
Abstract
Photosynthetic microbial mats possess extraordinary phylogenetic and functional diversity that makes linking specific pathways with individual microbial populations a daunting task. Close metabolic and spatial relationships between Cyanobacteria and Chloroflexi have previously been observed in diverse microbial mats. Here, we report that an expressed metabolic pathway for the anoxic catabolism of photosynthate involving Cyanobacteria and Chloroflexi in microbial mats can be reconstructed through metatranscriptomic sequencing of mats collected at Elkhorn Slough, Monterey Bay, CA, USA. In this reconstruction, Microcoleus spp., the most abundant cyanobacterial group in the mats, ferment photosynthate to organic acids, CO2 and H2 through multiple pathways, and an uncultivated lineage of the Chloroflexi take up these organic acids to store carbon as polyhydroxyalkanoates. The metabolic reconstruction is consistent with metabolite measurements and single cell microbial imaging with fluorescence in situ hybridization and NanoSIMS.
Similar content being viewed by others

Introduction
Linking biogeochemical processes observed in natural microbial communities with their associated metabolic pathways and assigning these pathways to specific groups remains a daunting task, particularly as community complexity increases (Kunin et al., 2008a). Photosynthetic microbial mats found in marine and hypersaline environments are stratified microbial communities that harbor extraordinary phylogenetic and functional diversity, harboring up to 42 phyla, including many candidate divisions (Nicholson et al., 1987; Ley et al., 2006; Kunin et al., 2008b; Dillon et al., 2009). Extensive measurements have been performed on these communities, documenting biogeochemical cycling of carbon, nitrogen and sulfur (Jorgensen and Des Marais, 1986; Canfield and Marais, 1993; Hoehler et al., 2001; Hoehler et al., 2002; Decker et al., 2005). The microbial populations involved in these biogeochemical cycles have been primarily inferred based on the physiology of isolates and expression of individual functional genes (Omoregie et al., 2004; Green et al., 2008; Orphan et al., 2008). However, a comprehensive understanding of element cycling in the mats requires reconstruction of complete metabolic pathways expressed by specific microbial groups.
Photosynthetic microbial mats perform light driven carbon fixation during daylight hours (Bebout et al., 2002). Photosynthate is accumulated, often as glycogen, through both oxygenic and anoxygenic photosynthesis (Nicholson et al., 1987). This photosynthate is fermented to organic acids and H2 under anoxic conditions at night (Hoehler et al., 2002; Burow et al., 2012). The flux through these fermentation products varies up to four orders of magnitude between day and night (Hoehler et al., 2001). Recent studies of mats collected at Elkhorn Slough, Monterey Bay, CA, USA, has provided persuasive evidence that Microcoleus spp., the most abundant cyanobacterial group in the mats, are the dominant fermenters (Burow et al., 2012). However, the pathways through which Microcoleus spp. ferment photosynthate are unknown, as are microbial groups and pathways that may further metabolize the organic acids derived from fermentation. Relative measures of activity comparing rRNA transcripts to genes in samples from the Elkhorn Slough mats have indicated that Chloroflexi are among the most active microbial group under dark, anoxic conditions (Burow et al., 2012). Studies in thermophilic mat systems collected in Yellowstone National Park, which are much less complex than marine and hypersaline mats, have suggested a link between anoxic fermentation of accumulated photosynthate by Cyanobacteria and carbon uptake by Chloroflexi (van der Meer et al., 2003, 2005, 2007). The intimate physical association of Cyanobacteria and Chloroflexi was demonstrated in hypersaline mats collected from Guerrero Negro, Baja California, where fluorescence in situ hybridization was used to visualize Chloroflexi filaments within the exopolysaccharide sheaths of Microcoleus spp. (Ley et al., 2006). The close links between Cyanobacteria and Chloroflexi observed in other microbial mat environments suggest that this association may be a general phenomenon observed in mats and that this association may play an important role in anoxic carbon cycling.
Metatranscriptomic analysis has been used to characterize active microbial communities in aquatic and terrestrial environments (Poretsky et al., 2005; Frias-Lopez et al., 2008; Gilbert et al., 2008; Urich et al., 2008; Turnbaugh et al., 2010). Gene expression from microbial assemblages has been correlated with biogeochemical data and the phylogeny of expressed genes affiliated with specific populations (Poretsky et al., 2009, 2010; McCarren et al., 2010; Hollibaugh et al., 2011; Liu et al., 2011; Mou et al., 2011; Shi et al., 2011). In engineered microbial systems where complexity is reduced, the expression of specific pathways has been defined and correlated with environmental conditions (He et al., 2010). Metatranscriptomics of microbial mats offer the possibility of defining key metabolic pathways in the biogeochemical cycling of carbon and of establishing a link in the carbon cycle between Cyanobacteria and Chloroflexi.
Here, we demonstrate that metatranscriptomic data obtained from Elkhorn Slough microbial mat samples collected under dark anoxic conditions were dominated by transcripts from Cyanobacteria and Chloroflexi. This density of sequenced transcripts allowed us to reconstruct pathways for anoxic carbon catabolism that link fermentation in the Cyanobacteria with organic acid uptake and carbon storage in the Chloroflexi. These metabolic pathway inferences were supported by 13C-acetate isotopic labeling studies of the Elkhorn Slough mat coupled with single cell microbial imaging.
Materials and methods
Sample site and collection
The sampling site was located in the Elkhorn Slough estuary in Central California, USA, at 36N 48′ 46.61″ (decimal degree Lat 36.81295) and −121S 47′ 4.89″ (decimal degree Long −121.7847). Microbial mats were sampled on 10 January 2009 and transported to a greenhouse facility at NASA Ames Research Center within 1–2 h. In the greenhouse, mat pieces were placed in acrylic aquaria transparent to ultraviolet radiation and covered with water for ca. 20 h before the beginning of a diel cycle study (Bebout et al., 2002; Burow et al., 2012). Diel cycle studies were carried out under natural solar irradiance, and the water temperature was kept constant at ca. 18 °C (in situ average). The data presented result from diel cycle studies carried out on the 12th/13th January and 13th/14th January 2009.
Biogeochemical assays
Replicate vials were prepared identically as follows: small subcores (11-mm diameter, 15-mm depth or vertically sectioned for depth profile analyses) were cut from whole sections of intact microbial mat and placed in serum vials with 4 ml of field site water. Serum vials were capped with butyl rubber stoppers. The 10.5-ml headspace of the serum vials was left as air for light/daytime incubations and was thoroughly flushed with N2 (gas and liquid phase degassed) for dark/nighttime incubations. H2 was allowed to accumulate in three replicate vials and was repeatedly sampled at specific time points over the course of the diel experiment. Organic acids were allowed to accumulate in replicate vials (separate to vials in which H2 was measured), with three vials sacrificed at each time point. Sections of microbial mat were also incubated individually in triplicate vials as described above after sectioning to identify the location of H2 production. Sectioning using a sterile scalpel blade yielding the following mat layers; 0–2 mm, 2–4 mm and 4–15 mm. To measure H2, 25 μl of headspace gas was withdrawn by volumetric syringe and injected directly onto a gas chromatograph with quantification by a mercuric oxide detector. To analyze organic acids, the entire liquid phase (4 ml) was sampled (with the associated incubation sacrificed). Liquid was filtered through 0.2 μm syringe-driven filters for storage in glass vials at –20 oC. Organic acids (C1–5) were quantified via high-pressure liquid chromatography (Albert and Martens, 1997).
Nucleic acid extraction from Elkhorn Slough mat cores
Ten mat cores of 1-cm diameter were flash frozen in liquid nitrogen at multiple time points during the diel cycle and stored at −80 °C until further processing. RNA of two samples (BN; 2100 hours, 12 January 2009; 4 h after sunset, and EN; 0700 hours, 13 January 2009, 20 min before dawn) was extracted from the uppermost 2 mm of 5 mat cores by combining phenol–chloroform extraction with parts of the RNeasyMini kit (Qiagen, Valencia, CA, USA). Per core, biomass was transferred in a tube containing 0.5 ml RLT buffer and homogenized using a rotor-stator homogenizer (Omni International, Kennesaw, GA, USA). The suspension was then bead-beated with zirconium beads (200 μm, OPS Diagnostics, Lebanon, NJ, USA) and the cell debris and beads pelleted. The supernatant was extracted with phenol–chloroform–isoamyl alcohol (125:24:1, pH 4.5, Ambion, Austin, TX, USA). The aqueous phase was run through the gDNA eliminator spin column to remove genomic DNA and further purified following the RNeasyMini kit protocol. Extracted RNA was treated with DNase using the TURBO DNA-free kit according to the protocol (Ambion). Amplification, sequencing and sequence analysis of 16S rRNA genes and transcripts is described in Supplementary Information.
Selective removal of rRNA and cDNA synthesis
The MICROBEnrich and MICROBExpress Kits (Ambion) were used to remove the ribosomal RNAs (rRNA) by a subtractive hybridization approach, thereby enriching the messenger RNA (mRNA) in the total RNA pool. In the MICROBEnrich protocol, seven additional eukaryotic capture probes, and in the MICROBExpress Kit, five additional cyanobacterial capture probes were used. The sequences can be found in Supplementary Information. Approximately 400 ng of mRNA were linearly amplified with the MessageAmp II-Bacteria Kit (Ambion) according to the manufacturer’s instructions. In all, 4 μg of the amplified, antisense RNA (aRNA) were converted to double-stranded cDNA with random hexamers in multiple replicates using the SuperScript Double-Stranded cDNA Synthesis Kit (Invitrogen, Carlsbad, CA, USA) and purified with DNA Purification Spin Columns.
The quality and quantity of total RNA, mRNA, aRNA and cDNA were verified by measurements on the NanoDrop-1000 Spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA), with the Qubit fluorometer (Life Technologies, Grand Island, NY, USA) and the Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA).
Metatranscriptomic sequencing and analysis
Double-stranded cDNA of the two samples (BN, 12 January 2009, 2100 hours and EN, 13 January 2009, 0700 hours) were sequenced at the Department of Energy Joint Genome Institute (JGI, Walnut Creek, CA, USA) on a 454 Genome Sequencer FLX Instrument (454 Life Sciences, Branford, CT, USA). After sequencing, ribosomal RNA reads were detected by BLAST search and removed from the data set. Non-rRNA sequences were analyzed for replicate sequences using the open-source program CD-HIT provided as a web-based tool at http://microbiomes.msu.edu/replicates according to a previously described protocol (Li and Godzik, 2006; Gomez-Alvarez et al., 2009). Replicates were defined as sequences sharing >99% nucleotide identity, with an allowable length difference of 2 bp, and a requirement that the first 3 bp of the replicate sequences be identical.
The BLASTX program in BLAST version 2.2.21 was used with dereplicated non-rRNA reads as query sequences against all amino-acid sequences in the microbial RefSeq release 45 (January 2011) database (Altschul et al., 1990; Pruitt et al., 2009). RefSeq BLAST hits with bitscores below 40 were removed, and the results were processed using MEGAN version 4.32 with default settings (Huson et al., 2007). Taxonomic assignments based on NCBI taxonomy and functional assignments based on SEED and KEGG were analyzed using graphical tools within MEGAN (Ogata et al., 1999; Overbeek et al., 2005).
Completeness of sampling estimates were made using Chao and ACE estimators in MOTHUR version 1.21.1 (Schloss et al., 2009). Read clusters used as input for MOTHUR were generated in two ways. First, reads with a top BLASTX hit to the same RefSeq protein sequence were considered part of the same mRNA transcript cluster and used to estimate absolute coverage. Second, reads unambiguously assigned by MEGAN to a single SEED functional category or species (as defined by NCBI taxonomy) were considered part of the same functional or species cluster and used to calculate functional and species sampling completeness, respectively.
FISH-SIMS
Small subcores (11-mm diameter, 15-mm depth) of the microbial mats were cut from whole sections of intact microbial mat and placed in serum vials with 4 ml water obtained from the field site. Serum vials were capped with butyl rubber stoppers and the headspace of the vials (10.5 ml) was thoroughly flushed with N2 (gas and liquid phase degassed) for dark/nighttime incubations. A stable isotope tracer, [2-13C]-acetate (99% 13C, Sigma, St Louis, MO, USA), was added to the liquid phase of vials containing live cores at a final concentration of 0.2 mM. As a control, cores pretreated with 4% paraformaldehyde (final concentration) were also incubated with 0.2 mM [2-13C]-acetate. The cores were incubated under dark, anoxic conditions for 10 h. The incubation was stopped by adding 4% paraformaldehyde to the live cores. Preliminary NanoSIMS analysis revealed that single cells in the live cores were labeled but not in the killed cores pretreated with paraformaldehyde. The method used to label Chloroflexi with fluorescent oligonucleotides was previously described (Woebken et al., 2012). Chloroflexi were targeted using equimolar amounts of the FISH (fluorescence in situ hybridization) probes CFX1223 and GNSB-941 (Björnsson et al., 2002). High-resolution secondary ion mass spectrometry (SIMS) was performed at LLNL with a Cameca NanoSIMS 50. The 13C/12C ratio was measured using 12C2 and 13C12C correced for the dimer abundances (Finzi-Hart et al., 2009).
Data deposit
The dereplicated metatranscriptomic sequences with rRNA sequences removed can be downloaded from IMG/M (http://img.jgi.doe.gov/cgi-bin/m/main.cgi) under the names Elkhorn Slough cyanobacterial mat night (2100 hours Metatranscriptome CGUI) (BN library) and Elkhorn Slough cyanobacterial mat day (0700 hours transcriptome CGUN) (EN library) at the tab ‘FASTA nucleic acid file for all scaffolds. 16S rRNA sequences obtained in this study are deposited under GenBank accession numbers JX002103– JX002655.
Results
Microbial mat characterization
H2 production was >10 × higher at night in the upper phototrophic layer (0–2 mm) in the Elkhorn Slough mat compared with H2 produced during the day or in the lower mat layers (Supplementary Figure 1a). Acetate, formate and propionate were concomitantly produced with H2, whereas lactate, butyrate and valerate were not detected (200 nM detection limit; Supplementary Figure 1b). H2 and organic acid measurements observed for the microbial mat sampled in January 2009 were similar to those previously reported from this field site in November 2009 (Burow et al., 2012).
The microbial community composition in the upper layer was assessed using amplicon pyrosequencing of the V8 region of SSU rRNA genes from a time point taken before sunrise (EN; 0700 hours, 13 January 2009); sequences grouping with 16 distinct phyla were observed (Supplementary Table 1 and Figure 1a). The active microbial community was investigated by amplicon pyrosequencing of the V8 region of SSU rRNA from cDNA derived from RNA extracted at the same time point (EN) and at the beginning of the dark period (BN; 2100 hours; 12 January 2009). The active communities were subsets of the total number of phyla (11 and 13 phyla, respectively) and dominated by Cyanobacteria. Chloroflexi sequences were present at low abundance in both the DNA and cDNA pyrotag libraries.
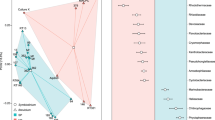
Nearly full-length 16S rRNA sequences were consistent with amplicon pyrosequencing of the cDNA (Figure 1a). Cyanobacterial sequences clustered into a few groups (Figure 2), of which the majority were related to Microcoleus chthonoplastes PCC 7420 and a cyanobacterium recently isolated from the Elkhorn Slough mats (ESFC-1) (Woebken et al., 2012). The Chloroflexi-related sequences were only distantly related to cultured representatives, but were closely related to sequences from uncultivated Chloroflexi recovered from hypersaline environment at Guerrero Negro, Baja California (Figure 3) (Ley et al., 2006).

(a) Comparison of nearly full-length Sanger and pyrotag amplicon 16S rRNA sequences of the phototrophic layer from Elkhorn Slough mat analyzed using the RDP classifier (Wang et al., 2007) with a confidence threshold of ⩾80%. Unassigned reads are sequences that could not be classified with a confidence threshold of ⩾80% on the phylum level. (b) Taxonomic classification of metatranscriptome reads analyzed by MEGAN using the RefSeq database (NCBI). Unassigned reads have an unknown taxonomic affiliation.

Neighbor joining tree of Cyanobacteria-related nearly full-length 16S rRNA sequences from cDNA samples recovered from BN (SIUS) and EN (SIUT) samples. OTU98 representatives of OTUs that contain more than one sequence (numbers of sequences per OTU in parenthesis) are depicted in the tree. Bootstrap values calculated with the PhyML algorithm that were ⩾50% are displayed in the tree. Scale bar represents 10% estimated sequence divergence.

Neighbor joining tree of Chloroflexi-related nearly full-length 16S rRNA sequences from cDNA samples BN (SIUS) and EN (SIUT). Elkhorn Slough mat sequences group with sequences retrieved from other microbial mat sites and are only distantly related with cultured Chloroflexi. Bootstrap values calculated with the PhyML algorithm that were ⩾50% are displayed in the tree. Scale bar represents 10% estimated sequence divergence.
Metatranscriptome sequence analysis
A total of 590 783 and 492 302 metatranscriptome sequence reads were generated for the BN and EN time points of which 43% (BN) and 32% (EN) were non-ribosomal sequences (Supplementary Table 2). After removal of these rRNA sequences and dereplication, 244 004 (BN) and 144 017 (EN) sequences remained for further analysis.
Coverage of the total pool of unique mRNA transcripts in the samples was analyzed in three ways: absolute, functional and species-level taxonomic. Absolute coverage was estimated by BLAST against the microbial RefSeq database, where reads yielding top BLAST hits from the same RefSeq amino-acid sequence were considered part of the same mRNA transcript. This RefSeq BLAST is likely to underestimate transcript diversity, as only 64.6% (BN) and 68.7% (EN) of reads for each sample showed significant similarity (bitscore >40) to any RefSeq sequence (Supplementary Table 3). Collector’s curves showed that sampling of the total number of unique mRNA transcripts is far from complete (Supplementary Figure 2a).
Estimations of functional and species-level coverage were obtained using the unambiguous assignment by MEGAN of reads to SEED functional categories or species. Although only 37.9–40.4% of reads were given a functional assignment and 15.6–22.9% given an unambiguous species-level assignment, both time points were sampled relatively completely with respect to the functional and species-level diversity presently described in RefSeq (Supplementary Table 3; Supplementary Figure 2b). These results are likely a reflection of the limited scope of the SEED and RefSeq databases compared with the full natural range of functions and species, rather than that of the true completeness of sampling. Moreover, although functions and species were well sampled independently, by no means were all the functions for every species sampled as well. However, these results do mean that these metatranscriptomic samples are broadly representative of those functions and species present in both the sample and SEED/RefSeq databases.
Abundant taxa in metatranscriptomes
The taxonomic composition of the microbial mat was assessed using the MEGAN software package for all protein-coding transcripts. Both metatranscriptomes were dominated by reads assigned to Cyanobacteria and Chloroflexi, the former constituting 22–47% of the reads, the latter 15–33% (Figure 1b). Proteobacteria and Bacteroidetes-affiliated reads were the next abundant groups. A large proportion of the reads were recruited to the M. chthonoplastes PCC 7240 genome. Of the 8294 predicted protein-coding genes in the M. chthonoplastes genome, 4030 (49%) were detected in the BN library and 1821 (24%) were detected in the EN library. Estimates of richness using Chao and ACE indices predicted the expression of 4978 and 4948 M. chthonoplastes protein-coding genes in the BN transcriptome, suggesting 81% (Chao) or 80% (ACE) coverage. These indices predicted 62% (Chao) and 48% (ACE) coverage in the EN transcriptome. Transcripts associated with Microcoleus spp. represented 24% (n=37 524) of all BN transcripts with matches to the RefSeq database (n=157 525) and 8% (n=7581) of all EN transcripts with matches to the RefSeq database (n=98 894). These reads are not necessarily all derived from a single Microcoleus relative, and may reflect a diverse pool of Microcoleus spp. that are more closely related to M. chthonoplastes PCC 7240 than to any other sequenced cyanobacterial genome. Unlike the Microcoleus spp., the Chloroflexi populations present in the mat samples did not have a closely related genome for fragment recruitment. Within the metatranscriptome reads affiliated with the phylum Chloroflexi, 96% were assigned to the class Chloroflexi at both time points, and ca. 85% of those reads further to the order Chloroflexales (BN; n=18 769 and EN; n=26 981, respectively; Supplementary Figure 3).
Anoxic carbon catabolism by Cyanobacteria and Chloroflexi
Transcript reads from Microcoleus spp. associated with carbohydrate metabolism were overrepresented when compared with carbohydrate reads observed for the whole community (BN; n=18% and EN; n=22%; Supplementary Figure 4). To elucidate the carbon catabolic pathways in the Microcoleus populations in the mat samples, fermentation pathways for conversion of glycogen were reconstructed from the M. chthonoplastes genome (Figure 4a; Supplementary Tables 4 and 5). Genes for glycogen phosphorylase and the EMP (Embden-Meyerhof-Parnas) pathway coded for proteins for the conversion of stored photosynthate, glycogen, to pyruvate. Genes for pyruvate transformation via three pathways were identified: to lactate (lactate dehydrogenase), to formate and acetyl-CoA (pyruvate-formate lyase) and to acetyl-CoA and CO2 (pyruvate-ferredoxin oxidoreductase). Genes to transform acetyl-CoA to ethanol (alcohol/aldehyde dehydrogenase) and acetate (acetate kinase) were identified. No annotated phosphotransacetylase was detected and no reads that cluster with other cyanobacterial phosphotransacetylases were identified in the transcript reads. The catalytic [NiFe] Hox hydrogenase was identified as part of a predicted seven gene operon with NAD(P)H dehydrogenase subunits (hoxEFUYH), a predicted aldehyde/alcohol dehydrogenase (adhE) and a hypothetical protein. Transcripts for all the genes described above were observed in the BN and EN libraries (Figure 5; Supplementary Table 6). Higher numbers of reads for the fermentative pathway genes were observed in the BN sample, consistent with the higher level of Microcoleus expression at the beginning of the dark period. The Microcoleus genes were the majority of all the cyanobacterial-associated reads assigned to genes in the fermentation pathway.

(a) Reconstruction of anoxic low-molecular-weight metabolite producing pathways based on the M. chthonoplastes genome. Abbreviations: ACK, acetate kinase; ADH, alcohol/aldehyde dehydrogenase; GP, glycogen phosphorylase; HOX, bidirectional [NiFe] hydrogenase; LDH, lactate dehydrogenase; PFL, pyruvate-formate lyase; PFR, pyruvate:ferredoxin oxidoreductase. Phosphotransacetylase (PTA) was not identified. (b). Reconstruction of glycogen to PHA conversion pathway in sequenced Chloroflexales genomes. Abbreviations: PDC, pyruvate dehydrogenase complex; PhaA, acetoacetyl-CoA transferase; PhaB, acetoacetyl-CoA reductase; PhaC, polyhydroxyalkonoate synthase. Detailed assignments for these genes are presented in the Supplementary Information (Supplementary Tables 4 and 5). Quantitative measurements of recruited reads from BN and EN data sets are presented in Figure 5 and Supplementary Table 6.

Comparison of reads involved in carbon catabolic pathways in BN and EN libraries. Reads representing transcripts for the Microcoleus spp. fermentation pathway and the Chloroflexales glycogen to PHA conversion. The reads are presented as a relative proportion of the total reads assigned to this SEED functional category. Pha (phaCAB and HOX (hoxEFUYH) represent the sum of multiple transcripts. Actual numbers of reads recruited to the genes in each category are presented in the Supplementary Information (Supplementary Table 6).
A number of Chloroflexales-associated reads were assigned as acetate- and lactate-permeases, consistent with the hypothesis that these microbes take up the organic acids excreted during fermentation by Microcoleus spp. (Figure 4b; Supplementary Tables 4–6). Organic acid uptake is often associated with production of polyhydroxyalkonoates (PHAs) that act as carbon storage compounds and uptake of acetate has been shown to be involved in the conversion of glycogen to PHAs in bacteria responsible for EBPR (enhanced biological phosphorus removal) (Mino et al., 1998). Pathways for the biosynthesis of PHAs were reconstructed from multiple Chloroflexales genomes and transcripts were identified for phaA (acetoacetyl-CoA transferase), phaB (acetoacetyl-CoA reductase) and phaC (PHA synthase) in both the BN and EN libraries (Figure 5). The identification of Chloroflexales-associated transcripts for glycogen phosphorylase in both libraries implies that reducing equivalents required for PHA production in these bacteria derive from the breakdown of glycogen. Transcripts for acetyl-CoA synthetase (acs), which converts acetate to acetyl-CoA, were also detected in both libraries. Accumulation of glycogen by the Chloroflexi is proposed to occur during anoxygenic photosynthesis to store carbon and reducing equivalents for dark, anoxic metabolism (Sirevag and Castenholz, 1979; Taffs et al., 2009). These reducing equivalents are generated by glucose metabolism through the EMP pathway to produce pyruvate, which is oxidized to acetyl-CoA by pyruvate dehydrogenase, which is detected in both libraries.
13 C-Acetate uptake by Chloroflexi observed by FISH-SIMS
The reconstructed pathway described above suggested that the Chloroflexi in the Elkhorn Slough mat assimilated organic acids under dark, anoxic conditions. Single cell analysis using NanoSIMS was used to test the specific hypothesis that acetate, the most abundant organic acid observed in the mats, was assimilated by the Chloroflexi. Mat samples were incubated with 0.2 mM 13C-acetate during the dark portion of the diel cycle and Chloroflexi cells in these samples were stained by the CARD-FISH procedure with Chloroflexi-specific oligonucleotide probes (Figure 6A) and identified by confocal laser scanning microscopy. This analysis revealed a morphological diversity consistent with the phylogenetic diversity detected in nearly full-length 16S rRNA sequences from Elkhorn Slough mats (Figure 3), in which the sequence identity of the Chloroflexi phylotypes ranged from 76.6% to 89.6%. The identified Chloroflexi were targets for NanoSIMS analysis measuring 13C/12C in individual bacterial cells. These FISH-SIMS experiments demonstrated specific uptake of 13C-lableled acetate into Chloroflexi cells (δ13C=260±35‰; n=40), with the observed 13C-label in Microcoleus (−15.8±3.0‰; n=12) and other cyanobacterial cells (−14.0±2.8‰; n=12) close to natural abundance (Figure 6B). Additional single cells were enriched in 13C above natural abundance, but did not stain with the Chloroflexi-specific probes.

(A) 13C-acetate uptake under dark, anoxic conditions within the microbial community of the Elkhorn Slough mat. (a) Scanning-electron micrographs (SEM). (b) CARD-FISH micrographs using CF mix probes (CFX1223 and GNSB-941) targeting Chloroflexi in green. (c) Ion micrographs of 13C/12C ratios of the same community members imaged by SEM and CARD-FISH. Scale bar=5 μm. (B) 13C-acetate uptake under dark, anoxic conditions by different microbial groups in the Elkhorn Slough mat. Chloroflexi (n=40) were identified using CF mix CARD-FISH probes. Microcoleus spp. (n=12) and other Cyanobacteria (n=12) were identified by their morphological characteristics. Error bars represent the standard error of the mean. Note that the probes do not distinguish Chloroflexi from Chloroflexales so these results are discussed at the phylum level.
Discussion
In this study, we have taken a highly diverse microbial system and used metatranscriptomic sequencing to identify genes in the two most abundant taxa, Cyanobacteria and Chloroflexi, which are responsible for anoxic catabolism of photosynthate. The success of this reconstruction is remarkable because of the relatively modest amount of sequencing (∼400 MB) used. It is highly unlikely that a comparable amount of metagenomic sequencing would have provided a resolved metabolic pathway because the diversity of the community would prevent assembly of large contiguous sequences (Kunin et al., 2008b). Metatranscriptomic sequencing focuses on the most highly expressed genes, which can resolve important metabolic processes. Central to the success of this strategy was the availability of isolate genome sequences for Microcoleus and the Chloroflexales that facilitated metabolic pathway reconstruction and fragment recruitment of transcripts. A notable discrepancy in the metatranscriptomic data obtained from Elkhorn Slough samples is the difference in relative abundance of the rRNA transcripts, which identify Cyanobacteria as the dominant active population, compared with the mRNA transcripts, which fluctuate between Cyanobacteria and Chloroflexi as the dominant populations. This phenomenon has been observed, though less dramatically, in metatranscriptomic studies of a microbial mat from Mushroom Spring in Yellowstone (Liu et al., 2011). The dominance of cyanobacterial rRNA may reflect the synthesis of large numbers of ribosomes by cyanobacterial cells, which have larger cell sizes in comparison to other bacterial cells. Cell size and ribosome content have previously been directly correlated in prokaryote cells (Ecker and Kokaisl, 1969). Alternatively, it could reflect an amplification bias in the PCR reaction with cDNA from the mats. However, since two different forward primers were used for pyrotag sequencing and Sanger clone libraries, this explanation is not very likely.
Combining the metatranscriptomic data and metabolic pathway reconstruction with metabolite and single cell measurements enabled us to propose an anoxic catabolic pathway for carbon fixed during the light period by oxygenic and anoxygenic photosynthesis. In this pathway, photosynthate accumulated during the day is stored as glycogen by both Microcoleus spp., derived from oxygenic photosynthesis and the Chloroflexi, derived from anoxygenic photosynthesis. Under dark anoxic conditions, Cyanobacteria depolymerize and ferment the glycogen, excreting H2 and organic acids and generating ATP for cellular maintenance. Chloroflexi depolymerize glycogen, generating reducing equivalents to transform organic acids cross-fed from the Cyanobacteria to PHAs, generating ATP for cellular maintenance. Our analysis does not exclude other pathways for anoxic metabolism of organic acids, such as sulfate reduction by δ-proteobacteria, but these pathways could not be completely reconstructed from the metatranscriptomic data sets (Risatti et al., 1994).
Reconstruction of fermentation pathways from the M. chthonoplastes genome identified multiple pathways for pyruvate fermentation, all of which were expressed in the mats samples analyzed by metatranscriptomics. Therefore, the predominant organic acid end products of pyruvate fermentation were predicted to be acetate, formate and lactate. Acetate and formate were observed, consistent with the predicted activity of pyruvate ferredoxin oxiodoreductase (PFR), pyruvate formate-lyase and pyruvate dehydrogenase complex. However, no lactate was observed, consistent with both the low number of observed lactate dehydrogenase transcripts and the detection of reads for lactate permeases from the Chloroflexi. Propionate production was also observed, which suggests that Microcoleus may have a pathway to ferment pyruvate to propionate, similar to pathways present in propionibacteria (Himmi et al., 2000). Interestingly, no genes in the Microcoleus genome or cyanobacterial transcripts in the two data sets could be assigned as a phosphotransacetylase despite the identification of multiple pathways in Microcoleus to produce acetyl-CoA and the production of high levels of acetate in the mat samples. This observation suggests that an unassigned protein may substitute for phosphotransacetylase in Microcoleus fermentation. The recruitment of metatranscriptomic reads provides persuasive evidence that H2 is generated by a NAD(P)H-dependent Hox hydrogenase, consistent with previous studies which utilized PCR and microarray-based methods (Burow et al., 2012, Marshall et al., 2012). The expression of PFR may imply that reducing equivalents for H2 are generated by oxidation of pyruvate to acetyl-CoA, as is observed for [FeFe] hydrogenases (Schut and Adams, 2009). However, the specific interactions between PFR and the Hox hydrogenase are unknown, despite the co-occurrence of PFR with Hox in all available cyanobacterial genomes (Carrieri et al., 2011).
An important hypothesis arising from analysis of the metatranscriptomic data was that the Chloroflexi should take up organic acids under dark, anoxic conditions. The linkage of organic acid uptake to anoxic conversion of glycogen to PHA has previously been observed in bacteria performing EBPR during wastewater treatment (Mino et al., 1998). The postulated link between fermentation products excreted by Microcoleus and carbon compounds taken up by Chloroflexi is consistent with observations of cross-feeding between these populations in other microbial mats. Glycogen fermentation and organic acid excretion by Cyanobacteria has been demonstrated in thermophilic Synechoccocus-dominated mats found in alkaline siliceous hot springs (Octopus Spring and Mushroom Spring) in Yellowstone National Park using both 14C-labeling and transcript analysis, which demonstrated upregulation of Synechoccocus fermentation genes (Nold and Ward, 1996, Steunou et al., 2006). In these mats, thermophilic Chloroflexales related to Roseiflexus castenholzii are also abundant and pulse labeling experiments with 13CO2 and 14CO2 provided evidence for the transfer of cyanobacterial-derived photosynthate from Synechoccocus to the Roseiflexus populations (van der Meer et al., 2005). The intermediacy of acetate was demonstrated by lipid analysis of Synechoccocus mat samples incubated with 13C-acetate. However, this acetate incorporation was observed during the morning, in contrast to observations with the Microcoleus-dominated mats described here, and may be directly linked to photoheterotrophic growth. The cross-feeding of Synechoccocus and Roseiflexus populations in the Yellowstone mats has served as the basis for developing in silico models of mat carbon metabolism that are broadly consistent with the anoxic carbon metabolism observed at Elkhorn Slough (Taffs et al., 2009). Though accumulation of PHAs by mat dwelling Chloroflexi has never been directly demonstrated, studies of microbial mats found in the Great Sippewissett Salt Marsh in Massachusetts, USA which contain Chloroflexi populations, have shown that PHAs accumulate at night and decrease during the day (Rothermich et al., 2000). The accumulation of PHA in the dark was putatively linked to bacterial breakdown of glycogen, which supports the model for the Elkhorn Slough mats derived from metatranscriptomic data. Additionally, studies of carbon metabolism in Chloroflexus auarantiacus have shown that this model organism produces both glycogen and PHA (Sirevag and Castenholz, 1979).
Acetate uptake and incorporation into PHA was demonstrated in single cells of uncultured Accumulibacter spp., a microbial population important in EBPR, by MAR-FISH using 14C-labeled acetate (Burow et al., 2008). Evidence for acetate uptake by single cells affiliated with the Chloroflexi was obtained by application of FISH-SIMS to the mat samples using 13C-labeled acetate and Chloroflexi-specific oligonucleotide probes. Other single cells that were not affiliated with the Chloroflexi were observed with enriched 13C/12C ratios, indicating that additional bacterial groups assimilated acetate under anoxic conditions. Future work will focus on cultivating Chloroflexi from the Elkhorn Slough mats to test the proposed linkage of acetate uptake to glycogen conversion to PHA and identifying other group involved in the anoxic metabolism of organic acids derived from fermentation.
In conclusion, metatranscriptomic sequencing of samples obtained from microbial mats has demonstrated that Cyanobacteria and Chloroflexi are the most active groups under dark, anoxic conditions. Genomic reconstruction and fragment recruitment of transcripts was used to reconstruct a pathway for the catabolism of photosynthate that linked fermentation by Microcoleus with PHA production by the Chloroflexi. This work highlights the utility of metatranscriptomics to define pathways for element cycling in complex microbial communities.
Accession codes
References
Albert DB, Martens CS . (1997). Determination of low-molecular-weight organic acid concentrations in seawater and pore-water samples via HPLC. Mar Chem 56: 27–37.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ . (1990). Basic local alignment search tool. J Mol Biol 215: 403–410.
Bebout BM, Carpenter SP, Des Marais DJ, Discipulo M, Embaye T, Garcia-Pichel F et al (2002). Long-term manipulations of intact microbial mat communities in a greenhouse collaboratory: simulating Earth's present and past field environments. Astrobiology 2: 383–402.
Björnsson L, Hugenholtz P, Tyson GW, Blackall LL . (2002). Filamentous Chloroflexi (green non-sulfur bacteria) are abundant in wastewater treatment processes with biological nutrient removal. Microbiology 148: 2309–2318.
Burow LC, Mabbett MN, Blackall LL . (2008). Anaerobic glyoxylate cycle activity during simultaneous utilization of glycogen and acetate in uncultured Accumulibacter enriched in enhanced biological phosphorus removal communities. ISME J 2: 1040–1051.
Burow LC, Woebken D, Bebout BM, McMurdie PJ, Singer SW, Pett-Ridge J et al (2012). Hydrogen production in photosynthetic microbial mats in the Elkhorn Slough estuary, Monterey Bay. ISME J 6: 863–874.
Canfield DE, Marais DJD . (1993). Biogeochemical cycles of carbon, sulfur, and free oxygen in a microbial mat. Geochim Cosmochim Acta 57: 3971–3984.
Carrieri D, Wawrousek K, Eckert C, Yu J, Maness PC . (2011). The role of the bidirectional hydrogenase in cyanobacteria. Bioresour Technol 102: 8368–8377.
Decker KLM, Potter CS, Bebout BM, Des Marais DJ, Carpenter S, Discipulo M et al (2005). Mathematical simulation of the diel O, S, and C biogeochemistry of a hypersaline microbial mat. FEMS Microbiol Ecol 52: 377–395.
Dillon JG, Miller S, Bebout B, Hullar M, Pinel N, Stahl DA . (2009). Spatial and temporal variability in a stratified hypersaline microbial mat community. FEMS Microbiol Ecol 68: 46–58.
Ecker RE, Kokaisl G . (1969). Synthesis of protein, ribonucleic acid, and ribosomes by individual bacterial cells in balanced growth. J Bacteriol 98: 1219–1226.
Finzi-Hart JA, Pett-Ridge J, Weber PK, Popa R, Fallon SJ, Gunderson T et al (2009). Fixation and fate of C and N in the cyanobacterium Trichodesmium using nanometer-scale secondary ion mass spectrometry. Proc Natl Acad Sci USA 106: 6345–6350.
Frias-Lopez J, Shi Y, Tyson GW, Coleman ML, Schuster SC, Chisholm SW et al (2008). Microbial community gene expression in ocean surface waters. Proc Natl Acad Sci USA 105: 3805–3810.
Gilbert JA, Field D, Huang Y, Edwards R, Li W, Gilna P et al (2008). Detection of large numbers of novel sequences in the metatranscriptomes of complex marine microbial communities. PLoS One 3: e3042.
Gomez-Alvarez V, Teal TK, Schmidt TM . (2009). Systematic artifacts in metagenomes from complex microbial communities. ISME J 3: 1314–1317.
Green SJ, Blackford C, Bucki P, Jahnke LL, Prufert-Bebout L . (2008). A salinity and sulfate manipulation of hypersaline microbial mats reveals stasis in the cyanobacterial community structure. ISME J 2: 457–470.
He SM, Kunin V, Haynes M, Martin HG, Ivanova N, Rohwer F et al (2010). Metatranscriptomic array analysis of ‘Candidatus Accumulibacter phosphatis’-enriched enhanced biological phosphorus removal sludge. Environ Microbiol 12: 1205–1217.
Himmi EH, Bories A, Boussaid A, Hassani L . (2000). Propionic acid fermentation of glycerol and glucose by Propionibacterium acidipropionici and Propionibacterium freudenreichii ssp shermanii. Appl Microbiol Biotechnol 53: 435–440.
Hoehler TM, Albert DB, Alperin MJ, Bebout BM, Martens CS, Des Marais DJ . (2002). Comparative ecology of H2 cycling in sedimentary and phototrophic ecosystems. Antonie Van Leeuwenhoek 81: 575–585.
Hoehler TM, Bebout BM, Des Marais DJ . (2001). The role of microbial mats in the production of reduced gases on the early Earth. Nature 412: 324–327.
Hollibaugh JT, Gifford S, Sharma S, Bano N, Moran MA . (2011). Metatranscriptomic analysis of ammonia-oxidizing organisms in an estuarine bacterioplankton assemblage. ISME J 5: 866–878.
Huson DH, Auch AF, Qi J, Schuster SC . (2007). MEGAN analysis of metagenomic data. Genome Res 17: 377–386.
Jorgensen BB, Des Marais DJ . (1986). Competition for sulfide among colorless and purple sulfur bacteria in cynobacterial mats. FEMS Microbiol Ecol 38: 179–186.
Kunin V, Copeland A, Lapidus A, Mavromatis K, Hugenholtz P . (2008a). A bioinformatician's guide to metagenomics. Microbiol Mol Biol Rev 72: 557–578.
Kunin V, Raes J, Harris JK, Spear JR, Walker JJ, Ivanova N et al (2008b). Millimeter-scale genetic gradients and community-level molecular convergence in a hypersaline microbial mat. Mol Syst Biol 4: 198.
Ley RE, Harris JK, Wilcox J, Spear JR, Miller SR, Bebout BM et al (2006). Unexpected diversity and complexity of the Guerrero Negro hypersaline microbial mat. Appl Environ Microbiol 72: 3685–3695.
Li W, Godzik A . (2006). CD-Hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22: 1658–1659.
Liu Z, Klatt CG, Wood JM, Rusch DB, Ludwig M, Wittekindt N et al (2011). Metatranscriptomic analyses of chlorophototrophs of a hot-spring microbial mat. ISME J 5: 1279–1290.
Marshall IP, Berggren DR, Azizian MF, Burow LC, Semprini L, Spormann AM . (2012). The Hydrogenase Chip: a tiling oligonucleotide DNA microarray technique for characterizing hydrogen-producing and -consuming microbes in microbial communities. ISME J 6: 814–826.
McCarren J, Becker JW, Repeta DJ, Shi Y, Young CR, Malmstrom RR et al (2010). Microbial community transcriptomes reveal microbes and metabolic pathways associated with dissolved organic matter turnover in the sea. Proc Natl Acad Sci USA 107: 16420–16427.
Mino T, Van Loosdrecht MCM, Heijnen JJ . (1998). Microbiology and biochemistry of the enhanced biological phosphate removal process. Water Res 32: 3193–3207.
Mou X, Vila-Costa M, Sun S, Zhao W, Sharma S, Moran MA . (2011). Metatranscriptomic signature of exogenous polyamine utilization by coastal bacterioplankton. Environ Microbiol Rep 3: 798–806.
Nicholson JAM, Stolz JF, Pierson BK . (1987). Structure of a microbial mat at Great Sippiwisset Marsh, Cape Cod, Massachusetts. FEMS Microbiol Ecol 45: 343–364.
Nold SC, Ward DM . (1996). Photosynthate partitioning and fermentation in hot spring microbial mat communities. Appl Environ Microbiol 62: 4598–4607.
Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M . (1999). KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 27: 29–34.
Omoregie EO, Crumbliss LL, Bebout BM, Zehr JP . (2004). Determination of nitrogen-fixing phylotypes in Lyngbya sp and Microcoleus chthonoplastes cyanobacterial mats from Guerrero Negro, Baja California, Mexico. Appl Environ Microbiol 70: 2119–2128.
Orphan VJ, Jahnke LL, Embaye T, Turk KA, Pernthaler A, Summons RE et al (2008). Characterization and spatial distribution of methanogens and methanogenic biosignatures in hypersaline microbial mats of Baja California. Geobiology 6: 376–393.
Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang HY, Cohoon M et al (2005). The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res 33: 5691–5702.
Poretsky RS, Bano N, Buchan A, LeCleir G, Kleikemper J, Pickering M et al (2005). Analysis of microbial gene transcripts in environmental samples. Appl Environ Microbiol 71: 4121–4126.
Poretsky RS, Hewson I, Sun S, Allen AE, Zehr JP, Moran MA . (2009). Comparative day/night metatranscriptomic analysis of microbial communities in the North Pacific subtropical gyre. Environ Microbiol 11: 1358–1375.
Poretsky RS, Sun S, Mou X, Moran MA . (2010). Transporter genes expressed by coastal bacterioplankton in response to dissolved organic carbon. Environ Microbiol 12: 616–627.
Pruitt KD, Tatusova T, Klimke W, Maglott DR . (2009). NCBI Reference Sequences: current status, policy and new initiatives. Nucleic Acids Res 37: D32–D36.
Risatti JB, Capman WC, Stahl DA . (1994). Community structure of a microbial mat—the phylogenetic dimension. Proc Natl Acad Sci USA 91: 10173–10177.
Rothermich MM, Guerrero R, Lenz RW, Goodwin S . (2000). Characterization, seasonal occurrence, and diel fluctuation of poly(hydroxyalkanoate) in photosynthetic microbial mats. Appl Environ Microbiol 66: 4279–4291.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB et al (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537–7541.
Schut GJ, Adams MWW . (2009). The iron-hydrogenase of Thermotoga maritima utilizes ferredoxin and NADH synergistically: a new perspective on anaerobic hydrogen production. J Bacteriol 191: 4451–4457.
Shi Y, Tyson GW, Eppley JM, DeLong EF . (2011). Integrated metatranscriptomic and metagenomic analyses of stratified microbial assemblages in the open ocean. ISME J 5: 999–1013.
Sirevag R, Castenholz R . (1979). Aspects of carbon metabolism in Chloroflexus. Arch Microbiol 120: 151–153.
Steunou AS, Bhaya D, Bateson MM, Melendrez MC, Ward DM, Brecht E et al (2006). In situ analysis of nitrogen fixation and metabolic switching in unicellular thermophilic cyanobacteria inhabiting hot spring microbial mats. Proc Natl Acad Sci USA 103: 2398–2403.
Taffs R, Aston JE, Brileya K, Jay Z, Klatt CG, McGlynn S et al (2009). In silico approaches to study mass and energy flows in microbial consortia: a syntrophic case study. BMC Syst Biol 3: 114.
Turnbaugh PJ, Quince C, Faith JJ, McHardy AC, Yatsunenko T, Niazi F et al (2010). Organismal, genetic, and transcriptional variation in the deeply sequenced gut microbiomes of identical twins. Proc Natl Acad Sci USA 107: 7503–7508.
Urich T, Lanzen A, Qi J, Huson DH, Schleper C, Schuster SC . (2008). Simultaneous assessment of soil microbial community structure and function through analysis of the metatranscriptome. PLoS One 3: e2527.
van der Meer MTJ, Schouten S, Bateson MM, Nubel U, Wieland A, Kuhl M et al (2005). Diel variations in carbon metabolism by green nonsulfur-like bacteria in alkaline siliceous hot spring microbial mats from Yellowstone National Park. Appl Environ Microbiol 71: 3978–3986.
van der Meer MTJ, Schouten S, Damste JSS, de Leeuw JW, Ward DM . (2003). Compound-specific isotopic fractionation patterns suggest different carbon metab lisms among Chloroflexus-like bacteria in hot-spring microbial mats. Appl Environ Microbiol 69: 6000–6006.
van der Meer MTJ, Schouten S, Damste JSS, Ward DM . (2007). Impact of carbon metabolism on C-13 signatures of cyanobacteria and green non-sulfur-like bacteria inhabiting a microbial mat from an alkaline siliceous hot spring in Yellowstone National Park (USA). Environ Microbiol 9: 482–491.
Wang QC, Garrity GM, Tiedje JM, Cole JR . (2007). Native Bayesian classifier for rapid assignment of rRNA sequences in new bacterial taxonomy. Appl Environ Microbiol 73: 5261–5267.
Woebken D, Burow LC, Prufert-Bebout L, Bebout BM, Hoehler TM, Pett-Ridge J et al (2012). Identification of a novel cyanobacterial lineage as active diazotrophs in a coastal microbial mat using NanoSIMS. ISME J 6: 1429–1437.
Acknowledgements
Funding was provided by the US Department of Energy (DOE) Genomic Science Program under contract SCW1039. Work at LLNL was performed under the auspices of the US Department of Energy at Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344. Work at LBNL was performed under the auspices of the US Department of Energy at Lawrence Berkeley National Laboratory under Contract DE-AC02-05CH11231. Pyrotag and metatranscriptomic sequencing were conducted by the Joint Genome Institute, which is supported by the Office of Science of the US Department of Energy under Contract No. DE-AC02-05CH11231. DW was funded by the German Research Foundation (Deutsche Forschungsgemeinschaft). We thank Jeff Cann, Associate Wildlife Biologist, Central Region, California Department of Fish and Game for coordinating our access to the Moss Landing Wildlife Area.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
Burow, L., Woebken, D., Marshall, I. et al. Anoxic carbon flux in photosynthetic microbial mats as revealed by metatranscriptomics. ISME J 7, 817–829 (2013). https://doi.org/10.1038/ismej.2012.150
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2012.150
Keywords
This article is cited by
-
Changes of bacterial community structure,monosaccharide composition and CO2 exchange along the successional stages of biological soil crusts
Environmental Geochemistry and Health (2023)
-
Integrated metagenomic and metatranscriptomic analyses of ultraviolet disinfection effects on antibiotic resistance genes and bacterial communities during wastewater treatment
Ecotoxicology (2021)
-
The microalga Haematococcus lacustris (Chlorophyceae) forms natural biofilms in supralittoral White Sea coastal rock ponds
Planta (2020)
-
Environmental Controls on CO2 Exchange along a Salinity Gradient in a Saline Boreal Fen in the Athabasca Oil Sands Region
Wetlands (2020)
-
Microbial community composition and dolomite formation in the hypersaline microbial mats of the Khor Al-Adaid sabkhas, Qatar
Extremophiles (2019)
