diagram of normalized AR protein counts across all resistomes. Edges connect sample nodes (squares) to AR function (circles). Edges and sample nodes are colored by sample ecology (green, soil; magenta, human gut; cyan, MDR soil isolates) and AR functions are colored by extent of sharing across ecologies (white, unique to ecology; gray, shared between two ecologies; black, shared across all three ecologies). Inset pie chart represents the percentage of AR Resfams families that belong to each group.
In order to determine the Resfams families that most discriminate antibiotic resistomes between ecologies, we used the supervised learning technique Random Forests (Knights et al., 2011). This analysis revealed that the separation of soil and gut resistomes is driven mainly by β-lactamase and tetracycline resistance functions (Figure 3). Subclass B3 β-lactamases (class B) are mainly associated with soil resistomes, whereas CblA (class A) and CMY/LAT/MOX (class C) β-lactamases are found mainly associated with the human gut. CblA β-lactamases are species-specific cephalosporinases that have been primarily identified in Bacteroides uniformis, a common resident of the human gut microbiota (Smith et al., 1994). Although this particular class A β-lactamase uniquely discriminates between soil and human gut resistomes, class A β-lactamases in general are prevalent across all environments and samples (94% of soil resistomes and 89% of human gut resistomes). The low ratio of class C β-lactamases to class B β-lactamases in the soil supports previous findings in studies of the soil resistome (Allen et al., 2008; Forsberg et al., 2014); however, our results indicate that this trend is opposite in the human gut resistome (class C/class B ratio: 0.42, soil; 4.3, gut). Whereas class A and class C β-lactamases have long been considered the most clinically important classes of β-lactamases, class B β-lactamases are a growing concern in the effective treatment of infectious disease (Rice and Bonomo, 2000), exemplified by the NDM-1 carbapenamase disseminating in MDR pathogens and environmental habitats (Walsh et al., 2011). Therefore, even though resistomes appear to be largely constrained by ecology, our results continue to emphasize the importance of environmental reservoirs of AR in the emergence of novel clinical resistance, particularly in this important class of AR genes.
In addition to Resfams families that discriminate resistance reservoirs, we sought to determine whether there was a core resistome across all samples and across all habitats. Highlighting the extreme diversity of AR genes, there was no single Resfams family that was shared across all samples, and only two Resfams families were shared across >50% of metagenomic samples (class A β-lactamases and MFS Antibiotic Efflux), and only six Resfams families were found in at least one sample from every habitat investigated (Supplementary Table S3).
Antibiotic resistance potential encoded in microbial genomes
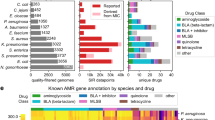
To understand the relative impacts of phylogenetic origin and ecological factors in shaping AR reservoirs, a greater appreciation for the relationship between AR function and bacterial community composition is needed. Accordingly, we used Resfams HMMs to predict the AR potential encoded in 6179 microbial genomes from the IMG database (Markowitz et al., 2012), representing diverse phylogenies and habitats (Supplementary Table S4). We summed all Resfams counts in a genome by the AR mechanisms listed in Supplementary Table S1 and calculated enrichment of a particular mechanism in (1) phyla, (2) habitat, as well as (3) phyla by habitat (Figure 4). Importantly, these results provide confirmation that Resfams accurately annotates AR function. For example, as was observed with functional selections, we found no significant difference in the percentage of total AR functions encoded in microbial genomes between habitats (Supplementary Figure S4), contradicting previous predictions using pairwise sequence alignment annotation methods (Hu et al., 2013). Further confirmation can be seen by examining resistance to glycopeptides, a class of antibiotics only active against Gram-positive bacteria because of their large molecular size that prevents their transport across outer membrane porins of Gram-negative bacteria (Pootoolal et al., 2002). Glycopeptide resistance mechanism distributions across phyla reflect this AR pressure as they were predicted exclusively in Gram-positive organism genomes.

Resistomes annotated by Resfams across phylogeny and habitats of 6179 sequenced bacterial isolate genomes. Binary heatmaps of genomes organized by (a) phylogeny, (b) habitat and (c) phylogeny within habitat. Sections of the heatmaps are colored if a particular AR mechanism is significantly enriched within a particular phyla or habitat (P<0.01, Fisher’s exact). Enrichment of β-lactamase Ambler class and tetracycline resistance functions is depicted across (d) phyla and (e) habitat (*P<0.01, Fisher’s exact).
Although AR is present in nearly all microbial genomes (84% of microbial genomes investigated encode at least one AR gene), there is significant AR mechanism enrichment by bacterial phyla and habitat (Figure 4 and Supplementary Table S5). For example, resistance to β-lactams, one of the most clinically important classes of antibiotics, is significantly enriched in Actinobacteria relative to other phyla (P<0.01, Fisher’s exact, Figure 4a), when summed across habitats. This is consistent with the Actinomycete class of bacteria being responsible for synthesis of the vast majority of natural β-lactam antibiotics, therefore requiring self-resistance. In addition, β-lactamases are enriched in soil bacteria versus other habitats (P<0.01, Fisher’s exact, Figure 4b), consistent with the vast majority of β-lactam-producing bacteria originating in the soil. However, our results show that β-lactamase resistance genes in the soil are distributed across all phyla with no significant enrichment in Actinobacteria. This suggests that many soil bacteria, regardless of phylogeny, have evolved to confer resistance to β-lactams, revealing a strong habitat by phylogeny relationship.
As resistance to tetracyclines represented some of the most discriminating Resfams functions between the soil and the human gut microbiota from our functional metagenomic analyses (Figure 3), we were prompted to further investigate tetracycline resistance mechanisms encoded by microbial genomes (Figures 4d and e). There are three known major mechanisms of tetracycline resistance: MFS efflux, ribosomal protection and drug inactivation. Our results suggest that the mechanism by which bacteria resist tetracycline antibiotics is heavily biased by habitat. Soil bacteria are significantly enriched for tetracycline MFS efflux pumps, whereas human-associated bacteria are significantly enriched for tetracycline ribosomal protection genes (P<0.01, Fisher’s exact). These results are consistent with our findings from functional metagenomic selections (Supplementary Figure S5). Conversely, pairwise sequence alignment to AR-specific databases incorrectly predicts enrichment of all tetracycline resistance mechanisms in the human gut versus soil (Figure 5b).

Annotation of antibiotic resistance across habitats using Resfams compared with pairwise sequence alignment. Predicted enrichment of β-lactamase Ambler classes and tetracycline resistance mechanisms (*P<0.01, Fisher’s exact) in sequenced genomes across habitats using (a, b) Resfams family profile HMMs compared with (c, d) BLAST to the ARDB and CARD databases.
Finally, we observed that pathogenic organisms are enriched for all surveyed AR mechanisms, excepting antibiotic inactivation and β-lactamases (Supplementary Figure S6). β-lactamases have long been recognized as one of the most widely distributed mechanism of AR and were found to be encoded in over 60% of bacterial genomes. Our results, however, emphasize that β-lactamases are commonly found in nonclinical environments and commensal organisms, and this has implications for understanding the further spread of β-lactamases to pathogens via nonclinical AR reservoirs (Humeniuk et al., 2002; Walsh et al., 2011).
The described resistome analysis would yield significantly different conclusions if microbial genomes were analyzed using pairwise sequence alignment to AR-specific databases. Not only would the total resistance potential of microbial genomes be significantly lower (P<0.001, Wilcoxon’s rank-sum test), but also the phylogenetic and ecological distribution of AR mechanisms would be significantly biased toward more heavily studied human-associated environments (Figure 5). Using the Resfams profile HMM database, we predict that a number of AR functions are enriched in the soil, consistent with evidence that antibiotic producers are primarily soil dwelling and that AR is ancient, having evolved in soil for over the past 30 000 years (D'Costa et al., 2011). Again, this result is not recapitulated using pairwise sequence alignment to AR-specific databases (Figure 5).
Conclusions
Using Resfams, we show that antibiotic resistomes cluster by ecology, with no core resistome shared between all samples. For example, while all communities display resistance to tetracycline, soil bacteria mainly resist tetracycline through MFS antibiotic efflux, whereas bacteria in the human gut microbiota typically resist tetracycline via ribosomal protection mechanisms. Importantly, these results are consistent between both the functional metagenomic data sets and the sequenced microbial genomes.
Our ability to accurately identify and annotate AR functions in microbial genomes and communities has important implications for our ability to fight infectious disease. It improves our understanding of the evolution, ecology and transmission of AR in pathogens, as well as has a direct impact on clinical diagnostics. Currently, the most common method used to characterize resistome composition from sequencing data is pairwise sequence alignment to AR-specific databases (Forslund et al., 2013; Hu et al., 2013). This approach biases toward human-associated organisms, vastly underestimating the potential impact of environmental resistance reservoirs on AR in pathogens. In order to address this problem, we developed and benchmarked a set of AR-specific gene families (Resfams) and associated profile HMMs and applied them to functional metagenomic data sets from the soil and human gut microbiota as well as to over 6000 sequenced microbial isolate genomes representing diverse phylogenies and habitats. By using a consensus model approach, we are able to significantly increase our ability to characterize highly diverse and understudied reservoirs of resistance while minimizing bias.
In order for the full potential of Resfams AR protein families to be realized, in-depth functional validation of genotype to phenotype predictions is necessary. In addition, Resfams AR protein families need to be continually updated and maintained in order to keep up with rapidly evolving bacterial AR. These challenges emphasize the importance of continued functional investigation of environmental and clinical AR reservoirs, and for these investigations to be intimately connected to the improvement of methods for annotation of AR phenotype from genotype.
References
Allen HK, Moe LA, Rodbumrer J, Gaarder A, Handelsman J . (2008). Functional metagenomics reveals diverse β-lactamases in a remote Alaskan soil. ISME J 3: 243–251.
Ambler RP . (1980). The structure of beta-lactamases. Philos Trans R Soc Lond B Biol Sci 289: 321–331.
Bateman A, Birney E, Durbin R, Eddy SR, Howe KL, Sonnhammer EL . (2000). The Pfam protein families database. Nucleic Acids Res 28: 263–266.
Benvenis R, Davies J . (1973). Aminoglycoside antibiotic-inactivating enzymes in actinomycetes similar to those present in clinical isolates of antibiotic-resistant bacteria. Proc Natl Acad Sci USA 70: 2276–2280.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336.
Cheng G, Hu Y, Yin Y, Yang X, Xiang C, Wang B et al. (2012). Functional screening of antibiotic resistance genes from human gut microbiota reveals a novel gene fusion. FEMS Microbiol Lett 336: 11–16.
D'Costa VM, King CE, Kalan L, Morar M, Sung WW, Schwarz C et al. (2011). Antibiotic resistance is ancient. Nature 477: 457–461.
Davies J, Davies D . (2010). Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev 74: 417–433.
Diaz-Torres ML, McNab R, Spratt DA, Villedieu A, Hunt N, Wilson M et al. (2003). Novel tetracycline resistance determinant from the oral metagenome. Antimicrob Agents Chemother 47: 1430–1432.
Diaz-Torres ML, Villedieu A, Hunt N, McNab R, Spratt DA, Allan E et al. (2006). Determining the antibiotic resistance potential of the indigenous oral microbiota of humans using a metagenomic approach. FEMS Microbiol Lett 258: 257–262.
Edgar RC . (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797.
Finley RL, Collignon P, Larsson DG, McEwen SA, Li XZ, Gaze WH et al. (2013). The scourge of antibiotic resistance: the important role of the environment. Clin Infect Dis 57: 704–710.
Finn RD, Clements J, Eddy SR . (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39: W29–W37.
Forsberg KJ, Reyes A, Wang B, Selleck EM, Sommer MOA, Dantas G . (2012). The shared antibiotic resistome of soil bacteria and human pathogens. Science (New York, NY) 337: 1107–1111.
Forsberg KJ, Patel S, Gibson MK, Lauber CL, Knight R, Fierer N et al. (2014). Bacterial phylogeny structures soil resistomes across habitats. Nature 509: 612–616.
Forslund K, Sunagawa S, Kultima JR, Mende DR, Arumugam M, Typas A et al. (2013). Country-specific antibiotic use practices impact the human gut resistome. Genome Res 23: 1163–1169.
Haft DH, Selengut JD, White O . (2003). The TIGRFAMs database of protein families. Nucleic Acids Res 31: 371–373.
Hu Y, Yang X, Qin J, Lu N, Cheng G, Wu N et al. (2013). Metagenome-wide analysis of antibiotic resistance genes in a large cohort of human gut microbiota. Nat Commun 4: 2151.
Humeniuk C, Arlet G, Gautier V, Grimont P, Labia R, Philippon A . (2002). Beta-lactamases of Kluyvera ascorbata, probable progenitors of some plasmid-encoded CTX-M types. Antimicrob Agents Chemother 46: 3045–3049.
Kazimierczak KA, Scott KP, Kelly D, Aminov RI . (2009). Tetracycline resistome of the organic pig gut. Appl Environ Microbiol 75: 1717–1722.
Knights D, Costello EK, Knight R . (2011). Supervised classification of human microbiota. FEMS Microbiol Rev 35: 343–359.
Liu B, Pop M . (2009). ARDB—Antibiotic Resistance Genes Database. Nucleic Acids Res 37: D443–D447.
Markowitz VM, Chen IM, Palaniappan K, Chu K, Szeto E, Grechkin Y et al. (2012). IMG: the Integrated Microbial Genomes database and comparative analysis system. Nucleic Acids Res 40: D115–D122.
Marshall CG, Lessard IA, Park I, Wright GD . (1998). Glycopeptide antibiotic resistance genes in glycopeptide-producing organisms. Antimicrob Agents Chemother 42: 2215–2220.
McArthur AG, Waglechner N, Nizam F, Yan A, Azad MA, Baylay AJ et al. (2013). The comprehensive antibiotic resistance database. Antimicrob Agents Chemother 57: 3348–3357.
Meyer F, Paarmann D, D'Souza M, Olson R, Glass EM, Kubal M et al. (2008). The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9: 386.
Moore AM, Patel S, Forsberg KJ, Wang B, Bentley G, Razia Y et al. (2013). Pediatric fecal microbiota harbor diverse and novel antibiotic resistance genes. PLoS One 8: e78822.
Mori T, Mizuta S, Suenaga H, Miyazaki K . (2008). Metagenomic screening for bleomycin resistance genes. Appl Environ Microbiol 74: 6803–6805.
Parsley LC, Consuegra EJ, Kakirde KS, Land AM, Harper Jr WF, Liles MR . (2010). Identification of diverse antimicrobial resistance determinants carried on bacterial, plasmid, or viral metagenomes from an activated sludge microbial assemblage. Appl Environ Microbiol 76: 3753–3757.
Pehrsson EC, Forsberg KJ, Gibson MK, Ahmadi S, Dantas G . (2013). Novel resistance functions uncovered using functional metagenomic investigations of resistance reservoirs. Front Microbiol 4: 145.
Pootoolal J, Neu J, Wright GD . (2002). Glycopeptide antibiotic resistance. Annu Rev Pharmacol Toxicol 42: 381–408.
Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C et al. (2012). The Pfam protein families database. Nucleic Acids Res 40: D290–D301.
Rice LB, Bonomo RA . (2000). beta-Lactamases: which ones are clinically important? Drug Resist Updat 3: 178–189.
Riesenfeld CS, Goodman RM, Handelsman J . (2004). Uncultured soil bacteria are a reservoir of new antibiotic resistance genes. Environ Microbiol 6: 981–989.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504.
Smith CJ, Bennett TK, Parker AC . (1994). Molecular and genetic analysis of the Bacteroides uniformis cephalosporinase gene, cblA, encoding the species-specific beta-lactamase. Antimicrob Agents Chemother 38: 1711–1715.
Sommer MO, Dantas G, Church GM . (2009). Functional characterization of the antibiotic resistance reservoir in the human microflora. Science 325: 1128–1131.
Spellberg B, Powers JH, Brass EP, Miller LG, Edwards Jr JE . (2004). Trends in antimicrobial drug development: implications for the future. Clin Infect Dis 38: 1279–1286.
Thai QK, Bos F, Pleiss J . (2009). The Lactamase Engineering Database: a critical survey of TEM sequences in public databases. BMC Genomics 10: 390.
Torres-Cortes G, Millan V, Ramirez-Saad HC, Nisa-Martinez R, Toro N, Martinez-Abarca F . (2011). Characterization of novel antibiotic resistance genes identified by functional metagenomics on soil samples. Environ Microbiol 13: 1101–1114.
Walsh TR, Weeks J, Livermore DM, Toleman MA . (2011). Dissemination of NDM-1 positive bacteria in the New Delhi environment and its implications for human health: an environmental point prevalence study. Lancet Infect Dis 11: 355–362.
Wright GD . (2010). Antibiotic resistance in the environment: a link to the clinic? Curr Opin Microbiol 13: 589–594.
Zhu W, Lomsadze A, Borodovsky M . (2010). Ab initio gene identification in metagenomic sequences. Nucleic Acids Res 38: e132.
Acknowledgements
Research reported in this publication was supported in part by the NIH Director’s New Innovator Award (http://commonfund.nih.gov/newinnovator/), the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK: http://www.niddk.nih.gov/) and the National Institute of General Medical Sciences (NIGMS: http://www.nigms.nih.gov/) of the National Institutes of Health under award numbers DP2DK098089 and R01GM099538 to GD. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. MKG is supported by a Mr and Mrs Spencer T. Olin Fellowship at Washington University. KJF received support from the NIGMS Cell and Molecular Biology Training Grant (GM 007067) and the NHGRI Genome Analysis Training Program (T32 HG000045). MKG and KJF are NSF graduate research fellows (award number DGE-11143954).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Author Contributions
MKG built and benchmarked Resfams profile HMM database, annotated functional selections and microbial genomes, performed ecological analysis and wrote the manuscript, with assistance from KJF and GD.
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Gibson, M., Forsberg, K. & Dantas, G. Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology. ISME J 9, 207–216 (2015). https://doi.org/10.1038/ismej.2014.106
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2014.106
This article is cited by
-
Longitudinal dynamics of farmer and livestock nasal and faecal microbiomes and resistomes
Nature Microbiology (2024)
-
Duplicated antibiotic resistance genes reveal ongoing selection and horizontal gene transfer in bacteria
Nature Communications (2024)
-
Collembolans maintain a core microbiome responding to diverse soil ecosystems
Soil Ecology Letters (2024)
-
Latent antibiotic resistance genes are abundant, diverse, and mobile in human, animal, and environmental microbiomes
Microbiome (2023)
-
Population-level impacts of antibiotic usage on the human gut microbiome
Nature Communications (2023)
