
Abstract
Whether fungal community structure depends more on historical factors or on contemporary factors is controversial. This study used culture-dependent and -independent (polymerase chain reaction-denaturing gradient gel electrophoresis (PCR-DGGE)) methods to assess the influence of historical and contemporary factors on the distributions of fungi in the wetland sediments at 10 locations along the Changjiang River and at 10 other locations in China. The culture-dependent approach detected greater species diversity (177 operational taxonomic units (OTUs)) than PCR-DGGE analysis (145 OTUs), and the species in the genera of Penicillium (relative frequency=16.8%), Fusarium (15.4%), Aspergillus (7.6%), Trichoderma (5.8%) and Talaromyces (4.2%) were dominant. On the basis of DGGE data, fungal diversity along the Changjiang River increased from upstream to downstream; altitude explained 44.8% of this variation in diversity. And based on the data from all 20 locations, the fungal communities were geographically clustered into three groups: Southern China, Northern China and the Qinghai-Tibetan Plateau. Multivariate regression tree analysis for data from the 20 locations indicated that the fungal community was influenced primarily by location (which explained 61.8% of the variation at a large scale), followed by total potassium (9.4%) and total nitrogen (3.5%) at a local scale. These results are consistent with the concept that geographic distance is the dominant factor driving variation in fungal diversity at a regional scale (1000–4000 km), whereas environmental factors (total potassium and total nitrogen) explain variation in fungal diversity at a local scale (<1000 km).
Similar content being viewed by others


Introduction
Those fungi with an extensive hyphal growth habit, a rapid growth rate and the ability to translocate nutrients through their hyphal network have fundamental roles in organic matter decomposition in all ecosystems, especially carbon cycling in peatlands (Thormann, 2006). Despite their importance, few studies have considered which factors generate and maintain fungal diversity and their distribution patterns (Mohamed and Martiny, 2011). As indicated by the classic dictum ‘everything is everywhere, but the environment selects’ (Beijerinck, 1913), environmental factors have long been considered to have a strong influence on fungal (Green et al., 2004) and bacterial biogeography (Fenchel and Finlay, 2005; Fierer and Jackson, 2006; Fuhrman et al., 2008; Schauer et al., 2010).
In surface sediments, microorganisms are sometimes assumed to disperse with riverine currents (Sutcliffe and Parks, 1989). The channel water in rivers and streams is diverted through subsurface flow paths and mixed with groundwater in close contact with geochemically sediment surfaces (Harvey and Fuller, 1998), which would lead to similar environmental conditions in the sediments along a river. Environmental factors are believed to select similar subsets of organisms from a ubiquitously dispersed bacterial assemblage (Hewson et al., 2007). However, there is little empirical evidence to support the hypothesis of ubiquitous dispersal. Recent research enriched our understanding of how environmental factors influence bacterial communities along river sediments, with one observation showing that bacterial assemblages were homogeneously dispersed along a 35-km transect of sediment under shallow water (Hewson et al., 2007), but other studies have documented habitat specificity for different types of bacteria, resulting in a heterogeneous spatial distribution in freshwater systems (Hewson and Fuhrman, 2004; Reche et al., 2005). Those results provide an essential reference for diversity and distribution of fungi in wetlands. Although fungi have large gradients in biogeochemical properties in wetlands, how factors shape fungal communities in sediments across a river and at the regional scale is overlooked and remains unexplored.
A large-scale riverine current provides contemporary environmental conditions and historical contingencies, which are considered to be the two main factors that shape fungal and bacterial communities in ecosystems (Martiny et al., 2006; O’Malley, 2007). However, the relative importance of these factors remains unclear. Several studies reported that geographic distance, which is considered to be representative of historical contingencies, significantly affected bacterial diversity (Ramette and Tiedje, 2007; Ge et al., 2008; Schauer et al., 2010), while other observations demonstrated that bacterial biogeography was significantly affected by contemporary environmental conditions (Fierer et al., 2007). In one study, bacterial biogeography was affected by geographic distance, while archaeal biogeography was influenced by contemporary environmental factors (Pagaling et al., 2009). For fungal biogeography, both contemporary environmental conditions and historical contingencies have been reported to have important roles (Green et al., 2004). The similarity of fungal communities colonizing Eucalyptus leaves in streams was related to the geographic distance between the streams and their temperature and altitude (Bärlocher et al., 2011). However, a recent study indicated that historical contingencies could be the dominant factor determining variation in bacterial diversity on a regional spatial scale (about 1000 km) and that certain contemporary disturbances also caused variation in soil bacterial diversity at a local spatial scale (Ge et al., 2008). At intermediate scales (10–3000 km), three of five studies reported a significant effect of distance, while one indicated a significant effect of contemporary environmental conditions (Martiny et al., 2006). Given that some results indicate the importance of environmental variations and others indicate the importance of historical contingencies, and that effects of environmental factors and historical contingencies on macroorganisms have been documented to be scale-dependent (Williams et al., 2002), we suspected that the effects of contemporary environmental conditions and historical contingencies on fungal communities might also be scale-dependent.
The Changjiang River in China, which is the third-longest river in the world, has many associated wetlands. We examined diversity and composition of fungi in 10 wetlands along the Changjiang River and 10 other independent wetlands around China by culture-dependent and polymerase chain reaction-denaturing gradient gel electrophoresis (PCR-DGGE) methods. The relative contributions of historical events and of environmental heterogeneity on fungal biogeography were evaluated for this data set comparing with the earlier published data on bacterial biogeography (Ge et al., 2008; Schauer et al., 2010). The objectives of this study were to: (i) describe the distribution of fungal diversity in wetlands along the Changjiang River and in other independent wetlands in China; (ii) determine whether fungal communities exhibit habitat specificity; and (iii) determine the relative importance of historical contingencies and contemporary environmental factors on fungal communities.
Materials and methods
Sites and soil collection
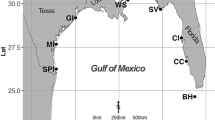
A total of 148 sediment samples involving 61 sampling areas from 20 wetlands were collected from typical wetlands along the Changjiang River and at other wetland locations throughout China in July 2009 (Figure 1 and Table 1). Of the 20 wetlands, 10 were part of the Changjiang River, and 10 were distant from the Changjiang River. Additional wetlands not associated with the Changjiang River were selected to achieve a wide geographic distribution across China and to obtain a range of wetland types. In addition, each wetland was considered typical, that is, representative of its type. Two to four sampling areas were randomly selected within each wetland, and two or three replicate sediment samples were collected from each sampling area. Sediment samples were collected along the bank of each wetland at depths of 0–40 cm. The samples were sealed in plastic bags, placed in a car refrigerator and immediately transported to the laboratory. The sediment samples were air-dried for soil physiochemical analysis and freeze-dried for the extraction of community DNA after passing through a 0.15-mm sieve. The longitude, latitude and altitude were recorded with an eXplorist 210 GPS (Magellan, San Dimas, CA, USA). The monthly mean temperatures were obtained from the China Meteorological Data Sharing Service System.

Geographic distribution of the wetland sites. Codes on the map are described in detail in Table 1. Pink triangles represent the sampling areas along the Changjiang River and yellow triangles represent wetlands at other sites in China. A full color version of this figure is available at the ISME Journal online.
Physiochemical analysis
The samples were assayed for organic carbon content, organic matter, total nitrogen, total potassium, total phosphorus and pH. Soil organic carbon content was analyzed by an H2SO4–K2Cr2O7 oxidation method (Nelson and Sommers, 1982); organic matter was determined by loss on ignition; total nitrogen was determined by carbon, hydrogen and nitrogen analysis; total potassium was analyzed by the sodium hydroxide melting method with flame photometers; total phosphorus was determined using the Murphy Riley method following a perchloric acid digestion (Gregorich and Carter, 2008); and pH was determined in 1:1 soil:water slurries with an acidometer (HANNA, Padova, Italy).
Fungal diversity as determined by a culture-dependent method
A 10-g subsample of each sediment was homogenized and serially diluted in sterile-distilled water to a final volume of 100 ml. After the suspensions were shaken for 30 min, 200-μl aliquots from the 10-, 100- and 1000-fold dilutions were plated onto 1/4 potato dextrose agar (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) containing 100 mg l−1 of streptomycin and 100 mg l−1 of chloromycetin. In all, 10 replicate plates were used for each dilution (30 plates per sample), and the cultures were kept at 25 °C for 1 week. Fungi with different colony morphologies were then selected and transferred to new potato dextrose agar plates with the antibiotics. After 10 days at 25 °C, isolates were identified based on morphological characteristics. Morphological identification was mainly based on the ‘Compendium of Soil Fungi’ (Domsch et al., 2007) and ‘Genera of Hyphomycetes’ (Seifert et al., 2011). The relative frequency of isolation, which was used as a measure of fungal species abundance, was calculated as the number of isolates of each taxon divided by the total number of isolates. Isolation rate was calculated as the number of isolates obtained from a specific wetland site divided by the total number of wetland samples. After colonies were identified based on morphology, hyphae were harvested for molecular identification as described in the next section.
Identification of fungal isolates based on ITS sequence analysis
After morphological identification of the sporulated isolates, the reliability of the initial morphological identifications was confirmed with phylogenetic analysis. For the sterile isolates, only the phylogenetic analysis was applied. After 10 days of growth on potato dextrose agar (see previous section), genomic DNA of each isolate was extracted with a simple and rapid ‘thermolysis’ method (Zhang et al., 2010), and the rDNA internal spacer region 1 (ITS1) regions were amplified with primers ITS1/ITS4 (White et al., 1990). The cycling conditions were conducted as described by Zhang et al. (2010).
Phylogenetic analysis was performed by comparing the ITS sequences in BLAST searches in GenBank with 97% homology for independent operational taxonomic unit (OTU). The best matching sequences were retrieved from the database and aligned, and a multiple sequence alignment was performed by Clustal X (European Bioinformatics Institute, Cambridge, UK) (Thompson et al., 1994). The phylogenetic tree was constructed using the neighbor-joining method implemented in Molecular Evolutionary Genetics Analysis (MEGA) 4.0 (The Biodesign Institute, Tempe, AZ, USA) (Tamura et al., 2007). The topologies of the resultant trees were evaluated by bootstrap analysis based on 1000 resamplings. Most OTUs were closely grouped with their BLAST database matches (Supplementary Figure S1). The rDNA sequences obtained from the isolates have been deposited in the GenBank database with accession nos. JX076933–JX077084.
Characterization of the fungal community based on a culture-independent method
Community DNA was extracted from the sediment samples using a bead-beater method with a mini-bead beater (Biospec Products, Bartlesville, OK, USA). The crude extracts were purified using the Gel Extraction Kit (Omega Bio-Tek, Norcross, GA, USA) according to the manufacturer’s instruction and were directly used for sequencing analysis. Then, the rDNA ITS1 regions were PCR amplified with the same protocol that was described for the culture-dependent method.
A 1-ml volume of diluted primary PCR product (1:500) was used as a PCR template in a nested PCR for DGGE analysis. The ITS1 fragment was amplified with primer ITS1-GC Clamp (Doaré-Lebrun et al., 2006) and ITS2 (White et al., 1990). The cycling conditions were followed as per methods outlined in Doaré-Lebrun et al. (2006).
DGGE analysis was performed using a Dcode Universal Mutation Detection System (Bio-Rad, Hercules, CA, USA) with a denaturing gradient of 30–50% in an 8% polyacrylamide gel, following the manufacturer’s instructions. The DGGE gels were run for 12 h at 80 V in 1 × TAE electrophoresis buffer (40 mM Tris-Cl, 1 mM EDTA, pH 8.0) at a constant temperature of 60 °C. After electrophoresis, the gels were stained with SYBR Green I nucleic acid gel stain (Invitrogen, Calsbad, CA, USA) for 30 min and were photographed with a Gel imaging system (Bio-Rad). The DGGE images were further analyzed using Quantity One (version 4.62; Bio-Rad, Munich, Germany) to determine the peak density of each band.
Community analysis
For assessment of fungal diversity and evenness in different sediments, the Shannon diversity index (H′), Simpson’s reciprocal index (1/D) and Pielou index (E) were calculated using the Biodap software (Gordon Thomas, Vancouver, BC, Canada). To represent graphically the evenness of fungal community in wetlands, Pareto-Lorenz (PL) evenness curves were constructed based on the DGGE profiles (Mertens et al., 2005). The functional organization (Fo), which is determined based on PL curves, is defined as ‘the ability of the community to organize in an adequate distribution of dominant microorganisms and resilient ones, condition that should assure the potentiality of counteracting the effect of a sudden stress exposure’ (Marzorati et al., 2008).
All data from DGGE profiles of different sediment samples were subjected to principal components analysis (PCA) to compare the molecular fingerprints of fungal communities in the sediments (Marzorati et al., 2008). For determination of the relationships between geochemical factors and fungal community structure based on DGGE profiles, redundancy analysis (RDA) was performed. Both PCA and RDA analyses were carried out using the Canoco Software (Version 4.5; Microcomputer Power, Ithaca, NY, USA).
Multivariate regression tree analysis
The single and combined effects of contemporary environmental conditions and historical contingencies on fungal diversity were explored by multivariate regression tree (MRT) analysis. A 1000 cross-validation process was used to decrease the structure complexity of MRT to predict the main relationship between multispecies data and environmental variables. Predictive accuracy is estimated from the cross-validated relative error, which varies from zero for a perfect tree to close to one for a poor tree (De’Ath, 2002). For the MRT analysis, dissimilarity among the response variables is defined as the total sum of squares of the response variable values, including H′, 1/D, E and Fo based on DGGE profiles. MRT analysis was carried out with the package ‘mvpart’ within the ‘R’ statistical programming environment.
The effect of geographic distance and the environmental parameters on fungal community composition was determined by analyzing the correlation between the geographic distance matrix and the fungal community dissimilarity matrix with a Mantel test (Mantel, 1967). The geographic distance matrix was calculated based on the latitude and longitude with the GRASS GI software (U.S. Army Corps of Engineers, Champaign, IL, USA) (Neteler, 1998). The dissimilarity matrix was calculated based on the DGGE profiles with SPSS 11.5 for Windows (SPSS Company, Chicago, IL, USA). The Mantel test was performed with XLSTAT 2012 for Windows (Pearson’s correlation, 10 000 permutations) with significance determined at P<0.05.
Results
Physiochemical characteristics of sediment samples
The sampling sites encompassed a wide range of stream characteristics across 4000 km. Average temperature of each sampling site ranged from 12.5 to 30.6 °C, with the lowest values in the sites located on the Qinghai-Tibetan Plateau and the highest in Guangdong Mangrove (Table 1). Sediment pH ranged from 5.9 to 8.7 and was lowest in Zimei Lake on the Qinghai-Tibetan Plateau (Table 1). Samples from Dali West Lake had the highest values of organic carbon content, organic matter, total nitrogen, C:N ratio and C:P ratio, and the lowest total potassium content (Table 1).
The relationship between altitude and temperature among wetlands was linear and negative (r2=0.71, P<0.001), but there were no significant relationship between any of the other environmental variables.
The fungal communities in wetland sediments as determined with a culture-dependent method
A total of 883 isolates were obtained and identified as belonging to 81 genera and 177 species based on morphological characteristics and phylogenetic analysis. The number of isolates ranged from 18 at Dali West Lake to 109 at Poyang Lake. Among the 177 species, 169 belonged to the Ascomycota, and the remaining species belonged to the Basidiomycota and the Zygomycota. The most frequently occurring genera were Penicillium (relative frequency=16.8%), followed by Fusarium (15.4%), Aspergillus (7.6%), Trichoderma (5.8%) and Talaromyces (4.2%); other genera accounted for 49.8% of the total fungal diversity (Supplementary Table S1).
Species of Penicillium, Fusarium and Aspergillus were dominant in sites along the Changjiang River, while species of Penicillium, Mortierella, Trichoderma, Cadophora and Dimorphospora were dominant in the Qinghai-Tibetan Plateau (Supplementary Figure S2). Although no species was detected in all sites, species of Penicillium, Fusarium, Aspergillus and Trichoderma were detected in 90.0, 75.0, 75.0 and 45.0%, respectively, of the sites. Of the 177 species, 89 were endemic and 40 were found in only two or three locations.
The fungal communities of wetland sediments as determined with a culture-independent method
The fungal communities were also analyzed by DGGE. OTU richness ranged from 14 at Dong Lake to 30 at Baiyangdian Lake, and fungal assemblages were very heterogeneous among sediments. In total, 145 distinct OTUs were detected by DGGE, but no OTU occurred in all sediments, which was consistent with results from the culture-dependent method. Of the 145 OTUs, 5% were detected in only one location, 10% were detected in two locations, and 14% in three locations.
Comparison of fungal communities along the Changjiang River vs those distant from the Changjiang River based on culture-independent data
When data from samples along the Changjiang River were analyzed by PCA ordination, the fungal communities formed three clusters (Figure 2), which closely corresponded to samples from upstream, midstream and downstream. The Changjiang River originates in the glaciers on the Qinghai-Tibetan Plateau and flows across Southern China before emptying into the East China Sea. The upstream samples, which were collected from sampling areas on the Qinghai-Tibetan Plateau, formed an independent cluster, while the samples from midstream and downstream were more similar to each other than they were to samples from upstream. Fungal diversity as determined by the Shannon index and the Simpson reciprocal index increased from upstream to downstream along the Changjiang River (Table 1).

PCA analysis based on DGGE profiles of the fungal communities along the Changjiang River. Each point indicates a sampling area in wetlands. Distances between any two points on the graph indicate the ecological distance between the community compositions. Numbers within parentheses are the percentage variance explained by each principal component. Circles, squares and triangles indicate samples from upstream, midstream and downstream locations, respectively, along the Changjiang River.
Samples from all 20 sites were used to investigate the relationships between environmental variables and fungal community composition by RDA. At least three general geographic clusters were recognized, that is, Southern China, Northern China and the Qinghai-Tibetan Plateau (Figure 3). The Qinghai-Tibetan Plateau is a separate elevated plateau in Western China and has an average elevation exceeding 4500 m; the samples from this area formed an independent cluster. Other samples were geographically divided into a Southern China cluster and a Northern China cluster by a line formed by the Huai River-Qin Mountains.

Relationships between fungal communities and environmental factors according to RDA based on DGGE profiles. Symbols indicate different sampling areas in wetlands, and the arrows indicate the types of environmental factors and their relative effects on fungal communities. The distance between symbols reflects their dissimilarity, and the relative position (in perpendicular distance) of a symbol to an arrow-line indicates the influence of the specified environmental factor on the fungal communities in different wetlands. Red, blue and green circles indicate samples from Southern China, Northern China and the Qinghai-Tibetan Plateau, respectively. A full color version of this figure is available at the ISME Journal online.
Biogeography: the relative influence of environmental and historical factors
MRT analysis was used to explain the relative effect of historical and contemporary environmental factors on the fungal communities from all samples (Figure 4). A visualized tree generated by MRT analysis showed 10 splits based on characters of sampling locations and soil chemical characters (cross-validated relative error=0.55). The tree explained 75.7% of the variance of the standardized diversity indices. All data were first clustered into two groups by status and this explained 43.3% of the variation in the original data set. One group contained the data from the Qinghai-Tibetan Plateau, and the other group contained the data from Southern and Northern China. The Qinghai-Tibetan Plateau group was split further by total potassium, which explained 9.4% of the variation. The groups from Southern and Northern China were then split by latitude into two groups that explained 4.5% variation. The samples from the shrub-dominated swamp of Guangdong Mangrove were separated, and then others were further grouped by latitude and split into Southern and Northern China, which explained 14.0% of the variation. Then, the diversity estimation of sediments from Southern China was split into six subgroups by total nitrogen (which explained 3.5% of the variation), phosphorus and potassium. The contributions of organic carbon content, C:N ratio, C:P ratio and pH were too small to be used in the MRT analysis.

MRT of the fungal diversity data associated with a total of 61 different sampling locations (Southern China, Northern China and the Qinghai-Tibetan Plateau) and environmental variables (organic carbon content, organic matter, total nitrogen, total potassium, total phosphorus, C:N ratio, C:P ratio, pH and temperature). Each split in the figure was represented graphically as a branch in a tree. Bar plots show the multivariate means of diversity estimates at each branch. The numbers of samples included in that splits are shown under bar plots. The average of the replications was applied for each sampling location. The standardized diversity estimates were used to construct the MRT. SC, Southern China; NC, Northern China; QTP, the Qinghai-Tibetan Plateau; OCC, organic carbon content; OM, organic matter; TN, total nitrogen; TK, total potassium; TP, total phosphorus; C/N, C:N ratio; C/P, C:P ratio. A full color version of this figure is available at the ISME Journal online.
MRT was also used to determine the effect of environmental factors on the patterns of fungal community structure along the Changjiang River (Figure 5). There were nine splits, and 91.0% of the variance was explained (cross-validated relative error=0.71). The tree showed that fungal diversity along the Changjiang River was first split by altitude, which explained 44.8% of the variation. Data from all upstream samples fell into one group, and data from mid- and downstream samples fell into another group. The diversity estimation of wetlands from upstream samples was then split by total potassium, and this split accounted for 14.3% of the variation in the data. The other group that included mid- and downstream samples was split by total phosphorus, which explained 11.6% of the variation.

MRT of environmental factors on the patterns of fungal community structure along the Changjiang River. Fungal community structure was indicated by H′, 1/D, E and Fo. The environmental factors included different sampling locations (upstream, midstream and downstream of the Changjiang River) and environmental variables (organic carbon content, organic matter, total nitrogen, total potassium, total phosphorus, C:N ratio, C:P ratio, pH and temperature). Each split in the figure was represented graphically as a branch in a tree. Bar plots show the multivariate means of diversity estimates at each branch. The numbers of samples included in that splits are shown under bar plots. The average of the replications was applied for each sampling location. U, M and D refer to upstream, midstream and downstream of the Changjiang River, respectively. OCC, organic carbon content; OM, organic matter; TN, total nitrogen; TK, total potassium; TP, total phosphorus; C/N, C:N ratio; C/P, C:P ratio. A full color version of this figure is available at the ISME Journal online.
Regardless of whether data from all 20 sites or from only the 10 Changjiang River sites were included, MRT analysis showed that the distribution of fungi was first clustered by sampling location at a relatively large scale. This finding was also well-supported by the Mantel test, which indicated that fungal community composition was positively correlated with geographic distance from all 20 sites (r=0.224, P=0.003), or from only the 10 Changjiang River sites (r=0.340, P=0.039). The environmental factors, such as total potassium, phosphorus and other physiochemical characteristics of the sediment samples determined fungal community composition at a local scale (<1000 km) but not at a large scale (4000 km).
Discussion
Community-level molecular techniques are widely used in comparative microbial ecology to assess the diversity of microbial communities and their response to changing environments (Marzorati et al., 2008), and DGGE is a well-established molecular tool for the study of microbial biogeography (Bates et al., 2012; Monroy et al., 2012; Schiaffino et al., 2011). Although culture-dependent methods are generally considered capable of detecting only a small portion of microbial communities (Marzorati et al., 2008), more fungi were detected in this study with the culture-dependent method than with DGGE. One explanation for this is the large number of plates (30) used for each sample processed by the culture-dependent method, which increased the probability of isolating fungi with low abundance. Among the 177 species isolated by the culture-dependent method, 65 were isolated only once, indicating that they were relatively rare. At the same time, three clones were sequenced from each main band clone library in different wetlands, and one band of DGGE may represent one, two or even three different taxa after BLAST in NCBI (data not shown). This may be another reason why the culture-independent method revealed fewer OTUs than the culture-dependent method in this study.
In addition to the potential for comigration of DNA fragments, DGGE has other limitations, that is, only relatively small fragments can be used (up to 500 bp), one species may form several bands, and only abundant species are detected (Marschner, 2007). In spite of the limitations, the parameters calculated from the DGGE fingerptints can be used as indicators of diversity in fungal or bacterial communities (Marzorati et al., 2008). Although high-throughput approaches (such as pyrosequencing and PhyloChip-based metagenomics) are probably superior to DGGE, the research described in this report was started before such approaches were readily available.
Biologists have generally assumed that species of microorganisms are essentially present everywhere but only proliferate under proper conditions (O’Malley, 2007); according to this view, the failure to detect a species at a particular location does not result from its absence but from inadequate conditions to support its proliferation. In a clone library of a sample, more clones sequenced, more OTUs detected (Buée et al., 2009). Although recent studies including this study have revealed that only a few fungal or bacterial taxa are ubiquitous, and that many OTUs occur only in particular environments or locations (Sogin et al., 2006; Pommier et al., 2007),we believe that many OTUs were widespread but were detected only in those locations due to the methodology shortcoming (Fuhrman, 2009).
Because a river carries sediment in its flows, it may have relatively similar environmental conditions and, consequently, similar microorganism assemblages along its length. Here, we hypothesized that there should be a gradual change of fungal communities along a river’s length as the sediment–river interface gradually changes. In this study, the fungal communities in upstream samples (most were located on the Qinghai-Tibetan Plateau) were substantially different from those in mid- and downstream samples from the Changjiang River based on the PCA and RDA analysis. The lack of gradual change between upstream and midstream samples may be attributed principally to the uplift of the Qinghai-Tibetan Plateau (Huang et al., 2008). Regression analysis indicated that altitude was negatively correlated with temperature (r2=0.71, P<0.001) and that altitude was negatively correlated with fungal diversity (r2=0.39, P<0.01). Previous studies have indicated that the uplift greatly influenced the climate and biogeography of the fauna and flora of the Qinghai-Tibetan Plateau (Shi et al., 1998) and that the Qinghai-Tibetan Plateau has high species richness and endemism (Huang et al., 2008; Wang et al., 2009). The intensive uplifts of the Qinghai-Tibetan Plateau are generally accepted to have significantly affected the development of the contemporary drainage systems (Li et al., 2000), which led to considerable habitat alteration that in turn greatly affected taxa in the region (Qi et al., 2007). The patterns of animal endemism, for example, were likely influenced by the recent uplift of the Qinghai-Tibetan Plateau (Huang et al., 2008). Sun and Liu (2008) found that the diversity of insect-associated fungi in the Qinghai-Tibetan Plateau is significantly different from that in other areas in China. We therefore suspect that the uplift of the Qinghai-Tibetan Plateau had a similar effect on fungal biogeography in this region, that is, the fungal communities of the Qinghai-Tibetan Plateau may be relatively isolated and therefore different from those in other parts of China. In contrast, if only the mid- and downstream parts of the Changjiang River are considered (Figure 2), the gradual change in the fungal communities is consistent with previously described changes in bacterial communities (Sekiguchi et al., 2002), which suggests that local environmental factors along a river may support similar assemblages if a physical barrier is absent (Hewson et al., 2007).
A long-standing topic in the study of biogeography is the relative influence of contemporary environmental factors vs the legacies of historical events on contemporary distribution patterns (Martiny et al., 2006). Recent studies have shown that the drivers of fungal and bacterial diversity differ with scale and microorganism type (Fierer et al., 2007; Pommier et al., 2007; Ge et al., 2008; Pagaling et al., 2009). MRT analysis in this study showed that the distribution of fungi in sediments along the Changjiang River and in other samples across China was significantly influenced by historical contingencies (sampling location). All historical factors accounted for 61.8% of the variation, indicating that geographic distance resulted in biogeographic provincialism of fungal diversity. The relationship between geographic distance and fungal community has been previously studied in arid soil, rhizosphere, leaves and other habitats (Green et al., 2004; Kivlin et al., 2011; van der Gast et al., 2011; Cordier et al., 2012) but not in wetlands. This study revealed that fungal community composition was positively correlated with geographic distance, and that geographic distance was a driver of diversity (r=0.224, P=0.003) at a large scale in wetlands. The effect of geographic distance on fungal biogeography in this study was consistent with results for bacterial biogeography in sediments (Pagaling et al., 2009; Schauer et al., 2010) and for the biogeography of macroorganisms in wetlands (Declerck et al., 2011). That the distributions of larger organisms are limited by mountains, large bodies of water and other physical barriers seems reasonable (Prosser et al., 2007). The results reported here indicate that physical barriers may also contribute to fungal geographic distribution.
While the fungal communities were different on the Qinghai-Tibetan Plateau than in the other locations, they also differed between Southern and Northern China, regions that are separated by the Huai River and Qin Mountains. A large body of literature has documented that the Qin Mountains act as a barrier and result in differences in climates (Jin et al., 2003), plant diversity (Zhang and Zhang, 1979) and human genetic diversity between Southern and Northern China (Xue et al, 2008). The Qin Mountains are sufficiently high to protect the area to the south from the cold to the north, resulting in different plant flora to the north and south (Kang and Zhu, 2007). Because plants often represent important sources of nutrition for fungi, the distribution of fungal communities may reflect the distribution of plant communities (Zachow et al., 2009).
Because they are generally larger, fungi are more likely to be restricted by geographical barriers than bacteria and archaea (Finlay, 2002). Although fungi can produce large numbers of spores, successful long-distance dispersal events of such spores are probably rare (Brown and Hovmoller, 2002) because most fungal spores are sensitive to environmental changes. Fungi are unlikely to survive for a long time after release and disperse over the Qin Mountains.
At a relatively small scale, bacterial communities were affected only by environmental factors or by both environmental and historical factors (Horner-Devine et al., 2004; Ramette and Tiedje, 2007). In this study, some environmental factors, such as total nitrogen, potassium and phosphorus, explained 13.8% of the variation in the fungal communities at the relatively small scale of <1000 km (Figure 4). This indicates that contemporary factors can influence fungal diversity within the overall pattern of provincialism caused by historical contingencies (Ge et al., 2008). Researchers have recently argued that the relative effects of historical contingencies and environmental disturbance on microorganisms are scale-dependent, which is similar to the case with macroorganisms (Williams et al., 2002). A recent study supported the view that historical events were the dominant drivers of bacterial diversity at a regional scale (>1000 km), but that environmental factors were the dominant drivers at a local scale (Ge et al., 2008). These results with fungi provide more evidence that the relative effects of environment vs history are scale dependent.
In summary, the effects of contemporary disturbances and historical contingencies on fungal communities have been infrequently studied. We conclude that fungal diversity in wetland sediments in China is high, and we infer that the variation in fungal diversity is driven by historical contingencies (natural barriers) at a regional scale (1000–4000 km) and by environmental factors at a small scale (<1000 km).
Accession codes
References
Bates ST, Nash TH, Garcia-Pichel F . (2012). Patterns of diversity for fungal assemblages of biological soil crusts from the southwestern United States. Mycologia 104: 353–361.
Beijerinck M . (1913) De Infusies en de Ontdekking der Backterien, Jaarboek van de Koninklijke Akademie v. Wetenschappen, Muller: Amsterdam, Netherlands.
Brown JKM, Hovmoller MS . (2002). Epidemiology—aerial dispersal of pathogens on the global and continental scales and its impact on plant disease. Science 297: 537–541.
Buée M, Reich M, Murat C, Morin E, Nilsson RH, Uroz S et al (2009). 454 Pyrosequencing analyses of forest soils reveal an unexpectedly high fungal diversity. New Phytol 184: 449–456.
Bärlocher F, Stewart M, Ryder DS . (2011). Analyzing aquatic fungal communities in Australia: impacts of sample incubation and geographic distance of streams. Czech Mycol 63: 113–132.
Cordier T, Robin C, Capdevielle X, Desprez-Loustau M-L, Vacher C . (2012). Spatial variability of phyllosphere fungal assemblages: genetic distance predominates over geographic distance in a European beech stand (Fagus sylvatica). Fungal Ecol 5: 509–520.
De'Ath G . (2002). Multivariate regression trees: a new technique for modeling species–environment relationships. Ecology 83: 1105–1117.
Declerck SAJ, Coronel JS, Legendre P, Brendonck L . (2011). Scale dependency of processes structuring metacommunities of cladocerans in temporary pools of High-Andes wetlands. Ecography 34: 296–305.
Doaré-Lebrun E, El Arbi A, Charlet M, Guérin L, Pernelle JJ, Ogier JC et al (2006). Analysis of fungal diversity of grapes by application of temporal temperature gradient gel electrophoresis—potentialities and limits of the method. J Appl Microbiol 101: 1340–1350.
Domsch KH, Gams W, Anderson TH (2007) Compendium of Soil Fungi. Academic Press: New York, NY, USA.
Fenchel T, Finlay JB . (2005). Bacteria and island biogeography. Science 309: 1997–1999.
Fierer N, Jackson RB . (2006). The diversity and biogeography of soil bacterial communities. Proc Natl Acad Sci USA 103: 626–631.
Fierer N, Morse JL, Berthrong ST, Bernhardt ES, Jackson RB . (2007). Environmental controls on the landscape-scale biogeography of stream bacterial communities. Ecology 88: 2162–2173.
Finlay BJ. . (2002). Global dispersal of free-living microbial eukaryote species. Science 296: 1061–1063.
Fuhrman JA, Steele JA, Hewson I, Schwalbach MS, Brown MV, Green JL et al (2008). A latitudinal diversity gradient in planktonic marine bacteria. Proc Natl Acad Sci USA 105: 7774–7778.
Fuhrman JA . (2009). Microbial community structure and its functional implications. Nature 459: 193–199.
Ge Y, He JZ, Zhu YG, Zhang JB, Xu Z, Zhang LM et al (2008). Differences in soil bacterial diversity: driven by contemporary disturbances or historical contingencies? ISME J 2: 254–264.
Green JL, Holmes AJ, Westoby M, Oliver I, Briscoe D, Dangerfield M et al (2004). Spatial scaling of microbial eukaryote diversity. Nature 432: 747–750.
Gregorich EG, Carter MR . (2008) Soil Sampling and Methods of Analysis. CRC Press: Boca Raton, FL, USA.
Harvey JW, Fuller CC . (1998). Effect of enhanced manganese oxidation in the hyporheic zone on basin-scale geochemical mass balance. Water Resour Res 34: 623–636.
Hewson I, Fuhrman JA . (2004). Richness and diversity of bacterioplankton species along an estuarine gradient in Moreton Bay, Australia. Appl Environ Microbiol 70: 3425–3433.
Hewson I, Jacobson-Meyers ME, Fuhrman JA . (2007). Diversity and biogeography of bacterial assemblages in surface sediments across the San Pedro Basin, Southern California Borderlands. Environ Microbiol 9: 923–933.
Horner-Devine MC, Lage M, Hughes JB, Bohannan BJM. (2004). A taxa–area relationship for bacteria. Nature 432: 750–753.
Huang XL, Lei FM, Qiao GX . (2008). Areas of endemism and patterns of diversity for aphids of the Qinghai-Tibetan Plateau and the Himalayas. J Biogeogr 35: 230–240.
Jin JH, Liao WB, Wang BS, Peng SL . (2003). Global change in Cenozoic and evolution of flora in China (in Chinese). Guihaia 23: 217–225.
Kang MY, Zhu Y . (2007). Discussion and analysis on the geo-ecological boundary in Qinling Range (in Chinese). Acta Ecol Sin 27: 2774–2784.
Kivlin SN, Hawkes CV, Treseder KK . (2011). Global diversity and distribution of arbuscular mycorrhizal fungi. Soil Biol Biochem 43: 2294–2303.
Li CG, Yin HF, Yu QW . (2000). Evolution of drainage systems and its developing trend in connection with tectonic uplift of Eastern Kunlun Mt. Chinese Sci Bull 45: 1904–1908.
Mantel N . (1967). The detection of disease clustering and a generalized regression approach. Cancer Res 27: 209–220.
Marschner P . (2007). Soil microbial community structure and function assessed by FAME, PLFA and DGGE—advantages and limitations. In: Varma A, Oelmüller R, (eds) Advanced Techniques in Soil Microbiology. Springer: USA, pp 181–200.
Martiny JBH, Bohannan BJM, Brown JH, Colwell RK, Fuhrman JA, Green JL et al (2006). Microbial biogeography: putting microorganisms on the map. Nat Rev Microbiol 4: 102–112.
Marzorati M, Wittebolle L, Boon N, Daffonchio D, Verstraete W . (2008). How to get more out of molecular fingerprints: practical tools for microbial ecology. Environ Microbiol 10: 1571–1581.
Mertens B, Boon N, Verstraete W . (2005). Stereospecific effect of hexachlorocyclohexane on activity and structure of soil methanotrophic communities. Environ Microbiol 7: 660–669.
Mohamed DJ, Martiny JBH . (2011). Patterns of fungal diversity and composition along a salinity gradient. ISME J 5: 379–388.
Monroy F, van der Putten WH, Yergeau E, Mortimer SR, Duyts H, Bezemer TM . (2012). Community patterns of soil bacteria and nematodes in relation to geographic distance. Soil Biol Biochem 45: 1–7.
Nelson DW, Sommers LE . (1982). Dry combustion method using medium temperature resistance furnace. In: Page AL, (ed) Methods of Soil Analysis. Part 2: Chemical and Microbial Properties 2nd edn. Soil Science Society of American and American Society: Madison, WI, USA, pp 539–579.
Neteler M . (1998) Introduction to GRASS GIS Software. University of Hannover: Hannover, Germany.
O’Malley MA . (2007). The nineteenth century roots of ‘everything is everywhere’. Nat Rev Microbiol 5: 647–651.
Pagaling E, Wang H, Venables M, Wallace A, Grant WD, Cowan DA et al (2009). Microbial biogeography of six salt lakes in Inner Mongolia China and a salt lake in Argentina. Appl Environ Microbiol 75: 5750–5760.
Pommier T, Canback B, Riemann L, Bostrom KH, Simu K, Lundberg P et al (2007). Global patterns of diversity and community structure in marine bacterioplankton. Mol Ecol 16: 867–880.
Prosser JI, Bohannan BJM, Curtis TP, Ellis RJ, Firestone MK, Freckleton RP et al (2007). Essay—the role of ecological theory in microbial ecology. Nat Rev Microbiol 5: 384–392.
Qi D, Guo S, Zhao X, Yang J, Tang W . (2007). Genetic diversity and historical population structure of Schizopygopsis pylzovi (Teleostei: Cyprinidae) in the Qinghai-Tibetan Plateau. Freshwater Biol 52: 1090–1104.
Ramette A, Tiedje JM . (2007). Multiscale responses of microbial life to spatial distance and environmental heterogeneity in a patchy ecosystem. Proc Natl Acad Sci USA 104: 2761–2766.
Reche I, Pulido-Villena E, Morales-Baquero R, Casamayor EO . (2005). Does ecosystem size determine aquatic bacterial richness? Ecology 86: 1715–1722.
Schauer R, Bienhold C, Ramette A, Harder J . (2010). Bacterial diversity and biogeography in deep-sea surface sediments of the South Atlantic Ocean. ISME J 4: 159–170.
Schiaffino MR, Unrein F, Gasol JM, Massana R, BalaguÉ V, Izaguirre I (2011). Bacterial community structure in a latitudinal gradient of lakes: the roles of spatial versus environmental factors. Freshwater Biol 56: 1973–1991.
Seifert KA, Morgan-Jones G, Gams W, Kendrick B . (2011) The Genera of Hyphomycetes. Centraalbureau voor Schimmelcultures: Utrecht, Netherlands.
Sekiguchi H, Watanabe M, Nakahara T, Xu B, Uchiyama H . (2002). Succession of bacterial community structure along the Changjiang River determined by denaturing gradient gel electrophoresis and clone library analysis. Appl Environ Microbiol 68: 5142–5150.
Shi YF, Li JJ, Li BY . (1998) Uplift and Environmental Change of Qinghai-Xizang (Tibetan) Plateau in the Late Cenozoic (in Chinese). Guangdong Science and Technology Press: Guangzhou, China.
Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, Neal PR et al (2006). Microbial diversity in the deep sea and the underexplored ‘rare biosphere’. Proc Natl Acad Sci USA 103: 12115–12120.
Sun BD, Liu XZ . (2008). Occurrence and diversity of insect-associated fungi in natural soils in China. Appl Soil Ecol 39: 100–108.
Sutcliffe JV, Parks YP . (1989). Comparative water balances of selected African wetlands. Hydrol Sci J 34: 49–62.
Tamura K, Dudley J, Nei M, Kumar S . (2007). MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) Software Version 4.0. Mol Biol Evol 24: 1596–1599.
Thompson JD, Higgins DG, Gibson TJ . (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673–4680.
Thormann MN . (2006). Diversity and function of fungi in peatlands: a carbon cycling perspective. Can J Soil Sci 86: 281–293.
van der Gast CJ, Gosling P, Tiwari B, Bending GD . (2011). Spatial scaling of arbuscular mycorrhizal fungal diversity is affected by farming practice. Environ Microbiol 13: 241–249.
Wang L, Abbott RJ, Zheng W, Chen P, Wang Y, Liu J . (2009). History and evolution of alpine plants endemic to the Qinghai-Tibetan Plateau: Aconitum gymnandrum (Ranunculaceae). Mol Ecol 18: 709–721.
White TJ, Bruns T, Lee S, Taylor J . (1990). Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, (ed) PCR Protocols: A Guide to Methods and Applications. Academic Press: New York, NY, USA, pp 315–322.
Williams SE, Marsh H, Winter J . (2002). Spatial scale, species diversity, and habitat structure: small mammals in australian tropical rain forest. Ecology 83: 1317–1329.
Xue F, Wang Y, Xu S, Zhang F, Wen B, Wu XS et al (2008). A spatial analysis of genetic structure of human populations in China reveals distinct difference between maternal and paternal lineages. Eur J Hum Genet 16: 705–717.
Zachow C, Berg C, Mueller H, Meincke R, Komon-Zelazowska M, Druzhinina IS et al (2009). Fungal diversity in the rhizosphere of endemic plant species of Tenerife (Canary Islands): relationship to vegetation zones and environmental factors. ISME J 3: 79–92.
Zhang XZ, Zhang ZY . (1979). A preliminary discussion on the northern boundary of subtropical zone in China: based on the distribution of broad-leaf woody evergreens on the Qingling Mountain (in Chinese). Acta Geograph Sin 34: 342–352.
Zhang YJ, Zhang S, Liu XZ, Wen HA, Wang M . (2010). A simple method of genomic DNA extraction suitable for analysis of bulk fungal strains. Lett Appl Microbiol 51: 114–118.
Acknowledgements
This research was jointly supported by the Natural Science Foundations of China (No. 41101238) and 973 Program (No. 2009CB522302). We thank Prof. Bruce Jaffee (University of California at Davis) for his critical comments and clarifications of the language.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
About this article
Cite this article
Wu, B., Tian, J., Bai, C. et al. The biogeography of fungal communities in wetland sediments along the Changjiang River and other sites in China. ISME J 7, 1299–1309 (2013). https://doi.org/10.1038/ismej.2013.29
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2013.29
Keywords
This article is cited by
-
Differences of bacterioplankton communities between the source and upstream regions of the Yangtze River: microbial structure, co-occurrence pattern, and environmental influencing factors
Brazilian Journal of Microbiology (2024)
-
Impact of environmental factors on diversity of fungi in sediments from the Shenzhen River Estuary
Archives of Microbiology (2023)
-
Fungal diversity notes 1611–1716: taxonomic and phylogenetic contributions on fungal genera and species emphasis in south China
Fungal Diversity (2023)
-
Seasonal variation significantly affected bacterioplankton and eukaryoplankton community composition in Xijiang River, China
Environmental Monitoring and Assessment (2022)
-
Distinct assembly mechanisms of microbial sub-communities with different rarity along the Nu River
Journal of Soils and Sediments (2022)

{kind=link}