
Abstract
Cyanobacterial harmful algal blooms (cyanoHABs) appear to be increasing in frequency on a global scale. The Cyanobacteria in blooms can produce toxic secondary metabolites that make freshwater dangerous for drinking and recreation. To characterize microbial activities in a cyanoHAB, transcripts from a eutrophic freshwater reservoir in Singapore were sequenced for six samples collected over one day-night period. Transcripts from the Cyanobacterium Microcystis dominated all samples and were accompanied by at least 533 genera primarily from the Cyanobacteria, Proteobacteria, Bacteroidetes and Actinobacteria. Within the Microcystis population, abundant transcripts were from genes for buoyancy, photosynthesis and synthesis of the toxin microviridin, suggesting that these are necessary for competitive dominance in the Reservoir. During the day, Microcystis transcripts were enriched in photosynthesis and energy metabolism while at night enriched pathways included DNA replication and repair and toxin biosynthesis. Microcystis was the dominant source of transcripts from polyketide and non-ribosomal peptide synthase (PKS and NRPS, respectively) gene clusters. Unexpectedly, expression of all PKS/NRPS gene clusters, including for the toxins microcystin and aeruginosin, occurred throughout the day-night cycle. The most highly expressed PKS/NRPS gene cluster from Microcystis is not associated with any known product. The four most abundant phyla in the reservoir were enriched in different functions, including photosynthesis (Cyanobacteria), breakdown of complex organic molecules (Proteobacteria), glycan metabolism (Bacteroidetes) and breakdown of plant carbohydrates, such as cellobiose (Actinobacteria). These results provide the first estimate of secondary metabolite gene expression, functional partitioning and functional interplay in a freshwater cyanoHAB.
Similar content being viewed by others


Introduction
The frequency of cyanobacterial harmful algal blooms (cyanoHABs) appears to be increasing on a global scale and is linked to nutrient pollution and changing regional climates (Sellner et al., 2003; Paerl et al., 2011). Anoxic dead zones, toxin production and foul odors that can require years to abate make CyanoHABs unsafe and undesirable (Paerl et al., 2011; Pan et al., 2011). Cyanobacterial blooms usually contain one prominent bloom-forming primary producer and a diverse mix of other photosynthetic and non-photosynthetic microorganisms that contribute to nutrient and energy cycling (Litchman et al., 2010). The cyanoHAB agent in the Kranji Reservoir, Singapore is Microcystis aeruginosa. This reservoir is a tropical, well-mixed, shallow eutrophic impoundment that experiences persistent cyanoHABs with detectable levels of microcystin toxin (Te and Gin, 2011). In the present work, the goal is to characterize the dynamics and variability of gene expression over a day/night cycle in the Kranji Reservoir for M. aeruginosa and other major taxa to gain insight into toxin synthesis and partitioning of functions for nutrients and energy cycling that affect water quality.
Functional and taxonomic annotation of expressed genes (mRNA) recovered from complex microbial communities (that is, metatranscriptomics) has enabled breakthrough insights into resource utilization and niche partitioning in microbial ecosystems (for example, Gifford et al., 2013; Ottesen et al., 2013; Vila-Costa et al., 2013). To date, comparatively few metatranscriptomic studies have been carried out in freshwater environments, and community gene expression in a freshwater cyanoHAB remains undescribed. Expression of toxin genes within cyanoHABs is of particular interest because of their effect on health. Cyanotoxins such as microcystin, anatoxin-a, aeruginosin, cylindrospermopsin and microviridin have been linked to symptoms from skin irritation to liver failure and death (Carmichael, 1992; Pouria et al., 1998; Carmichael, 2001; Ehrenreich et al., 2005; Stewart et al., 2006; Rounge et al., 2009; Dittmann et al., 2013). Although toxin production in HAB-forming cyanobacteria has been studied in detail using cultured isolates, little is known about the ecological role of these toxins, and the expression of toxin genes within blooms has not been measured. Through analysis of gene expression in the bloom community, it can be determined which toxin synthesis pathways are highly expressed and whether expression is variable as may be expected in response to transient environmental cues or consistent as may be expected for functions central to the physiology of the organism.
In this study, bacterioplankton from Kranji Reservoir were sampled over a 22-h period, and transcripts were sequenced. The distributions of expressed genes from different taxonomic groups in the bloom community were annotated based on function and analyzed to identify resource partitioning and functional interplay. In addition, with regards to the Microcystis population four question were addressed: (1) Which transcripts are most abundant? (2) Which transcripts change from day to night? (3) Are there differences in expression of genes from the core and flexible genome in Microcystis? and (4) what toxin-related biosynthetic pathways are detected, and how do their expression change over time? This is the first study to examine gene expression in a complex bacterioplankton community dominated by Microcystis over a diurnal cycle.
Materials and methods
Six grab samples were collected from Kranji Reservoir at 10-20 cm beneath a visible algal surface scum on 14 and 15 January 2010 at 1700, 2000, 0600, 0800, 1300 and 1500 hours followed by filtration onto a 0.22-μm Sterivex membrane and preservation in RNAlater (Invitrogen, Carlsbad, CA, USA; Wang, 2011). Environmental metadata for temperature, nutrient concentrations and dissolved microcystin toxin concentrations were measured for each sample. Detailed methods for sample processing and analysis are provided as an online supplement.
Briefly, RNA was extracted from filters and prepared for sequencing using a series of kits according to the manufacturers’ protocols. The Trizol reagent (Invitrogen) was used for RNA extraction, followed by depletion of tRNA, rRNA and eukaryotic mRNA using Ambion kits MegaClear, MicrobeEnrich and MicrobeExpress, respectively, followed by treatment with mRNA-only (Epicentre, Madison, WI, USA). RNA was amplified using MessageAmpII (Ambion, Austin, TX, USA), followed by double-stranded cDNA synthesis (Superscript, Invitrogen) and multiplexing for sequencing in a single lane of an Illumina GAII using Illumina (San Diego, CA, USA) adaptors ligated to custom barcodes. Sequence data have been submitted to the GenBank databases under accession PRJNA238448. Quantitative PCR (qPCR) was used to quantify cDNA transcripts using published primers and conditions (Nonneman and Zimba, 2002; Furukawa et al., 2006; Shao et al., 2009). Ribosomal RNA transcripts were identified by blastn (<1e-20, at >60% sequence identity and >50 bp alignment) to the SILVA database and classified in MG-RAST. Blastx against NR (bit-score>40) was used to taxonomically and functionally classify mRNA in MEGAN using the KEGG hierarchy. Expression of Microcystis genes was measured by recruiting reads to a M. aeruginosa pan-genome (>90 bp alignment; 90% identity) constructed from two publicly available genomes followed by calculation of RPKM (reads per kilobase per million mapped reds) in CLC Genomics Workbench to generate transcript profiles. Sample transcript profiles were log2 transformed, normalized and compared by Principal Component Analysis and Hierarchical Clustering in Multiexperiment Viewer (Saeed et al., 2006). Differentially expressed genes were identified through the Student’s t-test and with a multiple comparison test Significance Analysis of Microarrays (SAM) implemented in Multiexperiment Viewer. The differential distribution of KEGG pathways in Microcystis among different samples and among the four most abundant bacterial phyla was determined using a non-parametric Gene Set Enrichment Analysis (GSEA). Evidence for community polyketide and non-ribosomal peptide synthase (PKS and NRPS, respectively) expression was obtained by identifying ketosynthase (KS) and condensation (C) domains with the online tool NaPDoS (Ziemert et al., 2012).
Results
Sampling and sequencing
Six metatranscriptomes generated from the Kranji Reservoir contained 0.49 Gbp resulting in between 711 948 and 944 045 reads per sample. In all, 7–31% of these sequences represented rRNA as identified by blastn to the SILVA SSU database. Out of the remaining reads, 48–75% were assumed to be mRNA due to their match with proteins in the non-redundant protein database (Supplementary Table S1 and Supplementary Figure S1) and are hereafter referred to as ‘annotated mRNA’. The main environmental variables among collected metadata were light and temperature while nutrients and microcystin toxin did not vary significantly throughout the study period (Supplementary Figure S2).
Community structure of active bacterioplankton
The phylum Cyanobacteria accounted for the majority of all annotated mRNA and SSU rRNA reads (69% and 76%, respectively) followed by Proteobacteria (4% and 5%), Bacteroidetes (2% and 1%) and Actinobacteria (2% and 0.4%, Supplementary Figure S1). Taxonomic assignment of recovered SSU rRNA sequences revealed 533 co-existing bacterial genera in addition to microbial eukaryotes and grazers (for example, Daphnia, Rotifers) where Microcystis transcripts accounted for 63% of SSU rRNA and 39% of annotated mRNA (Supplementary Database S1 and S2). Additional observed genera each represented <0.5% of annotated mRNA transcripts, with the exception of the cyanobacterial genus Nostoc (1.2%).
Environmental expression of a M. aeruginosa pan-genome
M. aeruginosa transcripts from the six metatranscriptomes were identified by comparison to a Microcystis pan-genome constructed from strains PCC7806 and NIES-843 containing 8132 genes corresponding to core (in both strains) or flexible (in one strain) genes (Figure 1a). RPKM values for the Microcystis genes recA, psbA, prx, mcyA and mcyB were significantly correlated (Pearson’s R>0.87, P<0.01) to qPCR values that were normalized to the stably expressed housekeeping gene grpE (Shao et al., 2009) (Supplementary Table S2). In contrast, RPKM values were not well correlated with qPCR quantification of the same targets per ng of total cDNA consistent with the nature of RPKMs as a measure of relative expression within a genome rather than the absolute magnitude. Core genes are more highly represented and abundant among transcripts than flexible genes (Figure 1). All but a single core hypothetical gene is represented in the transcriptomes, while 93% of the 2902 flexible genes from M. aeruginosa NIES-843 and 76% of the 1868 flexible genes from M. aeruginosa PCC7806 were detected in at least one of the samples (Figure 1b).

Recruitment of transcripts from the Kranji Reservoir to a Microcystis aeruginosa pan-genome based on strains NIES-843 and PCC7806. (a) Venn diagram of the core (shared) and flexible (strain specific) genes that define the pan-genome. (b) Pie charts indicate the prevalence of transcripts from core or flexible genomes among the six metatranscriptomes. (c) Box plots present the median and percentile distribution in increments of 10% for M. aeruginosa transcript abundance (RPKM) for the six samples. Sample names Krb1-6 correspond with the collection time shown below the name.
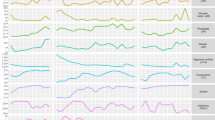
The most highly represented transcripts in the Kranji Reservoir Microcystis population stem from genes encoding photosynthesis, genetic information processing, gas vesicle synthesis, stress response, DNA repair, microviridin production and hypotheticals (Figure 2). GvpA (gas vesicle protein A) is the most highly represented transcript at all times except at 0800 hours when PsbA (photosystem II P680 reaction center D protein) transcripts are most abundant (Figure 2). Most highly expressed hypothetical genes are located next to gas vesicle (gvp) genes, photosynthetic pigment genes or mobile genetic elements (Figure 2) and may have related functions.

Highly represented transcripts from the M. aeruginosa pan-genome listed in descending order of average RPKM value. Heat map scale is the log2 of RPKM values. The 25 most abundant transcripts from each of the six samples correspond to 53 different genes. Gene numbers are given followed by a letter to indicate membership in the core or flexible genome (C=core, N=flexible NIES-843P=flexible PCC7806) and functions are given for PS=Photosynthesis, GV=Gas Vesicles, MGE=Mobile Genetic Element, GIP=Genetic Information Processing, aaM=amino acid metabolism and Chap=Chaperones. Hypothetical genes annotated based on gene neighborhoods or published studies (that is, microviridin, Ziemert et al., 2010) are marked with asterisk (*).
Diel changes in Microcystis transcript abundance
Principal Component Analysis of transcript abundance (RPKM) from the Microcystis pan-genome revealed a partitioning of samples into two major groups that coincided with collection during the daytime ‘day’ (0800, 1300 and 1500 hours) and early evening/night ‘night’ (1700, 2000, 0600 hours) (Figure 3). Hierarchical Clustering supported this partitioning (data not shown). Screening by SAM analysis and the Student’s t-test revealed 77 genes with significantly higher transcript abundance in day samples, including genes involved in energy metabolism, photosynthesis, oxidative phosphorylation, genetic information and processing, environmental information processing and metabolism of carbohydrates, amino acids (aa), nucleotides, cofactors and vitamins, amino sugars, nucleotide sugars and purines (SAM median false detection rate (FDR)=0.012 and t-test P<0.01; Supplementary Figure S3). Genes enriched at night participated in genetic information processing, metabolism of cofactors and vitamins, DNA replication and repair, mismatch repair and microcystin production (mcyA and mcyD), although differential expression of these single genes did not reach statistical significance after correction for multiple hypotheses or by SAM analysis (Supplementary Figure S4).

Principal component analysis of M. aeruginosa transcriptome profiles. The first two principal components shown account for 48% of the total variance of the data.
To further explore changes in Microcystis activity across the samples, genes were sorted into metabolic pathways based on the KEGG hierarchy and secondary metabolite gene clusters. Non-parametric GSEA revealed that KEGG pathways for photosynthesis and protein biosynthesis were upregulated during the day while multiple pathways for DNA synthesis and repair along with nitrogen metabolism, butanoate metabolism, aminoacyl-tRNA biosynthesis, porphyrin and chlorophyll metabolism and synthesis of the toxins microcystin and aeruginosin were enriched at night (P<0.001; FDR<0.25; Table 1).
Functional diversity and partitioning in the bacterioplankton community
Although Microcystis genes are dominant (45-63%) among annotated bacterial transcripts, the remaining proportion of bacterial transcripts originated from a diverse assemblage where an additional 29-34% had taxonomic assignments to four phyla: Cyanobacteria (excluding Microcystis), Proteobacteria, Actinobacteria, and Bacteroidetes (Supplementary Figure S1C). Functional gene expression based on the abundance of reads that recruit to KEGG categories was compared among the four majority phyla. Overall, 34% of the reads from the top four phyla could be assigned to a KEGG pathway. At the highest level of the KEGG function hierarchy, metabolism is most highly represented (54–71%), followed by genetic information processing (14–34%), environmental information processing (6–13%) and cellular processes (2–5%). GSEA identified 26 KEGG pathways significantly enriched in the Proteobacteria relative to the other top phyla (P<0.05, FDR<0.10), including motility/chemotaxis, secretion and metabolism of carbohydrates and amino acids and breakdown of several types of xenobiotics (Supplementary Table S3). Cyanobacteria are enriched in four KEGG pathways related to photosynthesis and vitamin metabolism. Bacteroidetes are enriched in pathways for glycosidases and glycan biosynthesis as well as degradation pathways shared with lysosomes. No enriched pathways were observed for Actinobacteria likely due to low statistical power from low sequence coverage.
Diversity and function of transporters and two-component systems
Expression of transporters and two-component systems within the major cyanoHAB taxa provides insight into their sensing and utilization of environmental resources and respective ecological niches. In the Kranji Reservoir, there is evidence for expression of 82 different ABC transporters and 94 different two-component systems the most abundant of which include the sensing and uptake of nutrients (all major Phyla), chemotaxis and cell motility through flagellar assembly (Proteobacteria), mechanisms for cell-cell interaction, including pilus formation (Cyanobacteria and Proteobacteria) and secretion systems (Proteobacteria) and efflux of unknown chemicals (all major Phyla) (Figures 4 and 5, Supplementary Figures S5-S7, Supplementary Tables S4, S6 and S7).

ABC transporters. Bar graph showing the number of reads matching ABC transporters in the top four phyla on the x axis. The y axis contains a description of the typical transported substrates followed by the names of the genes that make up the transporter based on KEGG annotation. See Supplementary Table S6 for KEGG identification numbers. Only transporters with at least 20 reads are shown. Black lines delineate the KEGG transporter categories: (1) saccharides and polyols, (2) phosphate and amino acids, (3) peptide and nickel, (4) mineral and organic ions, (5) metallic cations, iron-siderophore and vitamin B12 transport, (6) lipoprotein releasing, (7) lipooligosaccharide, (8) antibiotics, (9) heme, (10) cell division and (11) eukaryotic types.

Two-component systems. The number of reads matching two-component systems in the top four phyla are shown on the x axis. The y axis contains a description of the function followed by the gene names for the system based on KEGG annotation. The top five most common systems for each Phylum are shown.
Among the major phyla, high and variable proportions of transcripts for sensing and uptake of nitrogen and phosphorous from both inorganic and organic sources suggests resource partitioning and active recycling of nutrients within the bloom. In Cyanobacteria, the phosphate uptake system PstABCS is the most abundantly expressed ABC transporter. In contrast, uptake of nitrogen-rich peptides and amino acids are the top functions of ABC transport transcripts from Proteobacteria, Bacteroidetes and Actinobacteria (Figure 4), and sensing and responding to nitrogen availability is the top function among two-component system transcripts in Proteobacteria (GlnL/GlnG) (Figure 5). ABC transport of urea appears to be uniquely associated with Cyanobacteria, while utilization of organic phosphorous is evidenced by expression of the phosphonate transporter (Phn) in Proteobacteria and, to a much lesser extent, in Cyanobacteria, suggesting a likely role for heterotrophic Proteobacteria in recycling reduced organic phosphorous.
The intensity of nutrient limitation in the bloom may vary as a function of time of day. Samples collected at 1300 and 1500 hours revealed elevated expression of nutrient transporters in the Microcystis population relative to samples collected during the evening/night and morning (Supplementary Figure S8), pointing to the depletion of intracellular concentrations from sustained photosynthesis. At 1300 hours, maximal expression is evident for transporters of urea and ammonium as well as GlnB that regulates ammonia assimilation (Supplementary Figure S8). At both 1300 and 1500 hours, expression is elevated for the phosphate ABC transporter and alkaline phosphatase, which is responsible for cleaving phosphate groups off organic molecules.
Uptake of inorganic carbon is dominated by Cyanobacteria, corresponding to all annotated ABC transporter transcripts for bicarbonate. Uptake of organic carbon reveals potential for niche overlap and specialization. All phyla express transporters for uptake of peptides and branched chain amino acids. Proteobacteria are responsible for the majority of sorbitol and mannitol ABC transporters, and transcripts for the multi-component PhosphoTransferase System for neutral sugars were enriched in the Proteobacteria by GSEA (Supplementary Table S3 and S4). ABC transporters for cellobiose, xylose and alpha glucosides are almost exclusively annotated as actinobacterial, while in Bacteroidetes the peptide/nickel ABC transporter is represented by the most transcripts, far exceeding transcripts for uptake of other carbon substrates.
Analysis of secondary metabolites
To gain insight into the balance between synthesis and biodegradation of metabolites that negatively impact water quality, the metatranscriptomes were screened for secondary metabolite biosynthesis pathways and for biodegradation pathways of the toxin microcystin. The presence of KS and C domains, which were used as a proxy for identifying expressed NRPS and PKS pathways, were predominantly cyanobacterial (Figures 6a and b). A total of 73 different KS domains (1629 reads) and 82 different C domains (924 reads) from PKS and NRPS gene clusters, respectively, were identified that appear to originate from Cyanobacteria based on blastx. Of those cyanobacterial KS domains, 34 share >90% aa sequence identity with recruited reads including 25 KS domains from Microcystis strains, which strongly suggest that the corresponding PKS/NRPS clusters are expressed in the Reservoir. In contrast, only 8 of the 129 non-cyanobacterial KS domains have >90% sequence identity with recruited reads, and none match PKS or NRPS pathways with known products.

Taxonomic affiliation of Ketosynthase (KS) and condensation (C) domain transcripts matching the NaPDoS database of representative pathways linked to a known PKS or NRPS product.
The presence of C and KS domains and RPKM data indicates that all of the seven PKS and NRPS gene clusters in the Microcystis pan-genome were expressed in the reservoir (Table 2). Three of the seven clusters are known to produce the compounds microcystin (cluster 1), cyanopeptolin (cluster 8) and aeruginosin (cluster 9) and are in both reference genomes. Cluster 4 from NIES-843 has no known product, yet appears to be the most highly transcribed PKS/NRPS cluster in Kranji Reservoir in all samples (Table 2). Of the PKS/NRPS clusters in the two reference genomes, mean transcript RPKM from aeruginosin and microcystin clusters appear to vary significantly between the day and night (P=0.028 and P=0.017, respectively) with higher levels at night (Figures 7a and b) in agreement with GSEA analysis (Table 1) and qPCR (Supplementary Table S2). Eight additional PKS/NRPS gene clusters from other M. aeruginosa strains, as well as Planktothrix, Acaryochloris and Cyanothece, were observed in the Kranji Reservoir transcripts (Table 2) and shared >90% (aa) sequence identity to cyanobacterial domains in non-redundant protein database (Table 2).

Expression of secondary metabolite gene clusters, (a) clusters with a known product, (b) clusters that are not linked to a specific product. The error bars indicate 2 s.ds. from the mean. The P-values are for the test to reject null hypothesis that there is no difference between the mean night and day transcript abundance.
Ribosomally produced secondary metabolites were also observed in Kranji Reservoir, including the toxin microviridin and the odor-causing compound geosmin. The precursor peptide for microviridin was among the most highly expressed genes of Microcystis (Figure 2), whereas the precursor peptide of a second ribosomally synthesized toxin microcyclamide (Ziemert et al., 2008) was undetected. Five transcripts were recovered that matched the Nostoc geoA gene for geosmin synthase while none were recovered for genes for the synthesis of the odor compound MIB-2.
Biodegradation pathways for most secondary metabolites are currently unknown. Betaproteobacterial genera recently implicated in microcystin biodegradation (Mou et al., 2013) were observed among sequenced transcripts (Supplementary Database 1). No significant sequence similarity was observed for the mrlAB-mediated microcystin biodegradation pathway (Bourne et al., 2001; Mou et al., 2013).
Discussion
During the collection period, a green scum was apparent in the Kranji Reservoir, which has previously been identified as dominated by Microcystis cells. This study confirms the dominance and shows that approximately half of the bacterial transcripts are from Microcystis, exceeding transcripts from other cyanobacterial populations by at least 20-fold and non-cyanobacterial genera by at least 100-fold and enabling inference of trends at the population level for Microcystis and at the Phylum level for other taxa.
Genomic diversity of M. aeruginosa in Kranji Reservoir
Transcripts from a large majority of genes in the M. aeruginosa core and flexible genome were identified in the Kranji Reservoir. M. aeruginosa is a highly characterized cyanoHAB agent with high phenotypic and genetic diversity (Briand et al., 2008; Yoshida et al., 2008; van Gremberghe et al., 2011; Steffen et al., 2012; Humbert et al., 2013), including a variable distribution of toxin genes among different strains (Frangeul et al., 2008). Observation of flexible genes in Kranji Reservoir, Singapore from two M. aeruginosa strains isolated over broad geographic and temporal scales (that is, NIES-843 isolated in Japan in 1997 (Tanabe et al., 2007) and PCC7806 from the Netherlands in 1972 (Kappers, 1984)) is consistent with other studies documenting the global dispersal and recombination of genomes within this species (Tanabe et al., 2007; van Gremberghe et al., 2011; Humbert et al., 2013). Recently, 10 additional M. aeruginosa genomes were sequenced, refining the M. aeruginosa core genome from the 3362 genes considered in this study to a set of 2462 genes and extending the flexible genome by more than a factor of two (Humbert et al., 2013). Compared with this extended pan-genome, the majority of Kranji Reservoir Microcystis transcripts shared highest homology with strain NIES-843 included in the two-strain pan-genome (Supplementary Table S5). Interestingly, Lake Erie also showed higher recruitment of metagenome reads to strain NIES-843 over PCC7806 (Steffen et al., 2012). Flexible genes had lower transcript abundance than core genes, likely reflecting the existence of additional Microcystis genomes in the Kranji Reservoir with a common core and novel flexible genes. It is likely that further sequencing will reveal additional diversity, and work is currently in progress to characterize the genomic diversity of Microcystis in the Kranji Reservoir.
Highly expressed genes in M. aeruginosa and partitioning of functions between day and night
M. aeruginosa gene expression has been previously characterized under controlled experimental conditions (Straub et al., 2011; Harke and Gobler, 2013), and comparison with environmental populations (this study) highlights both similarities and differences potentially associated with environmental complexity. Strain PCC7806 examined by microarray exhibited common trends with the present study where the most highly expressed genes were for energy metabolism (photosynthesis) and were enriched during the day together with transcripts for carbon fixation, stress response and ribosome production (Straub et al., 2011). At night, the microarray study of PCC7806 revealed enrichment of the tricarboxylic acid-branched pathway, glycogen degradation, the oxidative pentose phosphate pathway and ammonium uptake (Straub et al., 2011). With the exception of ammonium uptake (Supplementary Figure S8), these observations are consistent with qualitative trends from individual genes in the present data set that do not reach statistical significance. In addition, in the reservoir population, there was significant nighttime enrichment of multiple additional pathways (Table 1) including DNA repair. Elevated transcription of DNA repair genes at night is also evident in marine Prochlorococcus (ProPortal Search, May 2013, (Kelly et al., 2012)). Low-light periods provide a reprieve from stresses associated with ultraviolet exposure and oxygenic photosynthesis. Nighttime repair of DNA damage incurred during the day may improve the overall fitness of environmental populations. In the reservoir, Microcystis transcripts for gas vesicle proteins (GvpA and GvpC) were expressed at a similar level as energy metabolism (photosynthesis) genes, indicating the dual importance of competition for surface colonization and light-harvesting for the success of bloom-forming Microcystis populations. Other genes highly expressed within the reservoir Microcystis population include the pilin subunit pilA and the two-component system regulator for Type IV pili that mediate cell-cell contact, twitching motility and have been implicated in lateral gene transfer in environmental populations (Figure 5) (Nakasugi and Neilan, 2005). Transcripts for stress-responsive genes for chaperon HspA and DnaK and oxidative stress mitigating peroxiredoxin were also highly represented in the reservoir population and may help the cells acclimate to dynamic environmental conditions.
Although previous experimental work has indicated that Kranji reservoir is phosphorous limited (Gin et al., 2011), the extent and nature of nutrient limitation may vary on a daily timescale. A recent study of M. aeruginosa clone LE-3 demonstrates increased expression of nitrogen or phosphorous uptake systems during experimentally induced limitation of the respective nutrients in lake water (Harke and Gobler, 2013). In the reservoir’s Microcystis population, the highest transcript abundance for nitrogen uptake (urea and ammonium) and phosphate uptake were observed at 1300 and 15 hours and may reflect nutrient limitation induced by elevated growth rates and competition for resources during high light and temperatures midday (Supplementary Figure S2).
Secondary metabolites and toxins
Secondary metabolite compounds are small molecules that can act as toxins, antibiotics, anti-cancer agents, surfactants, metal chelators and contribute to off-flavors and odors in freshwater (Di Lorenzo et al., 2004; Oliynyk et al., 2007; Fenical et al., 2009; Haft et al., 2010). Although the ecological function of many of these compounds remains unclear, recent studies have shed light on some potential roles within the context of a bloom community. The most highly expressed secondary metabolite gene from M. aeruginosa in Kranji Reservoir corresponds to the ribosomally produced serine-protease inhibitor microviridin (Ziemert et al., 2010), with maximal expression at 1700 and 2000 hours (Figure 2). A microviridin variant (type J) has been shown to be toxic to freshwater Daphnia; a grazer of Microcystis (Rohrlack et al., 2003, 2004), and production of toxins such as microviridin may represent an antipredatory strategy in bloom-forming Cyanobacteria. Other cyanotoxins such as microcystin can also be toxic to Daphnia (Rohrlack et al., 2001) although grazers adapted to high concentrations demonstrate resistance (Chislock et al., 2013). Microcystin may also function to enhance the aggregation of Microcystis cells as often occurs during bloom formation (Gan et al., 2012) and to provide a fitness advantage to Microcystis under oxidative stress (Zilliges et al., 2011). Despite transcript evidence that the microcystin biosynthetic operon was expressed throughout the day-night cycle, concentrations of dissolved microcystin toxin were measured near the limit of detection in this study (Supplementary Figure S2), potentially reflecting low synthesis activity, biodegradation (Bourne et al., 2001; Mou et al., 2013) and/or conversion of microcystin to insoluble conjugates that are not detected by the enzyme-linked immunosorbent assay-based method used in this study (Meissner et al., 2013).
The most highly expressed PKS/NRPS gene cluster was cluster 4 (Figure 6), part of the flexible genome of strain NIES-843. Although the product of cluster 4 has not been identified, bioinformatics analysis indicates that it is likely a hybrid polyketide/fatty acid product (Shulse and Allen, 2011). Interestingly, cluster 4 is related to a cluster previously identified in Anabaena sp. PCC7120 (Johnson et al., 2006) that has a role in heterocyst formation (Mella-Herrera et al., 2011), which is not a documented trait in Microcystis species (Watanabe, 1996).
The abundance of toxin-producing Microcystis in the reservoir (Te and Gin, 2011) represents a rare opportunity to examine temporal dynamics of expression of PKS/NRPS genes, which typically remains undetected without specific primers (Foerstner et al., 2008; Kaluzhnaya et al., 2012). Transcripts for all PKS/NRPS genes from Microcystis strains NIES-843 and PCC7806 were observed in the Kranji Reservoir throughout the day/night cycle, suggesting that they are continually expressed and thus are likely central to the physiology of the cells rather than produced in response to transient environmental cues. Enrichment of transcripts from PKS/NRPS gene clusters for microcystin and aeruginosin was apparent at night after normalization of transcript abundance to a housekeeping gene (qPCR) or to total gene expression (RPKM) (Table 1, Figure 6). Previous studies report higher transcript abundance of toxin genes during the day (Straub et al., 2011), which agrees with our findings for all qPCR targets quantified per ng cDNA in the samples (including microcystin biosynthesis genes mcyA and mcyB). However, normalization removes trends due to changes in baseline cellular activity such as growth and suggests that cells allocate a greater portion of their energy budget to toxin synthesis at night. This pattern was also observed for the toxin aeruginosin. We consider several hypotheses to explain this variation. First, the null hypothesis that these differences are due to chance alone was rejected (P<0.05) for GSEA; however, the low statistical power afforded by six samples precludes robust rejection as evidenced by a FDR of 18% (Table 1). Although the present study represents a level of replication on par with, or exceeding, recent metatranscriptome sequencing efforts (Ottesen et al., 2013; Vila-Costa et al., 2013), we identify the need to expand replication in future works. Alternately, enrichment of toxin gene transcripts at night may be due to heterogeneity in the distribution of Microcystis cells containing toxin genes, resulting in an increased proportion of toxigenic cells in samples collected at night. Finally, if enrichment is due to increases in toxin gene expression per cell at night, this may be an adaptation to ecological factors, such as enhanced nighttime grazing pressure. Indeed Daphnia grazers have been documented to carry out a diel vertical migration, rising to surface waters at dusk (Haupt et al., 2010). Elevated expression of genes for the antipredatory toxin microviridin in the 1700- and 2000- hour samples lends additional support to this hypothesis.
Partitioning of functions between phyla
The functions identified among metatranscripts show a phylum level partitioning lending to an apparent interplay within the HAB community. Based on ABC transporter profiles, Cyanobacteria have a major role in the uptake of phosphate, urea, neutral and branched chain amino acids and bicarbonate, likely to satisfy growth requirements during photosynthesis. At the same time, cells within the dominant cyanobacterial genus Microcystis produce gas vesicles that allow colonization of the upper water column and surface and secondary metabolites, including toxins with antipredatory activity such as microviridin and microcystin. Efficient competition for dissolved resources coupled with competition for space and defense against grazing likely contribute to the environmental fitness and competitive dominance of M. aeruginosa within the HAB community.
The profile of transporter substrates in the co-existing Proteobacteria largely overlap with both Cyanobacteria and Actinobacteria, with highest transcription levels observed for uptake of branched chain amino acids and including transcripts for uptake of organic phosphorous (phosphonate) compounds. Enrichment of proteobacterial genes for motility and chemotaxis suggest enhanced access to ephemeral nutrient patches, which may evolve during cell lysis and bloom decay, and expression of pili and contact-based secretion systems (Type III, Type VI) indicate cell-cell contact and molecular exchange with other members of the bloom community. Actinobacterial ABC transporter profiles reveal a diverse array of substrates and suggest specialization for the photosynthetically derived carbohydrates (cellobiose, xylose, maltose and alpha glucosides) that are not substrates for highly expressed transporters in other phyla. Bacteroidetes ABC transporter profiles indicate peptides are a major source of organic carbon while transcripts were enriched in pathways for hydrolysis and formation of glycosidic bonds, suggesting that they have a role in metabolism of complex sugars and dissolved proteins within the bloom community. Patterns in resource acquisition, utilization and secondary metabolite production are implicated as factors that support a bloom community where primary producers (Cyanobacteria) take up nutrients, inorganic carbon and monomers for primary production while producing a suite of small molecules with antipredatory activity while consumers (heterotrophic members of the Proteobacteria, Actinobacteria and Bacteroidetes) utilize photosynthate released from primary producers by exudation or cell lysis, thereby recycling the fixed energy and nutrients.
Conclusions and future directions
Water quality is an emergent property of a complex ecosystem composed of multiple interacting microbial populations. To understand the persistence of blooms and production of secondary metabolites that mediate water quality, the dynamics within the community must be considered. Several important limitations come with studying gene expression in a complex microbial community. We have used sequenced transcripts as a proxy for microbial community gene expression based on the assumption that biases introduced by collection, handling and sequencing are the same across samples and thus do not significantly impact conclusions based on comparison between samples. However, biases in database annotation are a serious concern for interpretation of sequence diversity. At the present time, genomes from freshwater microbes are under-represented in databases, limiting our ability to assign function and taxonomic affiliation to transcripts. Efforts are currently underway to sequence genomes from cultured isolates from the major phylogenetic groups of Kranji Reservoir and help address questions regarding the population genomics of Microcystis and the genomic diversity of co-occurring taxa. By integrating knowledge from isolates and model organisms with expression data from a complex ecosystem, we can gain a more realistic picture of the intricate microbial world.
Accession codes
References
Blom JF, Bister B, Bischoff D, Nicholson G, Jung G, Süssmuth RD et al (2003). Oscillapeptin J, a new grazer toxin of the freshwater cyanobacterium Planktothrix rubescens. J Nat Prod 66: 431–434.
Bourne DG, Riddles P, Jones GJ, Smith W, Blakeley RL . (2001). Characterisation of a gene cluster involved in bacterial degradation of the cyanobacterial toxin microcystin LR. Environ Toxicol 16: 523–534.
Briand E, Escoffier N, Straub C, Sabart M, Quiblier C, Humbert J-F . (2008). Spatiotemporal changes in the genetic diversity of a bloom-forming Microcystis aeruginosa (cyanobacteria) population. ISME J 3: 419–429.
Carmichael WW . (1992). Cyanobacteria secondary metabolites—the cyanotoxins. J Appl Microbiol 72: 445–459.
Carmichael WW . (2001). Health effects of toxin-producing cyanobacteria: “the CyanoHABs”. Hum Ecol Risk Assess 7: 1393–1407.
Chislock MF, Sarnelle O, Jernigan LM, Wilson AE . (2013). Do high concentrations of microcystin prevent Daphnia control of phytoplankton? Water Res 47: 1961–1970.
Christiansen G, Philmus B, Hemscheidt T, Kurmayer R . (2011). Genetic variation of adenylation domains of the anabaenopeptin synthesis operon and evolution of substrate promiscuity. J Bacteriol 193: 3822–3831.
Di Lorenzo M, Poppelaars S, Stork M, Nagasawa M, Tolmasky ME, Crosa JH . (2004). A nonribosomal peptide synthetase with a novel domain organization is essential for siderophore biosynthesis in Vibrio anguillarum. J Bacteriol 186: 7327–7336.
Dittmann E, Fewer DP, Neilan BA . (2013). Cyanobacterial toxins: biosynthetic routes and evolutionary roots. FEMS Microbiol Rev 37: 23–43.
Ehrenreich IM, Waterbury JB, Webb EA . (2005). Distribution and diversity of natural product genes in marine and freshwater cyanobacterial cultures and genomes. Appl Environ Microbiol 71: 7401–7413.
Fenical W, Jensen PR, Palladino MA, Lam KS, Lloyd GK, Potts BC . (2009). Discovery and development of the anticancer agent salinosporamide A (NPI-0052). Bioorg Med Chem 17: 2175–2180.
Foerstner KU, Doerks T, Creevey CJ, Doerks A, Bork P . (2008). A computational screen for type i polyketide synthases in metagenomics shotgun data. PLoS One 3: e3515.
Frangeul L, Quillardet P, Castets A-M, Humbert J-F, Matthijs H, Cortez D et al (2008). Highly plastic genome of Microcystis aeruginosa PCC 7806, a ubiquitous toxic freshwater cyanobacterium. BMC Genomics 9: 274.
Furukawa K, Noda N, Tsuneda S, Saito T, Itayama T, Inamori Y . (2006). Highly sensitive real-time PCR assay for quantification of toxic cyanobacteria based on microcystin synthetase a gene. J Biosci Bioeng 102: 90–96.
Gan N, Xiao Y, Zhu L, Wu Z, Liu J, Hu C et al (2012). The role of microcystins in maintaining colonies of bloom-forming Microcystis spp. Environ Microbiol 14: 730–742.
Gifford SM, Sharma S, Booth M, Moran MA . (2013). Expression patterns reveal niche diversification in a marine microbial assemblage. ISME J 7: 281–298.
Gin K, Ramaswamy U, Gopalakrishnan A . (2011). Comparison of nutrient limitation in freshwater and estuarine reservoirs in tropical urban Singapore. J Environ Eng 137: 913–919.
Haft D, Basu M, Mitchell D . (2010). Expansion of ribosomally produced natural products: a nitrile hydratase- and Nif11-related precursor family. BMC Biol 8: 70.
Harke MJ, Gobler CJ . (2013). Global transcriptional responses of the toxic Cyanobacterium, Microcystis aeruginosa, to nitrogen stress, phosphorus stress, and growth on organic matter. PLoS One 8: e69834.
Haupt F, Stockenreiter M, Reichwaldt ES, Baumgartner M, Lampert W, Boersma M et al (2010) Upward Phosphorus Transport by Daphnia diel Vertical Migration vol. 55. American Society of Limnology and Oceanography: Waco, TX, USA, ETATS-UNIS.
Humbert J-F, Barbe V, Latifi A, Gugger M, Calteau A, Coursin T et al (2013). A tribute to disorder in the genome of the bloom-forming freshwater Cyanobacterium Microcystis aeruginosa. PLoS One 8: e70747.
Jimbo M, Yamaguchi M, Muramoto K, Kamiya H . (2000). Cloning of the Microcystis aeruginosa M228 lectin (MAL) gene. Biochem Biophys Res Commun 273: 499–504.
Johnson MA, Peti W, Herrmann T, Wilson IA, Wüthrich K . (2006). Solution structure of Asl1650, an acyl carrier protein from Anabaena sp. PCC 7120 with a variant phosphopantetheinylation-site sequence. Protein Sci 15: 1030–1041.
Kaluzhnaya OV, Kulakova NV, Itskovich VB . (2012). Diversity of polyketide synthase (PKS) genes in metagenomic community of freshwater sponge Lubomirskia baicalensis. Mol Biol 46: 790–795.
Kappers FI . (1984) PhD Thesis, University of Amsterdam. University of Amsterdam: Amsterdam, The Netherlands.
Kelly L, Huang KH, Ding H, Chisholm SW . (2012). ProPortal: a resource for integrated systems biology of Prochlorococcus and its phage. Nucleic Acids Res 40: D632–D640.
Litchman E, de Tezanos Pinto P, Klausmeier C, Thomas M, Yoshiyama K . (2010). Linking traits to species diversity and community structure in phytoplankton. Hydrobiologia 653: 15–28.
Meissner S, Fastner J, Dittmann E . (2013). Microcystin production revisited: conjugate formation makes a major contribution. Environ Microbiol 15: 1810–1820.
Mella-Herrera RA, Neunuebel MR, Kumar K, Saha SK, Golden JW . (2011). The sigE gene is required for normal expression of heterocyst-specific genes in Anabaena sp. strain PCC 7120. J Bacteriol 193: 1823–1832.
Mou X, Lu X, Jacob J, Sun S, Heath R . (2013). Metagenomic identification of bacterioplankton taxa and pathways involved in microcystin degradation in Lake Erie. PLoS One 8: e61890.
Nakasugi K, Neilan BA . (2005). Identification of Pilus-like structures and genes in Microcystis aeruginosa PCC7806. Appl Environ Microbiol 71: 7621–7625.
Nishizawa A, Arshad AB, Nishizawa T, Asayama M, Fujii K, Nakano T et al (2007). Cloning and characterization of a new hetero-gene cluster of nonribosomal peptide synthetase and polyketide synthase from the cyanobacterium Microcystis aeruginosa K-139. J Gen Appl Microbiol 53: 17–27.
Nishizawa T, Ueda A, Nakano T, Nishizawa A, Miura T, Asayama M et al (2011). Characterization of the locus of genes encoding enzymes producing heptadepsipeptide micropeptin in the unicellular cyanobacterium Microcystis. J Biochem 149: 475–485.
Nonneman D, Zimba PV . (2002). A PCR-based test to assess the potential for microcystin occurence in channel catfish production ponds. J Phycol 38: 230–233.
Oliynyk M, Samborskyy M, Lester JB, Mironenko T, Scott N, Dickens S et al (2007). Complete genome sequence of the erythromycin-producing bacterium Saccharopolyspora erythraea NRRL23338. Nat Biotech 25: 447–453.
Ottesen EA, Young CR, Eppley JM, Ryan JP, Chavez FP, Scholin CA et al (2013). Pattern and synchrony of gene expression among sympatric marine microbial populations. Proc Natl Acad Sci 110: E488–E497.
Paerl HW, Hall NS, Calandrino ES . (2011). Controlling harmful cyanobacterial blooms in a world experiencing anthropogenic and climatic-induced change. Sci Total Environ 409: 1739–1745.
Pan G, Yang B, Wang D, Chen H, Tian B-h Zhang M-l et al (2011). In-lake algal bloom removal and submerged vegetation restoration using modified local soils. Ecol Eng 37: 302–308.
Pouria S, de Andrade A, Barbosa J, Cavalcanti RL, Barreto VTS, Ward CJ et al (1998). Fatal microcystin intoxication in haemodialysis unit in Caruaru, Brazil. Lancet 352: 21–26.
Rohrlack T, Dittmann E, Börner T, Christoffersen K . (2001). Effects of cell-bound microcystins on survival and feeding of Daphnia spp. Appl Environ Microbiol 67: 3523–3529.
Rohrlack T, Christoffersen K, Hansen P, Zhang W, Czarnecki O, Henning M et al (2003). Isolation, characterization, and quantitative analysis of microviridin J, a new microcystis metabolite toxic to Daphnia. J Chem Ecol 29: 1757–1770.
Rohrlack T, Christoffersen K, Kaebernick M, Neilan BA . (2004). Cyanobacterial protease inhibitor microviridin J causes a lethal molting disruption in Daphnia pulicaria. Appl Environ Microbiol 70: 5047–5050.
Rounge T, Rohrlack T, Nederbragt A, Kristensen T, Jakobsen K . (2009). A genome-wide analysis of nonribosomal peptide synthetase gene clusters and their peptides in a Planktothrix rubescens strain. BMC Genomics 10: 396.
Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N et al (2003). TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34: 374–378.
Sellner K, Doucette G, Kirkpatrick G . (2003). Harmful algal blooms: causes, impacts and detection. J Ind Microbiol Biotechnol 30: 383–406.
Shao J, Wu Z, Yu G, Peng X, Li R . (2009). Allelopathic mechanism of pyrogallol to Microcystis aeruginosa PCC7806 (Cyanobacteria): from views of gene expression and antioxidant system. Chemosphere 75: 924–928.
Shulse CN, Allen EE . (2011). Widespread occurrence of secondary lipid biosynthesis potential in microbial lineages. PLoS One 6: e20146.
Steffen MM, Li Z, Effler TC, Hauser LJ, Boyer GL, Wilhelm SW . (2012). Comparative metagenomics of toxic freshwater Cyanobacteria bloom communities on two continents. PLoS One 7: e44002.
Stewart I, Robertson I, Webb P, Schluter P, Shaw G . (2006). Cutaneous hypersensitivity reactions to freshwater cyanobacteria - human volunteer studies. BMC Dermatol 6: 6.
Straub C, Quillardet P, Vergalli J, de Marsac NT, Humbert J-F . (2011). A day in the life of Microcystis aeruginosa strain PCC 7806 as revealed by a transcriptomic analysis. PLoS One 6: e16208.
Swingley WD, Chen M, Cheung PC, Conrad AL, Dejesa LC, Hao J et al (2008). Niche adaptation and genome expansion in the chlorophyll d-producing cyanobacterium Acaryochloris marina. Proc Natl Acad Sci 105: 2005–2010.
Tanabe Y, Kasai F, Watanabe MM . (2007). Multilocus sequence typing (MLST) reveals high genetic diversity and clonal population structure of the toxic cyanobacterium Microcystis aeruginosa. Microbiology 153: 3695–3703.
Te SH, Gin KY-H . (2011). The dynamics of cyanobacteria and microcystin production in a tropical reservoir of Singapore. Harmful Algae 10: 319–329.
Tillett D, Dittmann E, Erhard M, von Dohren H, Borner T, Neilan BA . (2000). Structural organization of microcystin biosynthesis in Microcystis aeruginosa PCC7806: an integrated peptide-polyketide synthetase system. Chem Biol 7: 753–764.
van Gremberghe I, Leliaert F, Mergeay J, Vanormelingen P, Van der Gucht K, Debeer A-E et al (2011). Lack of phylogeographic structure in the freshwater cyanobacterium Microcystis aeruginosa suggests global dispersal. PLoS One 6: e19561.
Vila-Costa M, Sharma S, Moran MA, Casamayor EO . (2013). Diel gene expression profiles of a phosphorus limited mountain lake using metatranscriptomics. Environ Microbiol 15: 1190–1203.
Wang J . (2011). Dynamics of Community Gene Expression in a Freshwater Cyanobacterial Bloom Over a Day-Night Cycle. Massachusetts Institute of Technology: Cambridge, MA, USA.
Watanabe MF, Harada K-i, Carmichael WW, Fujiki H . (1996) Toxic Microcystis. illustrated edn. CRC Press: USA.
Welsh EA, Liberton M, Stöckel J, Loh T, Elvitigala T, Wang C et al (2008). The genome of Cyanothece 51142, a unicellular diazotrophic cyanobacterium important in the marine nitrogen cycle. Proc Natl Acad Sci USA 105: 15094–15099.
Yoshida M, Yoshida T, Satomi M, Takashima Y, Hosoda N, Hiroishi S . (2008). Intra-specific phenotypic and genotypic variation in toxic cyanobacterial Microcystis strains. J Appl Microbiol 105: 407–415.
Ziemert N, Ishida K, Quillardet P, Bouchier C, Hertweck C, de Marsac NT et al (2008). Microcyclamide biosynthesis in two strains of Microcystis aeruginosa: from structure to genes and vice versa. Appl Environ Microbiol 74: 1791–1797.
Ziemert N, Ishida K, Weiz A, Hertweck C, Dittmann E . (2010). Exploiting the natural diversity of microviridin gene clusters for discovery of novel tricyclic depsipeptides. Appl Environ Microbiol 76: 3568–3574.
Ziemert N, Podell S, Penn K, Badger JH, Allen E, Jensen PR . (2012). The natural product domain seeker NaPDoS: a phylogeny based bioinformatic tool to classify secondary metabolite gene diversity. PLoS One 7: e34064.
Zilliges Y, Kehr J-C, Meissner S, Ishida K, Mikkat S, Hagemann M et al (2011). The Cyanobacterial hepatotoxin microcystin binds to proteins and increases the fitness of Microcystis under oxidative stress conditions. PLoS One 6: e17615.
Acknowledgements
This research was funded by the National Research Foundation Singapore through the Singapore MIT Alliance for Research and Technology’s Center for Environmental Sensing and Modeling (CENSAM) research program. The National Science Foundation Postdoctoral Research Fellowship in Biology under Grant No. DBI-1202865 supports KP. We also thank the MIT Center for Environmental Health Sciences (US National Institute of Environmental Health Sciences NIEHS Grant P30-ES002109) for core facility support and the MISTI-Hayashi fund for travel support. We thank Dr Peter Shanahan, MIT, Professor Lloyd Chua at NTU and Dr Hans Eikaas and others at the Public Utilities Board of Singapore for help in coordinating sampling, Professor Libusha Kelly for advice on bioinformatic analysis and members of the laboratory of Professor Karina Gin at the National University of Singapore for helpful discussions regarding harmful algal blooms in the Kranji Reservoir.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Penn, K., Wang, J., Fernando, S. et al. Secondary metabolite gene expression and interplay of bacterial functions in a tropical freshwater cyanobacterial bloom. ISME J 8, 1866–1878 (2014). https://doi.org/10.1038/ismej.2014.27
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2014.27
Keywords
This article is cited by
-
Characterization and seasonal variation in biofilms attached to leaves of submerged plant
World Journal of Microbiology and Biotechnology (2024)
-
The ecological responses of bacterioplankton during a Phaeocystis globosa bloom in Beibu Gulf, China highlighted by integrated metagenomics and metatranscriptomics
Marine Biology (2022)
-
Coral mucus rapidly induces chemokinesis and genome-wide transcriptional shifts toward early pathogenesis in a bacterial coral pathogen
The ISME Journal (2021)
-
Competitive interactions as a mechanism for chemical diversity maintenance in Nodularia spumigena
Scientific Reports (2021)
-
Metabarcoding profiling of microbial diversity associated with trout fish farming
Scientific Reports (2021)
