
Abstract
Ecologists have long studied the temporal dynamics of plant and animal communities with much less attention paid to the temporal dynamics exhibited by microbial communities. As a result, we do not know if overarching temporal trends exist for microbial communities or if changes in microbial communities are generally predictable with time. Using microbial time series assessed via high-throughput sequencing, we conducted a meta-analysis of temporal dynamics in microbial communities, including 76 sites representing air, aquatic, soil, brewery wastewater treatment, human- and plant-associated microbial biomes. We found that temporal variability in both within- and between-community diversity was consistent among microbial communities from similar environments. Community structure changed systematically with time in less than half of the cases, and the highest rates of change were observed within ranges of 1 day to 1 month for all communities examined. Microbial communities exhibited species–time relationships (STRs), which describe the accumulation of new taxa to a community, similar to those observed previously for plant and animal communities, suggesting that STRs are remarkably consistent across a broad range of taxa. These results highlight that a continued integration of microbial ecology into the broader field of ecology will provide new insight into the temporal patterns of microbial and ‘macro’-bial communities alike.
Similar content being viewed by others


Introduction
Understanding how communities are structured in time is a fundamental pursuit in ecology. There is a long history of research on temporal dynamics in animal and plant communities (for example, Preston, 1960; Holling, 1973; McNaughton, 1977; Pimm, 1984; Ives et al., 2003; Ives and Carpenter, 2007), and this research has been integral to the development of a wide variety of concepts in ecology. In particular, research on long-term changes in plant and animal diversity have been instrumental in helping ecologists recognize successional dynamics (for example, Lockwood et al., 1997; Chase, 2003), identify relationships between community stability and biodiversity (Cottingham et al., 2001; White et al., 2006) and predict how communities may respond to disturbances, including longer-term global changes (for example, Fraterrigo and Rusak, 2008; Magurran et al., 2010).
Though microorganisms are ubiquitous, abundant and have critical roles in ecosystems, far less is known about the temporal dynamics exhibited by microbial communities relative to those exhibited by communities of larger organisms. A growing collection of site-specific studies in the microbial ecology literature suggests that microbial communities exhibit a wide range of discernable temporal patterns. For example, patterns of primary succession in an infant gut (Koenig et al., 2011) and on leaf surfaces (Redford and Fierer, 2009) as well as patterns of recurring seasonality in aquatic systems (Fuhrman et al., 2006; Shade et al., 2007; Eiler et al., 2012; Gilbert et al., 2012) have demonstrated that some microbial communities change directionally, according to environmental conditions. By contrast, patterns of stability in wastewater treatment systems suggest that some microbial communities are composed of core members that exhibit minimal temporal variability and rarer taxa that exhibit more pronounced fluctuations in abundance over time (Werner et al., 2011). Further, in a range of systems, including gut microbiota challenged with an antibiotic (Dethlefsen et al., 2008), lake microbial communities after water column mixing (Jones et al., 2008; Shade et al., 2012b) and soil communities of denitrifiers and nitrite oxidizers after exposure to increased temperature (Wertz et al., 2007), some microbial communities have the capacity to recover quickly after disturbance events, either to the pre-disturbance state or to an alternative stable state (Shade et al., 2012a). Overall, these and other time series highlight that microbial communities, like plant and animal communities, are dynamic and exhibit temporal patterns that can reflect underlying biotic and abiotic processes.
Because changes in microbial community composition are often associated with changes in the functional capabilities of those communities (for example, Bell et al., 2005; Fierer et al., 2007; Strickland et al., 2009), understanding microbial temporal patterns can be critical for understanding ecosystem processes. Despite this importance, our ability to generalize microbial community dynamics has been limited by a focus on site- or habitat-specific research. However, the ever-increasing accumulation of 16S rRNA gene sequence data allows for comparison of microbial communities across habitats. Though microbial communities from different habitats clearly differ in composition (Lozupone and Knight, 2007; Nemergut et al., 2011), it is unknown whether there are common patterns in the dynamics or variability of microbial communities through time and across habitats. To date, there has been no concerted analysis of microbial temporal dynamics across biomes, and thus understanding these dynamics and determining their commonalities are key challenges in microbial ecology.
Aquatic systems are perhaps the most frequently studied microbial communities through time, due, in part, to the relatively large number of long-term ecological studies conducted in aquatic systems. As a result, published time series of aquatic microbial communities have yielded key insights into the drivers of microbial dynamics in marine and freshwater systems. For instance, we know that physicochemical changes in the environment often drive shifts in aquatic microbial communities, as evident from the observation that marine, lake and river communities frequently exhibit pronounced seasonality (Hofle et al., 1999; Fuhrman et al., 2006; Shade et al., 2007; Nelson, 2008; Crump et al., 2009; Eiler et al., 2012). Furthermore, there is evidence that phytoplankton and bacterioplankton communities appear to be synchronous in some systems (Kent et al., 2007), potentially linked by the consumption of phytoplankton-specific exudates by heterotrophic bacterioplankton (Paver and Kent, 2010). These select examples of aquatic microbial time series (culled from hundreds in the literature), demonstrate the importance of time series analysis for identifying the important biotic and abiotic drivers of microbial community structure.
There are clear challenges to studying microbial communities through time. First, it is often difficult to characterize the micron-scale niches for microorganisms, and therefore the immediate environment experienced by many microbial communities remains unknown (Brock, 1987). Additionally, the timescales over which the greatest microbial community changes occur are typically unknown (Shade and Peter et al., 2013). Depending on the habitat, survey efforts may lack the temporal resolution to capture rapid community changes, particularly in systems with actively growing microbial populations, in which generation times may be on the order of minutes. Finally, surveying microbial diversity can be a daunting task. Individual samples often harbor hundreds to thousands of individual microbial taxa (Curtis and Sloan, 2004; Schloss and Handelsman, 2007; Quince et al., 2008), and the majority of microorganisms cannot be identified using traditional culture-based techniques (Pace, 1997; Schloss and Handelsman, 2007). Fortunately, the ongoing development of culture-independent tools and high-throughput sequencing technologies has made it feasible to describe the temporal dynamics of microbial communities at time scales and resolutions that were previously unattainable (Gonzalez et al., 2012).
We conducted a meta-analysis of newly available time series of microbial communities assessed via high-throughput sequencing of the 16S rRNA gene, which permits the detailed analysis of microbial community changes through time. Our objective was to characterize temporal dynamics of microbial communities from a suite of habitats, and when possible, to compare these dynamics with communities of larger organisms. To assess whether temporal patterns were common across both microbial and ‘macro’-bial organisms, we applied analyses previously applied to temporal patterns in plant and animal communities. We address the following outstanding questions: How variable are microbial communities over time, and how does this variability compare within habitats? What kinds of temporal patterns are often exhibited by microbial communities, and at what scales are these patterns most apparent? Do microbial communities exhibit species–time relationships (STRs), and if so, are those relationships similar to those for larger organisms?
Materials and methods
Data sets
We compiled bacterial and archaeal time series from 76 sites, spanning a wide range of study durations and habitats, including aquatic, air, brewery wastewater treatment, soil, plant- and human-associated communities. Fungal and other eukaryotic communities were not included in the meta-analysis. Each site had a minimum of five observations through time, and sites having a destructive sampling regime (for example, soils) were concatenated into a single time series. The compiled time series included eight seasonally sampled temperate bog lakes in Wisconsin, USA, each monitored at two locations: one sample from the upper, mixed layer (epilimnion) and one sample from the lower stratified layer (hypolimnion). These time series included between 1 and 3 years of approximately once or twice weekly observations during the ice-free period (Shade et al., 2008). Additional lake microbial communities were sampled intensively over the month of a whole-ecosystem disturbance experiment at three depths in the lake (Shade et al., 2012b). A second data set included air from near-surface troposphere samples from a mountaintop location in Colorado, USA (Bowers et al., 2012). The air communities were sampled continuously for hours over a few days, with samplings occurring approximately every month for about a year. Nine brewery wastewater treatment communities were sampled once per month for a year, and two of those sites had been previously sampled a few years before (Werner et al., 2011). The six-year English Channel time series, a coastal marine system, provided our longest series of monthly samples (Caporaso et al., 2012; Gilbert et al., 2012). Flower-associated microbial communities were sampled from six apple trees over the lifespan of the flowers (1 week), including five time points from before flowers opened until petal fall (Shade et al., 2013). In a study of human-associated microbiota, the palm, oral and gut microbial communities from two human subjects were collected approximately daily for a year for a male subject, and daily for 6 months for a female subject (Caporaso et al., 2011). Gut microbiota were also sampled from one infant across dietary shifts for the first 2.5 years of life (Koenig et al., 2011). Agricultural soils at the Kellogg Biological Station, Michigan, USA maintained under different management regimes were sampled monthly for 6 months (Lauber et al., 2013) and soils from National Ecological Observatory Network (NEON) sites in Hawaii and Florida, USA (www.neon.org) were sampled once per month for 3 months. Finally, six freshwater streams in Colorado, USA were sampled every 1–2 weeks for approximately 1 year (Portillo et al., 2012). Additional details about each data set are provided in Supplementary Table S1 and in the associated references.
Clearly, these sample sets vary widely with respect to their sampling intensity and study duration. However, this is unavoidable if we want to assess temporal dynamics for microbial communities from diverse environments, and, as has been demonstrated for meta-analyses of plant and animal community dynamics (Nekola and White, 1999; White et al., 2006; Soininen et al., 2007; Korhonen et al., 2010; White et al., 2010), we can still describe patterns that would not be evident if we were to restrict our analyses to a far more limited set of sample types.
Sequence analyses
The microbial communities in each of the 3431 individual samples were characterized by sequencing a portion of the 16S rRNA gene on either the Illumina (San Diego, CA, USA) or 454 (Branford, CT, USA) platforms. The 16S rRNA gene is widely used for determining the phylogenetic and taxonomic composition of bacterial communities and, in all the cases, the data were derived from PCR amplification of environmental DNA using primer pairs designed to amplify the gene region from all, or nearly all, known bacterial taxa. A closed reference operational taxonomic unit (OTU) picking protocol was applied to each data set separately (Caporaso et al., 2012). Briefly, OTUs were assigned based on 97% sequence identity to sequences in the Greengenes reference database (McDonald et al., 2012) preclustered at 97% identity (http://qiime.org/home_static/dataFiles.html). As we used the same reference-based OTU picking strategy for all samples, we could directly compare the relative abundances of taxa across samples. Furthermore, we sub-sampled each individual data set such that all samples from a given data set were compared at an equivalent sequencing depth (Supplementary Table S1). All analyses were performed on the rarefied OTU tables to permit comparisons of patterns in within- and between-community diversity. Our goal was not to quantify the absolute diversity found in any of the samples: this task is difficult, if not impossible, because individual samples may harbor thousands of rare taxa (Sogin et al., 2006). However, as recent work has demonstrated (Shaw et al., 2008; Kuczynski et al., 2010a), it is not necessary to characterize absolute diversity in order to accurately describe changes in within-sample and between-sample diversity within and between habitat types. Pielou’s evenness (Pielou, 1969), richness (number of OTUs) and Faith’s phylogenetic diversity (Faith, 1992) were used as within-sample (alpha) diversity metrics. Bray–Curtis was used as a taxon-based metric of differences in community composition (beta diversity), and the dissimilarities were calculated from the rarefied OTU tables in R using the vegan package (Oksanen et al., 2011; R Development Core Team, 2011). QIIME (version 1.2.1, (Caporaso et al., 2010) was used for constructing weighted and unweighted UniFrac distances. UniFrac is a commonly used phylogenetic distance metric to assess pairwise dissimilarity in community composition and incorporates information about differences in phylogenetic composition of community members (Lozupone and Knight, 2005; Lozupone et al., 2011), with weighted UniFrac accounting for differences in the relative abundances of community members.
Statistical analyses
All analyses were performed using the R environment for statistical computing (R Development Core Team, 2011), with the aid of the vegan and ggplot2 packages (Wickham, 2009; Oksanen et al., 2011). To compare temporal variability in diversity across habitats having inherently different diversities, we calculated the coefficient of variation (CV) in within-sample diversity for each community (Equation 1)

where σ is the s.d. and μ is the mean.
We calculated median absolute deviation (MAD) to compare variability in between-sample diversity (Equation 2).

Finally, we calculated z-scores of richness to examine the step-wise variability of richness through time, across data sets (Equation 3).

where χ is the raw value of richness, μ is the mean of the sample, and σ is the s.d. around the mean.
In using the coefficient of variation, median absolute deviation and z-scores of richness over time, we compared variability in diversity rather than absolute measures of diversity, which was most appropriate for comparing communities from different habitats that were assessed using different protocols for 16S rRNA short-read sequencing. To assess whether there were patterns in community structure that could be described by time between observations, we related community similarity/distance to time elapsed using Mantel tests with Pearson’s correlation on 999 permutations.
We evaluated the decay of community similarity over time (time-decay) using the same methods for calculating decay of community similarity over space (distance-decay; Nekola and White, 1999; Soininen et al., 2007). Though assessment of distance-decay is common in the literature, it is less common to assess time-decay. To assess time-decay in microbial communities, we used a similar approach adopted by Korhonen et al. (2010) to the meta-analysis of aquatic community time-decay. A log-linear model was fitted between the change in community structure (assessed by pair-wise similarities or distances, including Bray–Curtis, UniFrac and unweighted UniFrac) and days elapsed. Community dissimilarities were converted to similarities by subtracting from one. Similarities were log-transformed. The slope of the log-linear model is a rate of community change, sometimes referred to as turnover (Nekola and White, 1999). For plotting time-decay examples from each biome, we applied lowess smoothing over windows the length of 5% of the total series. Because time-decay can be sensitive to the duration of the study, we also performed a simple analysis of how quickly microbial communities change at temporal scales from 1 week to 1 month, from 1 to 6 months, from 6 months to 1 year, from 1 to 2 years, from 2 to 3 years, from 3 to 4 years, from 4 to 5 years and from 5 to 6 years. These windows encompass the breadth of time series durations included in the meta-analysis. For all pairs of observations within a community’s time series, a rate of change was calculated by dividing Bray–Curtis dissimilarity by the time between observations. Next, all pairs of observations were partitioned into the appropriate temporal window (determined by the time between observations) and an average rate of change for each window was calculated. The global average rate of change was summarized across sites from the same habitat (reported in Supplementary Table S2).
STRs for each site were constructed in R by calculating richness using the moving window approach of White et al. (2006). This approach involves partitioning a time series into as many window subsets as possible given the number of observations and fitting the STR model (the power-law relationship between time and richness) at each window. For example, a 250-time point series could divided into one 250-point window, two 249-point windows, and so on. The power function, rather than the lognormal function, was used so that our results would be directly comparable with the results from communities of larger organisms reported in White et al. (2006). There is some debate regarding which function is most appropriate for describing the STR (McGill, 2003; White et al., 2006), but White et al. (2006) found that the log and power functions produced identical patterns. We compared STR patterns in microbial communities with patterns for communities of larger organisms. However, microbial communities are widely considered to be more diverse than communities of larger organisms, and it remains difficult to make a direct comparison, as discussed at length previously (Fierer and Lennon, 2011).
There are caveats to the data sets used here that are worth highlighting. First, high-throughput sequencing of 16S rRNA genes can introduce biases in the determination of microbial diversity; these limitations have been described elsewhere (for example, Kunin et al., 2010; Haas et al., 2011; Quince et al., 2011; Soergel et al., 2012). Second, because sequencing depth varied across the sample sets (Supplementary Table S1), it is difficult to directly compare the temporal dynamics of individual taxa across the data sets (particularly taxa that are relatively rare). For this reason, we have focused our meta-analyses entirely on the overarching patterns in within- and between-sample diversity, which should be reasonably robust to differences in sequencing depth (Kuczynski et al., 2010b); however, we also tested whether inter-biome differences were a byproduct of differences in sequencing depth. Third, we picked OTUs using a closed-reference database protocol to compare data sets generated using different primers that target different variable regions of the 16S rRNA gene (as done in Caporaso et al., 2011). Although this method allowed us to directly compare data sets using the same reference phylogeny, any sequences not matching the well-curated database were discarded from the analyses. Therefore, communities that had better representation of taxa in the database (for example, human-associated communities) had a higher proportion of taxon assignments than, for instance, flower communities, where a larger percentage of the sequences lack a close match to those found in the database. However, we found no relationship between the proportion of sequences that matched the reference database and patterns of temporal variability, suggesting that frequency of matches to the reference database did not bias overall patterns.
Results and discussion
Temporal variability in within-sample diversity
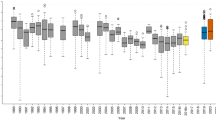
We used coefficients of variation calculated over each site’s time series to compare the variability in community evenness (equitability of representation of taxa), richness (number of taxa) and phylogenetic diversity (breadth of lineages). Variability in these diversity metrics was generally consistent across sites representing the same habitat (Figure 1a). Evenness exhibited less variability than richness or phylogenetic diversity over time. This is likely due to the exceedingly uneven nature of microbial communities (for example, Dethlefsen et al., 2008; Quince et al., 2008), where there are few very prevalent community members and a relatively large number of rare members (a ‘long tail’ distribution). Variability in community structure (beta diversity), measured as the median absolute deviation in weighted UniFrac distances, exhibited similar patterns as the variability in other diversity metrics (Figure 1b). Supplementary Figure S1 additionally provides for each time series a visualization of every time point’s deviation from the mean richness.

Temporal variability in (a) Pielou’s evenness (equitability of representation of taxa), richness (number of taxa), Faith’s phylogenetic diversity (breadth of lineages) and (b) weighted UniFrac distance, a community distance metric (between-sample diversity). Solid lines are the mean within each habitat. (c) Correlations in variability for each measure of diversity. All R>0.72 and P<0.001. Coefficient of variation (CV) and median absolute deviation (MAD) are unit-less metrics. WWT, brewery wastewater treatment.
Variability in evenness, richness, phylogenetic diversity and community structure were all highly correlated (Figure 1c), demonstrating that both phylogenetic (phylogenetic diversity and UniFrac) and taxonomic (evenness and richness) approaches to community diversity revealed similar overarching patterns in temporal variability across sites. This was surprising because we did not necessarily expect phylogenetic metrics, such as phylogenetic diversity, and taxonomic metrics, such as richness, to vary consistently with one another in time. This result suggests that the diversity metric used to investigate temporal variability may not matter, allowing robust comparisons of microbial diversity measured in different ways across different studies. Furthermore, differences in temporal variability across sites and habitats were not likely an artifact of differences in survey effort (the number of sequences per sample) or an artifact of our ability to identify taxa against the reference database (Figure 2, Pearson’s correlation, all P-values >0.10). These results suggest that there are often clear differences in the temporal variability in both within- and between-sample diversity within bacterial communities from different environment types.

Temporal variability in diversity is not dependent on sequencing depth or the proportion of sequences that could be matched to a reference database for taxonomic assignment (all P>0.10). Coefficient of variation (CV) and median absolute deviation (MAD) are unit-less metrics and are used here to describe temporal variability in richness, evenness, phylogenetic diversity, and UniFrac distance.
Overall, soil and brewery wastewater treatment communities were consistently less variable than other community types (Figure 1). Though we cannot know a priori the drivers governing the observed rates of change, we can generate reasonable hypotheses based on knowledge of the biology and ecology of a microbial habitat. For example, we may expect a relatively low degree of variability in soil communities for two reasons. First, soil environmental conditions may simply be less variable at the timescale of the included studies (6 months) than some of the other habitats considered in this study. Second, soil communities contain a large proportion of dormant organisms (Lennon and Jones, 2011) and because our sequencing approach could not discriminate between active and inactive members, the communities may appear to change relatively little over the given time scale. Soils also have high spatial heterogeneity, which can mask changes in local communities with time because of high community variability across micro-sites. However, brewery wastewater treatment communities may vary minimally for different reasons. Wastewater treatment facilities have been engineered to perform a function (for example, nutrient removal Curtis and Sloan, 2006; Martín et al., 2006; Harris et al., 2012). Thus, a wastewater treatment system may represent an environmental filter for microorganisms that survive in wastewater treatment processes while system performance is maintained. However, operating conditions, such as feeding rate, and other environmental variables (including temperature) change over time in wastewater treatment facilities, as was true for the environmental conditions in the system studied here (Werner et al., 2011). Therefore, these communities may have low variability simply because of an environmental filter, rather than because of dormancy or invariant environmental conditions.
Stream bacterioplankton communities, the infant gut community, flower communities and human palm communities were highly variable in diversity (Figure 1). Again, we can generate hypotheses as to the drivers of this variability based on the knowledge of the microbial habitat. Streams in the sub-alpine region studied here are flow-through systems with pronounced physicochemical instability. They experience frequent and rapid shifts in environmental conditions as well as shifts in bacterial inputs into the stream channel (from sediment, soil and biofilms) that are likely driven by pulse flashes in hydrological conditions (Portillo et al., 2012). Human palm sites were previously noted to be highly variable (Fierer et al., 2008; Caporaso et al., 2011), and this was attributed to frequent exposure to bacterial inocula from a diverse array of touched surfaces and a high degree of temporal heterogeneity in environmental conditions (likely driven, in part, by the disturbances associated with washing events). In this way, palm and stream communities both experience environmental conditions that can vary considerably over time.
The time series that represent cases of microbial primary succession, the infant gut and flower surfaces, also exhibited a high degree of temporal variability in diversity. In both cases, there was microbial colonization of a sterile (or nearly sterile) habitat and rapid replacement of members through time as the environment is altered (Fierer et al., 2010). Thus, just as primary succession in plant communities can often lead to rapid changes in diversity and community composition over time, we would also expect a high degree of variability in diversity for microbial communities undergoing succession.
Predictability in microbial community changes over time and across temporal scales
We next asked whether communities sampled close together in time were, on average, more similar in composition than communities sampled further apart. Approximately 26–40% of the 76 sites had community changes that were correlated with time over the entirety of study duration (Figure 3). Specifically, there were 34 significant relationships using Bray–Curtis, 26 relationships for unweighted UniFrac and 21 relationships for weighted UniFrac out of the 76 sites. Thus, in less than half of the sites did we find that samples collected at shorter intervals harbored communities that were consistently more similar than those collected at longer intervals. However, there was at least one site from each biome that exhibited community changes that were significantly correlated with time (Figure 3). Overall, these results suggest that temporal variability is not necessarily predictable solely by considering the time between sampling events.

(a–c) Changes in community structure were related to time between community observations. The x axes labels give the number of significant relationships over the total sites from the habitat. WWT is brewery wastewater treatment. All points shown had Mantel P<0.05 on 999 permutations; tests that were not significant are not shown. For example, all six flower communities (green points) were significant using Bray–Curtis, one was significant using weighted UniFrac and none were significant using unweighted UniFrac metrics.
Those communities that consistently exhibited temporal dependence included air, marine, human and lake communities. The air, marine and lake community time series were sampled over a duration that included seasonal changes, and thus temporal trends are expected for these sites (Shade et al., 2008; Bowers et al., 2012; Gilbert et al., 2012). Further, human-associated microbial communities sampled consecutively over time have been shown to be more similar than those sampled more distantly in time (Costello et al., 2009), which is consistent with our detection of a temporal trend for these communities. These results suggest that overall temporal signals exist for communities from certain habitats and that, for communities from these habitats, the relationship between community structure and time is insensitive to the choice of metric used for evaluating community structure (Figure 3).
Several habitats had no or few communities that exhibited correlations between time and community structure (Figure 3). For example, depending on the metric used, anywhere from five to none of the 25 soil time series exhibited significant temporal dependence (Figure 3). For soil communities, inter-annual changes would not be evident in this data set, as the study durations were ⩽6 months. Thus, it may be that changes in soil communities are not correlated with time at the time scale included but that such relationships could become evident if we had time series extending across full years or multiple years.
It is also not surprising that the structure of brewery wastewater treatment communities was often not correlated with time, as any changes in operation would not necessarily be related to time. For the flower communities, the taxonomic metric, Bray–Curtis, detected a significant temporal signal (Figure 3a) where the phylogenetic metrics did not (Figures 3b and c), likely because closely related taxa were replacing one another (Shade et al., 2013), a change to which a phlyogenetic metric would be less sensitive. Only one of the stream communities was correlated with time, and this correlation was evident only when using the weighted UniFrac distance metric (Figure 3c). Because environmental conditions in the sampled streams are flashy with irregular pulse distances (Portillo et al., 2012), changes in stream communities may not be correlated with time but rather with the occurrence of these infrequent events.
Plant and animal communities often (but not always) become less similar with increasing time or geographical distance, a phenomenon known as similarity-decay (Nekola and White, 1999). To determine whether there was decay of microbial community structure over time (time-decay), we fit log-linear models to community similarity over differences in time between sample collections (Nekola and White, 1999; Korhonen et al., 2010; Figure 4). The rate of temporal change in community structure is the slope of the model, and this slope is a measure of community turnover. A community that is not changing over time would have a slope of zero. More negative, steeper slopes indicate a faster rate of change than less negative slopes. In Figure 4, very gradual time-decay is evident, even for microbial communities that exhibit seasonality, such as the coastal marine and lake sites, as well as for sites that have high temporal variability, such as the stream community. Overall, 31–40% of microbial communities exhibited significant time-decay in community similarity (Supplementary Figure S2, P<0.05). Slope was not correlated with richness (all P>0.05 for all similarity metrics), suggesting that differences in the number of taxa detected in individual samples did not influence outcomes. Slopes were barely negative in most cases. However, one flower (Gala 2), one soil (HI_R12) and one brewery wastewater treatment (E1) had slopes between −0.02 and −0.03. Aside from these exceptions, the consistency of microbial time-decay suggests that community turnover is generally quite slow.

(a–h) An example of time-decay from each microbial habitat. White chart background shows that the model was fit using weighted UniFrac distances and gray shows that the model was fit using unweighted UniFrac distances. Note differences in x axis ranges. The blue line is the smoothed series of community similarities, the dotted red line is the overall mean similarity and the dashed black line is the log-linear time-decay model. NS, not significant.
Time-decay can be sensitive to a study’s duration. Therefore, we asked at what temporal scale communities changed the most quickly for the study duration and sampling intensities of observations that were available to us. To do this, we calculated a simple rate of change by dividing pair-wise Bray–Curtis dissimilarity by the time between observations and then partitioned observations into nine broad temporal ranges, from 1 day to 6 years (Supplementary Figure S3), representing the breadth of the available time series. This analysis also showed a consistent trend of slower change with longer durations of time, and additionally reveals gaps in the currently available time series, and points to the temporal resolutions that are yet unknown for certain habitats (Supplementary Table S2).
Notably, just because microbial community turnover is very gradual at long study durations does not mean that there is no change occurring in these communities. Rather, it suggests that there are not drastic or new changes occurring, such as the addition of new or phylogenetically distinct taxa. Also, it is possible that some of the communities may be changing around a ‘baseline’ of normal variability, following the conceptual model of ball and urn for alternative stable states (Beisner et al., 2003).
STRs
STRs describe the accumulation of richness in a community, over increasingly longer durations (for example, Preston, 1960). The exponent of the STR provides an indication of the rate at which new taxa are observed in a community over time; the higher the exponent the more new taxa are introduced over time. Depending on the scale of the study and the community of interest, STRs can be explained by sampling, ecological or evolutionary effects (Preston, 1960; White et al., 2006, 2010). Over short-time periods, incomplete sampling effort (duration or intensity) often drives STRs, but evolutionary processes such as speciation and extinction become more important for STRs over longer-time periods. Between these extremes, ecological processes such as meta-community dynamics are important for STR, including dispersal of transient tourists into a regional species pool (Magurran and Henderson, 2003). Though the STR spatial equivalent, the species–area relationship, remains actively investigated in microbial ecology (for example, Horner-Devine et al., 2004; Fierer and Jackson, 2006; Green and Bohannan, 2006; Martiny et al., 2006; Woodcock et al., 2006; Bell, 2010), STRs are less often documented for microorganisms. However, STRs have been reported for communities from diverse environments, such as on leaf surfaces (Redford and Fierer, 2009), in streams (Portillo et al., 2012) and in bioreactors (Van Der Gast et al., 2008), hinting that STRs may generally apply to microbial communities.
We found that all communities had significant STRs (P<0.05, Figure 5a). Microbial STRs were not related to study duration or sequencing depth (Pearson’s correlation P=0.81 and 0.36, respectively), suggesting limited, if any, influence of sampling. However, our analysis does not distinguish stochastic processes (for example, random presence and absence of taxa) from deterministic properties in driving the STR, and both of these likely contribute. There was a very consistent taxa–time relationship across microbial communities, ranging from 0.24 to 0.61. Communities had comparable STR exponents within biomes, again highlighting the within-biome consistency. Furthermore, differences in the STR exponents across biomes could be explained by known ecological attributes of the communities. For example, the air and stream communities, with high flow-through and heterogeneity, had high STR. The infant gut and flower primary succession communities also had high STR. The soil communities had the lowest STR exponents, demonstrating that despite their high levels of diversity (for example, Elshahed et al., 2008), the taxa present at a given location do not change appreciably over time (a pattern also noted in Fierer and Jackson, 2006). These results are consistent with the general trends in diversity over time as seen in Figure 1 and suggest that fluctuating abiotic conditions drive higher microbial STRs. These environmental fluctuations potentially allow for time series to effectively encompass a wider range of microbial niches (Shurin, 2007).

STR exponent, w, from power-law fit to the accumulation of taxa detected with time, as per White et al. (2006). (a) Microbial community STR across habitats. The dashed line indicates the average exponent value for each habitat. Error bars are s.e. but, in most cases, are too small to visualize on the chart. WWT is brewery wastewater treatment. (b) Microbial STR exponent for phyla detected across all habitats: Actinobacteria, Bacteroidetes, Firmicutes, Proteobacteria and Verrucomicrobia. White circles are samples that had STR values beyond range times the interquartile range (potential statistical outliers). (c) Average STR exponent by taxonomic group for larger organisms, reproduced from Figure 3 of White et al. (2006). Error bars are exponent ranges for each taxonomic group.
We next asked whether STRs could be distinguished at the phylum level. For each community, we calculated STRs for common phyla observed within each habitat (Actinobacteria, Bacteroidetes, Firmicutes, Proteobacteria and Verrucomicrobia), at each site. We found a range of STRs at the phylum-level, but the exponents had comparable means across phyla (Figure 5b). This suggests that microbial phyla do not have inherently different rates of replacement but that the local ecology drives the observed STR within a community. One exception to this may be the Firmicutes, which had a slightly higher mean STR and wider upper range than the other phyla. We speculate that this pattern may be a product of many taxa affiliated with Firmicutes being capable of sporulation (Onyenwoke et al., 2004) and may either have higher rates of dispersal into communities or are able to persist in communities in very low abundances when conditions are unfavorable and bloom when conditions become favorable. However, taxa affiliated with other phyla are also capable of persisting in dormant states, and more evidence is needed to address this hypothesis.
It can be difficult to directly compare STR patterns of microbial communities with communities of larger organisms. First, most microbes probably have generation times shorter than those of larger organisms, and the microbial time series investigated here likely included far more microbial generations than comparable study durations of plant and animal taxa. In addition, species definitions as applied to microbes are distinct from how plant and animal species are typically defined (Stackebrandt et al., 2002; Gevers et al., 2005). Finally, microbial communities are often more diverse than communities of larger organisms, with individual samples harboring hundreds to thousands of taxa as compared with the tens of taxa reported in White et al., (2006). However, despite these caveats, the range of STRs for microbial communities was the same as that reported in White et al., (2006) meta-analysis of STR for larger organisms (Figures 5a and c). On average, STR exponents are higher for microorganisms than for larger organisms, and the exponents were less variable within communities. Though White et al. (2006) found a relationship between STRs and richness in their meta-analysis, we found no such relationship for microbial communities (P=0.19), suggesting that microbial community STRs are better explained by changes in environmental conditions than by local richness. However, the slightly higher average STR for microbial communities over macrobial communities may be due to the generally higher richness of microbial communities. Random changes in the occurrences of these community members may partly be driven by the stochastic process of drift (Vellend, 2010), which could also contribute to the STR. Nonetheless, the overall consistency in STRs across microbial and macrobial communities is striking, suggesting that this may be an ecological pattern that is ubiquitous across scales, phyla and habitats.
Conclusions and future directions
From this meta-analysis, we gain a preliminary understanding of how temporal patterns in microbial communities compare with each other and with those of communities of larger organisms. There was consistent variability in diversity and STRs across communities from similar habitats. This consistency can be leveraged when making logistical decisions about sampling regimes and for understanding baseline levels of temporal variability. Within a habitat, these temporal patterns may represent the equilibrium from which disturbance events may alter microbial dynamics. This information is important for identifying when a microbial community is experiencing a disturbance, anticipating how quickly a community may recover from such events, and determining if and when a community has recovered post-disturbance (Allison and Martiny, 2008; Shade et al., 2012a; Shade and Peter et al., 2013).
A challenge in conducting this meta-analysis was the availability of directly comparable microbial time series collected across habitats. The same protocols and standards of quality across data sets were necessary for this undertaking, and though there are 76 microbial communities that span 8 distinct microbial biomes, there are obviously many habitats that were not represented in the meta-analysis simply because data were not available. Further, some of the available time series were limited in duration or sampling intensity, and therefore, it cannot be determined whether temporal patterns were not discovered because they truly do not exist or because of a limitation in the sampling effort. For example, we may observe seasonality in soil microbial communities sampled over 6 years instead of over 6 months. With the increased availability of high-throughput sequencing, the number of higher-resolution, longer-term time series will only increase. For example, the Earth Microbiome Project is leading a concerted effort to collect and curate high-quality microbial community sequencing data and corresponding contextual data from diverse environments (Gilbert et al., 2010). Placing microbial community dynamics within the rich context of environmental dynamics will provide insight into the key drivers of those communities, help to explain discrepancies in patterns across communities from similar habitats, and allow ecologists to begin predicting microbial dynamics. Efforts like these will help to build theory for microbial ecology, advancing beyond system-specific observations (Prosser et al., 2007). Additional discussion about challenges with sampling microbial communities in time (including defining a microbial community, sampling the same community longitudinally and accounting for micro-scale spatial heterogeneity) is available as a Supplementary Discussion.
This meta-analysis highlights that microbial communities, like plant and animal communities, are variable with time; that microbial temporal dynamics are dependent on the habitat type in question; and, furthermore, that microbial temporal dynamics are often predictable. Perhaps more importantly, this work demonstrates the utility of time course analyses in microbial ecology. Likewise, this meta-analysis highlights that microbial communities represent a useful system for studying temporal dynamics in communities, dynamics that would be very difficult to explore in plant and animal communities where generation times are often far longer. For example, it is often easier to execute disturbance experiments with microbial communities than with plant and animal communities, and microbial responses can be detected over relatively short-time periods (for example, Dethlefsen et al., 2008; Shade et al., 2012b). These kinds of directed, in situ experiments with microbial communities may provide key empirical validation (or invalidation) of theoretical paradigms.
Just as comparing the spatial patterns exhibited by microorganisms versus larger organisms has yielded interesting findings that have allowed us test paradigms in biogeography (for example, Horner-Devine et al., 2004; Fuhrman et al., 2008; King et al., 2010), paradigms almost wholly derived from research on plant and animal communities, we can use microbial communities to build a more comprehensive understanding of time–biodiversity relationships. The continued integration of microorganisms into the broader field of ecology will clearly be advantageous for both ‘macro’-bial and microbial ecologists, providing rich insight into the common forces that shape patterns of distribution and diversity for organisms of all sizes.
References
Allison SD, Martiny JBH . (2008). Resistance, resilience, and redundancy in microbial communities. Proc Natl Acad Sci USA 105: 11512–11519.
Beisner B, Haydon D, Cuddington K . (2003). Alternative stable states in ecology. Front Ecol Environ 1: 376–382.
Bell T . (2010). Experimental tests of the bacterial distance-decay relationship. ISME J 4: 1357–1365.
Bell T, Newman JA, Silverman BW, Turner SL, Lilley AK . (2005). The contribution of species richness and composition to bacterial services. Nature 436: 1157–1160.
Bowers RM, McCubbin IB, Hallar AG, Fierer N . (2012). Seasonal variability in airborne bacterial communities at a high-elevation site. Atmos Environ 50: 41–49.
Brock TD . (1987). The study of microorganisms in situ: progress and problems. In M Fletcher, TRG Gray, JG Jones (Eds.),. Forty-first symposium of the Society for General Microbiology. University of St. Andrews: Cambridge University Press.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336.
Caporaso JG, Lauber CL, Costello EK, Berg-Lyons D, Gonzalez A, Stombaugh J et al (2011). Moving pictures of the human microbiome. Genome Biol 12: R50.
Caporaso JG, Paszkiewicz K, Field D, Knight R, Gilbert JA . (2012). The Western English Channel contains a persistent microbial seed bank. ISME J 6: 1089–1093.
Chase JM . (2003). Community assembly: when should history matter? Oecol 136: 489–498.
Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R . (2009). Bacterial community variation in human body habitats across space and time. Science 326: 1694.
Cottingham KL, Brown BL, Lennon JT . (2001). Biodiversity may regulate the temporal variability of ecological systems. Ecol Lett 4: 72–85.
Crump BC, Peterson BJ, Raymond PA, Amon RMW, Rinehart A, McClelland JW et al (2009). Circumpolar synchrony in big river bacterioplankton. Proc Natl Acad Sci USA 106: 21208–21212.
Curtis TP, Sloan WT . (2004). Prokaryotic diversity and its limits: microbial community structure in nature and implications for microbial ecology. Curr Opin Microbiol 7: 221–226.
Curtis TP, Sloan WT . (2006). Towards the design of diversity: stochastic models for community assembly in wastewater treatment plants. Water Sci Technol 54: 227–236.
Dethlefsen L, Huse S, Sogin ML, Relman DA . (2008). The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol 6: e280.
Eiler A, Heinrich F, Bertilsson S . (2012). Coherent dynamics and association networks among lake bacterioplankton taxa. ISME J 6: 330–342.
Elshahed MS, Youssef NH, Spain AM, Sheik C, Najar FZ, Sukharnikov LO et al (2008). Novelty and uniqueness patterns of rare members of the soil biosphere. Appl Environ Microbiol 74: 5422–5428.
Faith DP . (1992). Conservation evaluation and phylogenetic diversity. Biol Conserv 61: 1–10.
Fierer N, Bradford MA, Jackson RB . (2007). Toward an ecological classification of soil bacteria. Ecology 88: 1354–1364.
Fierer N, Hamady M, Lauber CL, Knight R . (2008). The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc Natl Acad Sci USA 105: 17994.
Fierer N, Jackson RB . (2006). The diversity and biogeography of soil bacterial communities. Proc Natl Acad Sci USA 103: 626–631.
Fierer N, Nemergut D, Knight R, Craine JM . (2010). Changes through time: integrating microorganisms into the study of succession. Res Microbiol 161: 635–642.
Fierer N, Lennon J . (2011). The generation and maintenance of diversity in microbial communities. Am J Bot 98: 439–448.
Fraterrigo JM, Rusak JA . (2008). Disturbance-driven changes in the variability of ecological patterns and processes. Ecol Lett 11: 756–770.
Fuhrman JA, Hewson I, Schwalbach MS, Steele JA, Brown MV, Naeem S . (2006). Annually reoccurring bacterial communities are predictable from ocean conditions. Proc Natl Acad Sci USA 103: 13104–13109.
Fuhrman JA, Steele JA, Hewson I, Schwalbach MS, Brown MV, Green JL et al (2008). A latitudinal diversity gradient in planktonic marine bacteria. Proc Natl Acad Sci USA 105: 7774.
Gevers D, Cohan FM, Lawrence JG, Spratt BG, Coenye T, Feil EJ et al (2005). Re-evaluating prokaryotic species. Nat Rev Microbiol 3: 733–739.
Gilbert JA, Meyer F, Antonopoulos D, Balaji P, Brown CT, Desai N et al (2010). Meeting report: the terabase metagenomics workshop and the vision of an Earth Microbiome project. Stand Genomic Sci 3: 243.
Gilbert JA, Steele JA, Caporaso JG, Steinbrück L, Reeder J, Temperton B et al (2012). Defining seasonal marine microbial community dynamics. ISME J 6: 298–308.
Gonzalez A, King A, Robeson MS, Song SJ, Shade A, Metcalf JL et al (2012). Characterizing microbial communities through space and time. Curr Opin Biotechnol 23: 431–436.
Green J, Bohannan BJM . (2006). Spatial scaling of microbial biodiversity. Trends Ecol Evol 21: 501–507.
Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G et al (2011). Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res 21: 494–504.
Harris J, Baptista J, Curtis T, Nelson A, Pawlett M, Ritz K et al (2012). Engineering difference: matrix design determines community composition in wastewater treatment systems. Ecol Eng 40: 183–188.
Hofle MG, Haas H, Dominik K . (1999). Seasonal dynamics of bacterioplankton community structure in a eutrophic lake as determined by 5S rRNA analysis. Appl Environ Microbiol 65: 3164–3174.
Holling CS . (1973). Resilience and stability of ecological systems. Annu Rev Ecol Syst 4: 1–23.
Horner-Devine MC, Lage M, Hughes JB, Bohannan BJM . (2004). A taxa-area relationship for bacteria. Nature 432: 750–753.
Ives AR, Carpenter SR . (2007). Stability and diversity of ecosystems. Science 317: 58–62.
Ives AR, Dennis B, Cottingham K, Carpenter SR . (2003). Estimating community stability and ecological interactions from time-series data. Ecol Monogr 73: 301–330.
Jones SE, Chiu CY, Kratz TK, Wu JT, Shade A, McMahon KD . (2008). Typhoons initiate predictable change in aquatic bacterial communities. Limnol Oceanogr 53: 1319–1326.
Kent AD, Yannarell AC, Rusak JA, Triplett EW, McMahon KD . (2007). Synchrony in aquatic microbial community dynamics. ISME J 1: 38–47.
King AJ, Freeman KR, McCormick KF, Lynch RC, Lozupone C, Knight R et al (2010). Biogeography and habitat modelling of high-alpine bacteria. Nat Commun 1: 53.
Koenig JE, Spor A, Scalfone N, Fricker AD, Stombaugh J, Knight R et al (2011). Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci USA 108: 4578.
Korhonen JJ, Soininen J, Hillebrand H . (2010). A quantitative analysis of temporal turnover in aquatic species assemblages across ecosystems. Ecology 91: 508–517.
Kuczynski J, Costello EK, Nemergut DR, Zaneveld J, Lauber CL, Knights D et al (2010a). Direct sequencing of the human microbiome readily reveals community differences. Genome Biol 11: 210.
Kuczynski J, Liu Z, Lozupone C, McDonald D, Fierer N, Knight R . (2010b). Microbial community resemblance methods differ in their ability to detect biologically relevant patterns. Nat Methods 7: 813–819.
Kunin V, Engelbrektson A, Ochman H, Hugenholtz P . (2010). Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ Microbiol 12: 118–123.
Lauber CL, Ramirez KS, Aanderud Z, Lennon J, Fierer N . (2013). Temporal variability in soil microbial communities across land-use types. ISME J e-pub ahead of print 4 April 2013; doi: 10.1038/ismej.2013.50.
Lennon JT, Jones SE . (2011). Microbial seed banks: the ecological and evolutionary implications of dormancy. Nat Rev Microbiol 9: 119–130.
Lockwood JL, Powell RD, Nott MP, Pimm SL . (1997). Assembling ecological communities in time and space. Oikos 80: 549–553.
Lozupone C, Knight R . (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71: 8228–8235.
Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R . (2011). UniFrac: an effective distance metric for microbial community comparison. ISME J 5: 169–172.
Lozupone CA, Knight R . (2007). Global patterns in bacterial diversity. Proc Natl Acad Sci USA 104: 11436–11440.
Magurran AE, Baillie SR, Buckland ST, Dick JMP, Elston DA, Scott EM et al (2010). Long-term datasets in biodiversity research and monitoring: assessing change in ecological communities through time. Trends Ecol Evol 25: 574–582.
Magurran AE, Henderson PA . (2003). Explaining the excess of rare species in natural species abundance distributions. Nature 422: 714–716.
Martiny JBH, Bohannan BJM, Brown JH, Colwell RK, Fuhrman JA, Green JL et al (2006). Microbial biogeography: putting microorganisms on the map. Nat Rev Microbiol 4: 102–112.
Martín HG, Ivanova N, Kunin V, Warnecke F, Barry KW, McHardy AC et al (2006). Metagenomic analysis of two enhanced biological phosphorus removal (EBPR) sludge communities. Nat Biotechnol 24: 1263–1269.
McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A et al (2012). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6: 610–618.
McGill B . (2003). Strong and weak tests of macroecological theory. Oikos 102: 679–685.
McNaughton SJ . (1977). Diversity and stability of ecological communities: a comment on the role of empiricism in ecology. Am Nat, 515–525.
Nekola JC, White PS . (1999). The distance decay of similarity in biogeography and ecology. J Biogeogr 26: 867–878.
Nelson CE . (2008). Phenology of high-elevation pelagic bacteria: the roles of meteorologic variability, catchment inputs and thermal stratification in structuring communities. ISME J 3: 13–30.
Nemergut DR, Costello EK, Hamady M, Lozupone C, Jiang L, Schmidt SK et al (2011). Global patterns in the biogeography of bacterial taxa. Environ Microbiol 13: 135–144.
Oksanen JF, Blanchet G, Kindt R, Legendre P, Minchin PR, O’Hara R et al (2011), vegan: Community Ecology Package, R package version 2.0, 2nd edn..
Onyenwoke RU, Brill JA, Farahi K, Wiegel J . (2004). Sporulation genes in members of the low G+C Gram-type-positive phylogenetic branch (Firmicutes). Arch Microbiol 182: 182–192.
Pace NR . (1997). A molecular view of microbial diversity and the biosphere. Science 276: 734–740.
Paver S, Kent A . (2010). Temporal patterns in glycolate-utilizing bacterial community composition correlate with phytoplankton population dynamics in humic lakes. Microb Ecol 60: 406–418.
Pielou EC . (1969) An Introduction to Mathematical Ecology. Wiley-Interscience: New York, NY, USA.
Pimm SL . (1984). The complexity and stability of ecosystems. Nature 307: 321–326.
Portillo MC, Anderson SP, Fierer N . (2012). Temporal variability in the diversity and composition of stream bacterioplankton communities. Environ Microbiol 14: 2417–2428.
Preston F . (1960). Time and space and the variation of species. Ecology 41: 611–627.
Prosser JI, Bohannan BJM, Curtis TP, Ellis RJ, Firestone MK, Freckleton RP et al (2007). The role of ecological theory in microbial ecology. Nat Rev Microbiol 5: 384–392.
Quince C, Curtis TP, Sloan WT . (2008). The rational exploration of microbial diversity. ISME J 2: 997–1006.
Quince C, Lanzen A, Davenport RJ, Turnbaugh PJ . (2011). Removing noise from pyrosequenced amplicons. BMC Bioinformatics 12: 38.
R Development Core Team (2011) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria.
Redford AJ, Fierer N . (2009). Bacterial succession on the leaf surface: a novel system for studying successional dynamics. Microb Ecol 58: 189–198.
Schloss PD, Handelsman J . (2007). The last word: books as a statistical metaphor for microbial communities. Annu Rev Microbiol 61: 23–34.
Shade A, Jones SE, McMahon KD . (2008). The influence of habitat heterogeneity on freshwater bacterial community composition and dynamics. Environ Microbiol 10: 1057–1067.
Shade A, Kent AD, Jones SE, Newton RJ, Triplett EW, McMahon KD . (2007). Interannual dynamics and phenology of bacterial communities in a eutrophic lake. Limnol Oceanogr 52: 487–494.
Shade A, McManus PS, Handelsman J . (2013). Unexpected diversity during community succession in the apple flower microbiome. mBio 4: e00602–e00612.
Shade A, Peter H, Allison SD, Baho D, Berga M, Buergmann H et al (2012a). Fundamentals of microbial community resistance and resilience. Front Microbiol 3: 417.
Shade A, Peter H, Allison SD, Baho DL, Berga M, Bürgmann H et al (2013). Fundamentals of microbial community resistance and resilience. Front Microbiol 3: 417.
Shade A, Read JS, Youngblut ND, Fierer N, Knight R, Kratz TK et al (2012b). Lake microbial communities are resilient after a whole-ecosystem disturbance. ISME J 6: 2153–2167.
Shaw AK, Halpern AL, Beeson K, Tran B, Venter JC, Martiny JBH . (2008). It’s all relative: ranking the diversity of aquatic bacterial communities. Environ Microbiol 10: 2200–2210.
Shurin JB . (2007). How is diversity related to species turnover through time? Oikos 116: 957–965.
Soergel DA, Dey N, Knight R, Brenner SE . (2012). Selection of primers for optimal taxonomic classification of environmental 16S rRNA gene sequences. ISME J 6: 1440–1444.
Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, Neal PR et al (2006). Microbial diversity in the deep sea and the underexplored ‘rare biosphere’. Proc Natl Acad Sci USA 103: 12115–12120.
Soininen J, McDonald R, Hillebrand H . (2007). The distance decay of similarity in ecological communities. Ecography 30: 3–12.
Stackebrandt E, Frederiksen W, Garrity GM, Grimont PAD, Kampfer P, Maiden MCJ et al (2002). Report of the ad hoc committee for the re-evaluation of the species definition in bacteriology. Int J Syst Evol Microbiol 52: 1043–1047.
Strickland MS, Lauber C, Fierer N, Bradford MA . (2009). Testing the functional significance of microbial community composition. Ecology 90: 441–451.
Van Der Gast CJ, Ager D, Lilley AK . (2008). Temporal scaling of bacterial taxa is influenced by both stochastic and deterministic ecological factors. Environ Microbiol 10: 1411–1418.
Vellend M . (2010). Conceptual synthesis in community ecology. Q Rev Biol 85: 183–205.
Werner JJ, Knights D, Garcia ML, Scalfone NB, Smith S, Yarasheski K et al (2011). Bacterial community structures are unique and resilient in full-scale bioenergy systems. Proc Natl Acad Sci USA 108: 4158–4163.
Wertz S, Degrange V, Prosser JI, Poly F, Commeaux C, Guillaumaud N et al (2007). Decline of soil microbial diversity does not influence the resistance and resilience of key soil microbial functional groups following a model disturbance. Environ Microbiol 9: 2211–2219.
White EP, Adler PB, Lauenroth WK, Gill RA, Greenberg D, Kaufman DM et al (2006). A comparison of the species–time relationship across ecosystems and taxonomic groups. Oikos 112: 185–195.
White EP, Ernest S, Adler PB, Hurlbert AH, Lyons SK . (2010). Integrating spatial and temporal approaches to understanding species richness. Philos Trans R Soc Lond B Biol Sci 365: 3633–3643.
Wickham H . (2009) ggplot2: Eelegant Graphics for Data Analysis. Springer: New York, NY, USA.
Woodcock S, Curtis TP, Head IM, Lunn M, Sloan WT . (2006). Taxa-area relationships for microbes: the unsampled and the unseen. Ecol Lett 9: 805–812.
Acknowledgements
This work would not have been possible without the generosity and collaborative spirit of the many primary authors of the data sets included in the meta-analysis. AS is a Gordon and Betty Moore Foundation Fellow of the Life Sciences Research Foundation. We thank Petr Keil for insightful discussions. We thank Jack Gilbert for the Western English Channel dataset, and Jack Gilbert and Trina McMahon for the lakes dataset. NF was supported by funding from the National Science Foundation and the US Department of Agriculture. RK was supported, in part, by the Howard Hughes Medical Institute and the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Shade, A., Gregory Caporaso, J., Handelsman, J. et al. A meta-analysis of changes in bacterial and archaeal communities with time. ISME J 7, 1493–1506 (2013). https://doi.org/10.1038/ismej.2013.54
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2013.54
Keywords
This article is cited by
-
Soil bacterial community composition is more stable in kiwifruit orchards relative to phyllosphere communities over time
Environmental Microbiome (2023)
-
Organic fertilizer and hydrothermal conditions change the distribution of Medicago sativa L. productivity and soil bacterial diversity in coastal saline soil
Chemical and Biological Technologies in Agriculture (2023)
-
Distinct and Temporally Stable Assembly Mechanisms Shape Bacterial and Fungal Communities in Vineyard Soils
Microbial Ecology (2023)
-
The microbial environment modulates non-genetic maternal effects on egg immunity
Animal Microbiome (2022)
-
Terrestrial connectivity, upstream aquatic history and seasonality shape bacterial community assembly within a large boreal aquatic network
The ISME Journal (2022)

{kind=link}
{kind=link}