
Abstract
Bacterioplankton communities are deeply diverse and highly variable across space and time, but several recent studies demonstrate repeatable and predictable patterns in this diversity. We expanded on previous studies by determining patterns of variability in both individual taxa and bacterial communities across coastal environmental gradients. We surveyed bacterioplankton diversity across the Columbia River coastal margin, USA, using amplicon pyrosequencing of 16S rRNA genes from 596 water samples collected from 2007 to 2010. Our results showed seasonal shifts and annual reassembly of bacterioplankton communities in the freshwater-influenced Columbia River, estuary, and plume, and identified indicator taxa, including species from freshwater SAR11, Oceanospirillales, and Flavobacteria groups, that characterize the changing seasonal conditions in these environments. In the river and estuary, Actinobacteria and Betaproteobacteria indicator taxa correlated strongly with seasonal fluctuations in particulate organic carbon (ρ=−0.664) and residence time (ρ=0.512), respectively. In contrast, seasonal change in communities was not detected in the coastal ocean and varied more with the spatial variability of environmental factors including temperature and dissolved oxygen. Indicator taxa of coastal ocean environments included SAR406 and SUP05 taxa from the deep ocean, and Prochlorococcus and SAR11 taxa from the upper water column. We found that in the Columbia River coastal margin, freshwater-influenced environments were consistent and predictable, whereas coastal ocean community variability was difficult to interpret due to complex physical conditions. This study moves beyond beta-diversity patterns to focus on the occurrence of specific taxa and lends insight into the potential ecological roles these taxa have in coastal ocean environments.
Similar content being viewed by others


Introduction
Microbial biogeographical studies describe spatial and temporal distribution patterns of microbes, and shed light on the degree of diversity, patterns of dispersal, and levels of species interactions in different environments (Martiny et al., 2006). We know from biogeographical studies that there are differences in the dominant microbial taxa across habitats and over seasons (Nemergut et al., 2011; Caporaso et al., 2012). In aquatic systems, microbial communities vary spatially with water mass characteristics including depth, salinity, temperature, pH, hydrological conditions, organic matter, phytoplankton interactions, and other biological and physical factors (Field et al., 1997; Crump et al., 2003; Kent et al., 2007; Lozupone and Knight, 2007; Fuhrman et al., 2008; Ghiglione et al., 2008; Lauber et al., 2009; Galand et al., 2010; Fortunato and Crump 2011). On temporal scales, seasonal succession and annual reassembly of communities have been shown in both fresh and marine waters (Morris et al., 2005; Fuhrman et al., 2006; Carlson et al., 2009; Crump et al., 2009; Nelson, 2009; Andersson et al., 2010).
Biogeography of aquatic microbial communities has been well described across spatial and seasonal scales (Gilbert et al., 2009; Galand et al., 2010; Kirchman et al., 2010; Crump et al., 2012; Fortunato et al., 2012), and although most studies discuss changes in community composition and structure, little is known about the specific taxa that distinguish one community from another. In one long-term study of the English Channel, Gilbert et al. (2012) showed strong repeatable seasonal patterns in the general bacterioplankton community and in specific taxonomic groups, and found that seasonal variability was influenced by length of day, temperature, nutrients, and photosynthetically active radiation, rather than biomass of zooplankton species. Eiler et al. (2012) found complex associations within and between bacterioplankton communities and synchronous temporal changes for specific taxonomic groups in a freshwater lake. These studies described variation in specific taxa over time relative to variation in environmental conditions, but both studies were conducted at one or a handful of fixed locations, and did not consider the effect of dispersal.
Several studies of common marine bacterial taxa, such as SAR11, SAR86, and Prochlorococcus, describe spatial and temporal distributions of these groups over different conditions (Carlson et al., 2009; Treusch et al., 2009; Malmstrom et al., 2010; Morris et al., 2012). Malmstrom et al. (2010) showed consistency in the depth distribution of ecotypes of Prochlorococcus over a 5-year period in the Pacific. The depth distribution of SAR11 ecotypes has also been well described, with distinct populations observed in the epipelagic and upper mesopelagic at the long-term Bermuda Atlantic Time Series (BATS) (Carlson et al., 2009) and across gradients in nutrient, chlorophyll, and organic carbon concentrations in the South Atlantic (Morris et al., 2012). What is not fully described in these studies is how patterns in the abundance of these taxonomic groups are related to patterns of community distribution in these environments.
We expanded on these studies by describing patterns of variability in both individual taxa and bacterial communities across a broad range of coastal aquatic environments, and by identifying key taxa that define specific environments. Using 16S rRNA gene amplicon pyrosequencing, we characterized bacterioplankton communities from 596 water samples collected across the Columbia River coastal margin from 2007 to 2010. The coastal waters of the Pacific Northwest are highly productive due to nutrients supplied by seasonal upwelling and freshwater inputs (Hickey and Banas, 2003). The biological and physical processes of these waters are complex due to variable winds, remote wind forcing, shelf width, and submarine canyons (Hickey and Banas, 2008; Hickey et al., 2010), which in turn may differentially affect the composition of bacterioplankton communities along the Oregon and Washington coasts (Fortunato and Crump, 2011). The Columbia River is the second largest river in the United States with a mean annual discharge of 7300 m3 s−1 (Hickey et al., 1998). This significant release of freshwater has a strong impact on the chemical, physical, and biological characteristics of the river plume and coastal ocean (Hickey et al., 2010).
An earlier study of 300 water samples collected in 2007 and 2008 showed bacterioplankton communities of the Columbia River coastal margin separated into seven environments: river, estuary, plume, epipelagic, mesopelagic, shelf bottom and slope bottom (Fortunato et al., 2012). Here we expand on this work and focus on population-level analyses to determine the key taxa in each environment and describe how the relative abundance of these taxa shifts with changing environmental conditions. We hypothesized that there exists seasonal shifts of dominant taxa in each environment and reassembly of the same environment-specific communities each year. Our results confirmed both these characteristics of bacterioplankton communities in river-influenced environments, and we identified indicator taxa that characterize each season. In contrast, the coastal ocean communities varied with environmental conditions that were not linked to seasonality but instead tended to show strong spatial variability.
Materials and methods
Water samples were collected across the Columbia River coastal margin (latitude 44.652 to 47.917, longitude −123.874 to −125.929) as part of the NSF-funded Center for Coastal Margin Observation and Prediction (CMOP). Samples were collected between 2007 and 2010 on 14 cruises aboard the R/V Wecoma, R/V New Horizon, and R/V Barnes. Aboard the R/V Wecoma, water samples were collected from the Columbia River, estuary, plume, and three coastal lines (Columbia River line, Newport Hydroline, La Push line) in April, August, and November of 2007; April, June, July, and September of 2008; and May and July of 2010. In 2009, the same set of samples was collected aboard the R/V New Horizon in May and September (Supplementary Figure S1). Samples were collected using an instrumented 12- or 24-bottle rosette equipped with a Seabird 911+ conductivity-temperature-depth sensor or a high-volume low-pressure pump as described previously (Fortunato et al., 2012).
DNA samples (1–6 l per sample) were collected from 10 l Niskin bottles, preserved and extracted as described previously (Fortunato and Crump, 2011), and PCR-amplified for amplicon pyrosequencing using primers targeting bacterial 16S rRNA genes (Hamady et al., 2008) as described previously (Fortunato et al., 2012). Pyrosequencing was completed in two runs at Engencore at the University of South Carolina (http://engencore.sc.edu/). The first run of 2007–2008 samples was performed on a Roche-454 FLX pyrosequencer (454 Life Sciences, Banford, CT, USA) and has been published (Fortunato et al., 2012). The second run of 2009–2010 samples was completed using Titanium chemistry.
Water was also collected and analyzed for environmental parameters including nutrients, pigments, and rate of bacterial production as described previously (Fortunato and Crump, 2011) or as described below. Particulate organic carbon (POC) and particulate nitrogen (PN) were collected by filtering 250–1000 ml of water onto 25 mm diameter combusted (4.5 h at 500 °C) GF/F filters (Whatman). Filters were folded, placed in plastic collection bags and stored at −20 °C. POC and PN content of the suspended particulate matter on acid-fumed filters (Hedges and Stern, 1984) were determined at the University of California, Davis using a Carlo Erba NA-1500 Elemental Analyzer system (CE Elantech, Lakewood, NJ, USA) as described by (Verardo et al., 1990). For each day that samples were collected, we determined several basic physical characteristics of the Columbia River coastal margin. Columbia River flow (m3 s−1) at Bonneville dam (upstream of furthest river site) was measured by the US Army Corps of Engineers (USACE, http://www.nwd-wc.usace.army.mil). Columbia River estuary residence time (in days) was calculated from river discharge records by USACE (http://www.nwd-wc.usace.army.mil). Upwelling index at latitudes 45°N and 48°N (m3s−1 per 100 m) as determined by Schwing and Mendelssohn (1997) was reported by the US National Oceanographic and Atmospheric Administration (NOAA, http://las.pfeg.noaa.gov/).
Pyrosequences were quality controlled using the AmpliconNoise pipeline (Quince et al., 2011) with recommended procedures for FLX (Clean204.pl, PyroNoise, SeqDist, SeqNoise) and Titanium (CleanMinMax.pl, PyroNoiseM, SeqDistM, SeqNoiseM) chemistry. Maximum sequence length was set to 250 bp (Parse.pl), and chimera were identified and removed (PerseusD). Sequences were clustered into operational taxonomic units (OTUs) using QIIME (v1.4.0, Caporaso et al., 2010). Sequences from each sample were unweighted (unweight_fasta.py), concatenated and primers were removed. OTUs were identified using uclust (pick_otus.py), and representative sequences were selected (pick_rep_set.py). The taxonomy of OTUs was determined in MOTHUR (Schloss et al., 2009) using the Greengenes 2011 database (McDonald et al., 2012). Taxonomic assignments with <80% confidence were marked as unknown. A total of 608 samples were analyzed but reduced to 596, as samples with low number of sequences were omitted.
Relative abundances were calculated for OTUs in each sample, and pairwise similarities among samples were calculated using the Bray–Curtis similarity coefficient (Legendre and Legendre, 1998). Similarity matrices were visualized using multiple dimensional scaling (MDS) ordination. Analysis of similarity statistics (ANOSIM) was calculated to test the significance of differences among a priori sampling groups based on environmental parameters. These analyses were carried out using PRIMER v6 for Windows (PRIMER-E Ltd, Plymouth, UK).
As found in Fortunato et al. (2012), bacterioplankton communities separated into seven environments (ANOSIM, P<0.001): river, estuary, plume, epipelagic, mesopelagic, shelf bottom, and slope bottom. The plume group consisted of surface samples with salinity <31, the epipelagic group included surface and chlorophyll maximum samples (average depth=9 m), the mesopelagic group consisted of samples within and below the thermocline (average depth=56 m), the shelf bottom group consisted of bottom samples with depth <350 m and the slope bottom group included bottom samples deeper than 850 m. These seven environmental categories were used for further analyses. To better determine the variability within each environment, OTUs were assigned to a specific location based on the maximum average relative abundance of each OTU. For example, if OTU 1 was most abundant in the river, then it was called a river OTU.
To identify the specific OTUs that characterize each of the environments, we used Indicator Species Analysis run in R (v2.14.0, R Development Core Team, 2011), using the package labdsv (http://ecology.msu.montana.edu/labdsv/R) and test indval (Dufrene and Legendre, 1997). Indicator values (IV) range from 0 to 1, with higher values for stronger indicators. Only OTUs with IV >0.3 and P<0.05 were considered good indicators (Dufrene and Legendre, 1997). This approach was also used to identify key OTUs for seasonal groups within river, estuary, and plume communities, and for spatial groups within the epipelagic community.
Environmental data were compiled from all years and tested for normality (Shapiro–Wilkes test, P>0.05). Variables that were not normally distributed were transformed to as close to normality as possible. Variables included were as follows: depth, salinity, temperature, dissolved oxygen (DO), nitrate, nitrite, ammonium, ortho-phosphate, silicic acid (DSi), dissolved organic nitrogen, phosphorus and carbon (DON, DOP, DOC), POC and PN, chlorophyll a (Chl a), bacterial production rate, Columbia River flow, Columbia River estuary residence time, and coastal upwelling index at latitudes 45°N and 48°N. Analyses were completed with a reduced set of 312 samples due to missing environmental data. The number of samples in each environment was: slope bottom=16, shelf bottom=72, mesopelagic=23, epipelagic=95, plume=52, estuary=46, river=8. We also removed OTUs that were only present in one sample as environmental analyses may be skewed by rare taxa, which resulted in a reduced set of 5038 OTUs.
Our analysis used a two-step approach. First, in Primer v6 (PRIMER-E Ltd), BV-STEP analysis was used to identify sets of variables that potentially influenced community variability (ρ>0.95, Δρ<0.001, 10 random starting variables, 100 restarts; Clarke and Ainsworth, 1993). BIO-ENV was used to rank each individual environmental variable by degree of association with community variability. BV-STEP and BIO-ENV use the Spearman rank correlation coefficient (ρ) to determine the degree of association between similarity matrices of 16S amplicon sequences (Bray–Curtis similarity) and environmental data (Euclidean distances). Then, using the environmental variables identified by BV-STEP, we performed a canonical correspondence analysis (CCA) to determine the percent of community variability explained by environmental variables (ter Braak, 1986). When community data varied linearly along environmental gradients (instead of unimodal), we ran redundancy analysis (RDA) instead of CCA (Legendre and Anderson, 1999). CCA/RDA analyses were run in R (v2.14.0), using the vegan package (http://vegan.r-forge.r-project.org/), for each environment and for abundant taxonomic groups within each environment. We reduced the size of the environmental dataset by removing highly correlated variables (ρ>0.90). Depth was included as a proxy variable for conditions that change in the vertical, including light and pressure.
Sequences are deposited in the NCBI Sequence Read Archive (SRA) under the accession numbers SRP006412 for 2007–2008 and SRA058065 for 2009–2010. Tables of sample metadata and representative sequences for each OTU are provided in Supplementary Tables S3 and S4.
Results
Sequence analysis of the full data set (n=596) yielded 11 082 OTUs comprising 428 372 sequences. Bacterioplankton communities separated into the seven environment-specific groups described previously (Fortunato et al., 2012; ANOSIM, P<0.001): river, estuary, plume, epipelagic, mesopelagic, shelf bottom, and slope bottom along gradients of salinity and depth (Supplementary Figure S2).
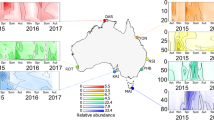
Within each of the seven environments, bacterioplankton communities were variable across space and over time. Seasonal variability and annual reassembly of bacterioplankton communities in the Columbia River, estuary, and plume were evident in the MDS diagrams (Figures 1a, 2a, and 3a). For analysis of seasonal patterns, only OTUs specific for each environment were used to create MDS plots in order to specifically address the variability of the dominant members of the local communities. In the river, communities shifted along a seasonal continuum from April to November, and separated into two significant groups: early year (April–July) and late year (August–November) (ANOSIM, P<0.001). Actinobacteria made up a larger percentage (32%) of the late-year community than the early-year community (12%). A freshwater SAR11 group was present in the late-year community (7%), but was absent in early-year samples (Figure 1b, Supplementary Table S1). Comparing river community variability with environmental conditions, we found that the concentration of POC and PN correlated with river community composition (ρ=0.686, Table 1). The correlation increased to ρ=0.857 with inclusion of ammonium, DOC, Chl a, and DO (Table 1). Early in the year, during spring and freshet periods, the river had higher flow and higher concentrations of POC, PN, DOC, and some inorganic nutrients.

(a) Seasonal multidimensional scaling diagram of river samples, produced with Bray–curtis similarity values for communities that only included river-specific OTUs (Stress=0.11). Samples separated into two significantly different communities, early (April–July) and late (August–November) (ANOSIM, P<0.001). (b) Taxonomy of the two seasonal groups. (c, d) Spatial distribution of a top indicator for early (IV: 0.74) and late (IV: 0.94), by average relative abundance, across the ordination of the river MDS (IV=Indicator value).

(a) Seasonal multidimensional scaling diagram of estuary samples, produced with Bray–curtis similarity values for communities that only included estuary-specific OTUs (Stress=0.19). Samples separated into two significantly different communities, early (April–July) and late (August–November) (ANOSIM, P<0.001). (b) Taxonomy of the two seasonal groups. (c, d) Spatial distribution of a top indicator for early (IV: 0.96) and late (IV: 0.87), by average relative abundance, across the ordination of the estuary MDS.

(a) Seasonal multidimensional scaling diagram of plume samples, produced with Bray-Curtis similarity values for communities that only included plume-specific OTUs (Stress=0.17). Samples separated into two significantly different communities, non-summer (April–July, November) and summer (August–September) (ANOSIM, P<0.001). (b) Taxonomy of the two seasonal groups. (c, d) Spatial distribution of the top indicator for non-summer (IV: 0.76) and summer (IV: 0.67), by average relative abundance, across the ordination of the plume MDS.
In the estuary, there was a significant difference between early- and late-year communities (ANOSIM, P<0.001, Figure 2a), and community composition correlated with seasonality of river flow and residence time (ρ=0.550, Table 1). Including temperature, DO, ortho-phosphate, and PN increased the correlation to ρ=0.638. The early community had a larger percentage of Oceanospirillales, whereas the late community was dominated by Flavobacteria (Figure 2b). Indicator analysis showed the top indicator for the early estuary was an Oceanospirillales OTU, which in some samples comprised up to 34% of the community (Figure 2c, Supplementary Table S1). This same OTU was found in the estuary in a previous study (Crump et al., 1999). The top late year indicator was a Rhodobacteraceae OTU, which made up 9.6% of the community.
The plume community also appeared to be influenced by river inputs, but did not correlate as strongly with environmental variables. The plume community varied seasonally, and correlated with temperature (ρ=0.465) and bacterial production rate (ρ=0.317; Table 1). These variables were elevated in the late summer months (August and September) compared with the rest of the year with average temperature increasing from 11.5 to 15.6 °C and average bacterial production rate from 0.59 to 0.96 μgC l−1h−1. Changes in environmental conditions corresponded to the development of a significantly distinct summer bacterioplankton community (August and September) that assembled each year in the plume (ANOSIM, P<0.001, Figure 3a). In this community, 31% of sequences were from Polaribacter and Rhodobacteraceae, which made up only 2% of non-summer communities. In non-summer months, 72% of the community was represented by unknown Flavobacteria. Indicator analysis showed that the top indicator for summer plume was a Rhodobacteraceae OTU (Figures 3b and d, Supplementary Table S1).
In the slope bottom environment, in deep waters far off shore, DO (ρ=0.606) and temperature (ρ=0.587, Table 1) were most important to community variability. In the shallower shelf bottom environment, which includes samples from nearshore (<20 miles to shore) and offshore (>20 miles from shore), and a depth range of 18–350 m, the community varied with differences in depth, temperature, DO, and rate of bacterial production (ρ=0.618, Table 1). In the epipelagic there was little variation in the depth of samples, but this group extended over large longitudinal and latitudinal scales. The MDS plot of the epipelagic community showed no significant difference across the latitudinal gradient but there was a significant difference between nearshore and offshore communities (ANOSIM, P<0.001). The top indicators were a Flavobacteria OTU for the nearshore and a Pelagibacter OTU for the offshore community (Supplementary Table S2). Community variability was not strongly correlated to any environmental variable (Table 1).
Within each of the seven environments, we found distinct taxonomic assemblages. Figure 4a shows the complete taxonomy of the seven environments, which each displayed a unique taxonomic fingerprint consisting of different percentages of major taxonomic groups. The phylum SAR406 and the Gammaproteobacteria family SUP05 were most prevalent in the deep ocean environment and mixed out in shallower water. The opposite can be said for the common surface bacteria groups SAR86 and Rhodobacteraceae, which were prevalent in the estuary, plume, and epipelagic. The ubiquity of SAR11 was evident, as it made up a large percentage of the community in each of the seven environments.

Taxonomic composition of each environment. (a) Taxonomy for all OTUs in each environment. (b) Taxonomy only for OTUs that were assigned to each environment based on the maximum average relative abundance.
Figure 4b depicts the taxonomy of OTUs specific to each environment. Focusing on environment-specific OTUs, it is evident that one or two taxonomic groups dominate each environment. In the mesopelagic, SAR11 sequences accounted for over 60% of the community, while Flavobacteria dominated the plume, specifically Polaribacter sequences, which made up 15% of the community. Many of the top indicators corresponded to the dominant taxonomic group in each environment. In the slope bottom environment, top indicators included a SAR406 (IV: 0.94) and a gammaproteobacterium OTU (IV: 0.91), both large percentages of the deep ocean community (Figure 4b, Figure 5). In the mesopelagic, indicators included a SUP05 and a SAR11 OTU, whereas in the epipelagic, surface ocean taxa SAR86 and Prochlorococcus were top indicators (Figure 5). In the plume, a Polaribacter OTU was the top indicator (IV: 0.42) and comprised up to 30% of the sequences in some plume samples. Top estuary indicators included a Flavobacteria and a Rhodobacteraceae OTU, whereas in the river, common freshwater taxa from the class Betaproteobacteria were top indicators (Figure 5). In a graph of average relative abundance of OTUs in samples where they occur vs occupancy (i.e., percentage of samples containing each OTU), top indicator OTUs for epipelagic, mesopelagic, and plume appeared to be generalist taxa, as defined in Barberan et al. (2012), with high relative abundance across a large number of samples. The top river, estuary, and slope bottom indicators, however, tended towards specialist taxa, with high relative abundance but detection in fewer samples (Supplementary Figure S3).

Bubble plot of top indicator OTUs in each environment. The size of the bubble indicates the average relative abundance (%) of each OTU in each of the seven environments. Black shaded bubbles show the environment for which each OTU is an indicator. Indicator values are displayed next to each OTU.
The environmental variables that strongly correlated with the variability of the dominant taxonomic groups in their respective environments were, with differing degrees, the same variables correlating with overall community variability of that environment (Table 2, Figure 6). In the slope bottom environment, variability of the SAR406 community was correlated with changes in DO (ρ=0.472), whereas in the shelf bottom, variability of the SUP05 community was correlated with temperature and DO (ρ=0.432, Table 2, Figures 6a and b). For the families Rhodobacteraceae and Flavobacteria in the estuary, river flow and residence time were the top factors that correlated with the variability of these taxonomic groups (ρ=0.660 and 0.550, respectively, Table 2). In the river, variability of the freshwater SAR11 community was correlated with inorganic and organic nutrients, particularly POC and PN (ρ=0.760, Table 2).

Correlations of the relative abundance of top indicator OTUs and environmental variables for each of the seven environments (a–g). Spearman correlation coefficient (ρ) and P-values are indicated for each relationship.
Discussion
Recent biogeographical studies comparing abundance of bacterial taxa with environmental conditions have either focused on the temporal variation of one common taxonomic group (Carlson et al., 2009; Malmstrom et al., 2010) or have studied multiple taxa but on a limited spatial scale (Eiler et al., 2012; Gilbert et al., 2012). In this study we assessed the variability of communities over 4 years, in seven environments spanning broad environmental gradients, and identified key taxa that characterize each of these environments. Results showed seasonal reassembly of bacterioplankton communities in the river, estuary, and plume, with identification of taxa that characterize specific seasonal conditions in each environment, and correlation of community variability with seasonally-influenced variables such as river flow and residence time. Seasonality was not detected in the coastal ocean, where communities were more variable on a spatial scale, and community composition was complicated by seasonal and annual fluctuations in physical factors like mixing and upwelling.
The physical mixing of communities is evident throughout the Columbia River coastal margin, as taxonomic groups increase and decrease across salinity and depth gradients (Figure 4). In each of the seven environments specific niches are created, which lead to the prevalence of dominant taxonomic groups. This is evident in Figure 4a, in which the overall community shows gradual shifts in these communities across space, but in contrast, Figure 4b depicting the environment-specific communities shows sharp differences among environments and shows there are certain taxa that dominate each environment. OTUs from these dominant taxonomic groups were also the top indicators for each environment, suggesting these dominant groups are key to shaping community composition and indicating that the variability within each environment is driven by shifts in the most abundant taxa.
These key indicator taxa also displayed starkly different ecological strategies across environments, as described by (Barberan et al., 2012). An abundance vs occupancy plot showed that top indicator OTUs for upper water-column environments (plume, epipelagic, mesopelagic), including Rhodobacteraceae, SAR86, and SAR11 OTUs, displayed generalist taxa qualities, with high relative abundance and occurrence in a high number of samples outside their indicator environment (Supplementary Figure S3). Conversely, river, estuary, and slope bottom indicator OTUs, including Limnohabitans, Oceanospirillales, and SAR406, were more specialized to their respective environments and were abundant in fewer samples outside their indicator environment (Supplementary Figure S3 and Supplementary Table S5). As mixing occurred across the Columbia River coastal margin, generalist indicator OTUs sustained a high level of abundance across many environments, while specialist indicator OTUs were abundant in fewer samples and more specific to their respective indicator environment (Supplementary Figure S3 and Supplementary Table S5).
Seasonality of communities in the river, estuary, and plume environments is linked to the environment of the Columbia River. In the river community, seasonal fluxes of inorganic and organic nutrients, especially POC and PN, appear to shape the changes in the bacterioplankton community over an annual cycle. Higher river flow generally brings more particulate matter downstream, with high POC concentrations during the spring freshet and low concentrations during summer months. The relative abundance of OTU 11923, an Actinobacteria, and OTU 2500, a freshwater SAR11, both negatively correlated with POC concentrations (ρ=−0.664 and −0.616, respectively, Figures 5 and 6g), such that these taxa are most abundant in the summer months when river flow and nutrient concentrations are low. The opposite was found for OTU 7389, a Burkholderiales OTU, where periods of higher POC concentrations resulted in higher abundance (ρ=0.527). The sequences of these three river OTUs match, with 100% identity, clones from freshwater environments around the globe (unpublished data from NCBI). OTU 7389 also matched a clone from low salinity bottom waters of the Chesapeake Bay estuary (Shaw et al., 2008), suggesting that this OTU favors a particle rich, high nutrient environment.
Community composition in the estuary also was influenced by upstream river conditions, specifically river flow and residence time. In a river-dominated system like the Columbia River estuary the amount and composition of river water and the time that water spends in the estuary greatly affects the development of the bacterioplankton community. In this study, the early estuary community (April–July) was more river-dominated because river flow was higher (average=6553 m3s−1) and residence time was shorter (average=5.70 d) compared with the late community (August–November), when flow was lower (average=2899 m3s−1) and residence time was longer (average=11.5 d). Indicator analysis showed that the top indicator for the early estuary was an Oceanospirillales OTU (OTU 627), which made up over one-third of the sequences in some estuarine samples. A BLAST search of this OTU sequence revealed it was identical to both free-living and particle-attached clones from a previous study of the Columbia River estuary (Crump et al., 1999). In Crump et al. (1999), the estuary was sampled in May, which is the month of peak abundance for OTU 627, indicating this Oceanospirillales taxon is a well-established population in the early months of the year in the Columbia River estuary. BLAST results also showed this sequence was 100% identical only to clones from the Columbia River estuary. The indicator for the late year, and more coastal ocean-influenced community was a Rhodobacteraceae OTU (OTU 8288). This sequence was identical to clones from the Delaware Bay (Shaw et al., 2008) and was nearly identical (99%) to Roseobacter sp. clones from the Gulf of Mexico (Pinhassi et al., 2005), demonstrating that this taxon is prevalent in coastal ocean-influenced environments as seen in later months in the Columbia River estuary.
In the plume, the community composition was less correlated with river specific variables like river flow and residence time, and more with seasonal variables like temperature, leading to a distinct bacterioplankton community in the summer months. Of the top ten indicators for the plume environment, seven were Flavobacteria OTUs, with Flavobacteria sequences making up 46% and 72% of sequences in summer and non-summer months, respectively. This class of bacteria was previously shown to be prevalent in productive environments like phytoplankton blooms (Simon et al., 1999) and upwelling zones (Alonso-Saez et al., 2007). The Columbia River plume is highly productive with seasonal upwelling supplying nutrients to the surface waters to fuel production (Hickey et al., 1998; Hickey et al., 2010). The top plume indicator was OTU 9443, a Polaribacter taxon, which is a genus of class Flavobacteria. One study of bacterioplankton communities along a coast to open ocean transect in the North Atlantic Ocean found Polaribacter taxa to be the most prevalent of all Flavobacteria, with higher abundances in coastal samples (Gomez-Pereira et al., 2010). A comparison of all Polaribacter OTUs in the plume to environmental variables showed correlation with temperature, DO, bacterial production rate, DOC, and upwelling (ρ=0.576, Table 2). Additionally, the relative abundance of the top indicator, Polaribacter OTU 9443, was positively correlated with bacterial production (ρ=0.557, Figure 6e) and this OTU closely matched sequences of clones from coastal upwelling systems, including the Brazilian (Cury et al., 2011) and Chilean coasts (Pommier et al., 2007), as well as along the Oregon coast during a diatom bloom (Morris et al., 2006), further demonstrating the prevalence of this taxon in highly productive coastal environments like the Columbia River plume.
Seasonality of environmental conditions shaped communities across the river to ocean gradient, but in the coastal ocean, the variability within each environment was correlated to spatial differences in environment. In the slope bottom, where samples ranged in depth from 600 to 2900 m, community composition correlated strongly with DO concentration (ρ=0.607, Table 1). DO ranged from 0.26 to 2.67 mg l−1 with lower concentrations found in shallower oxygen-minimum zone samples (∼1000 m) and higher concentrations found in deeper waters (∼2900 m). The top indicator for the slope bottom environment was a SAR406 OTU (OTU 4871). The relative abundance of this SAR406 OTU was negatively correlated with oxygen concentrations (ρ=−0.486, Figure 6a), indicating an adaptation to low-oxygen environments. Moreover, this OTU identically matched sequences from oxygen-minimum zones in the eastern North Pacific (Walsh et al., 2009) and the Hawaii Ocean Timeseries (HOT) Station ALOHA (Swan et al., 2011). In the shallower shelf bottom (18–350 m), community composition varied with proximity to shore, location along the coast, temperature, DO, and bacterial production rates. One of the top indicators for this environment was a very abundant OTU (up to 38% of sequences) taxonomically identified as SUP05 (OTU 14135). However, a BLAST search showed this OTU closely matched sequences identified as the ARCTIC96BD-19 group, a closely related Gammaproteobacteria group to SUP05 (Walsh et al., 2009). Both of these groups are important to chemolithoautotrophy in oxygen-minimum zones (Walsh et al., 2009). Swan et al. (2011) showed single amplified genomes of ARCTIC96BD-19 from HOT Station ALOHA have both Rubisco and sulfur oxidation genes, including an isolate that was 99% identical to OTU 14135. This similarity, coupled with the abundance of OTU 14135, suggests chemolithoautotrophic processes may be important in low-oxygen bottom environments on the Oregon and Washington shelf.
In the epipelagic, there was little correlation with any of the environmental variables that we measured and no seasonal patterns were visible. The heterogeneity of the epipelagic from mixing, upwelling, river flow, and other physical and biological factors made it difficult to define a set of environmental variables that best explained community composition. This contrasts with the river, estuary, and plume environments where seasonal patterns of community composition were more clearly defined and communities appeared to be driven by seasonally fluctuating environmental conditions. Throughout the year, the river is inoculated by the same upstream sources of bacteria and flows into the estuary, which then influences the plume, creating a consistent, predicable bacterioplankton community that changes seasonally and reassembles annually. In the coastal ocean environmental factors are less consistent and predicable as winds, upwelling, and runoff from land can change on variable time-scales. Thus, the epipelagic community is inoculated by different sources depending on various physical conditions, which create a more complicated community where taxa increase and decrease frequently. Although there were no strong correlations between the epipelagic community and the physical and biogeochemical parameters in this study, there are many other environmental factors including top-down controls such as grazing and viral infection, as well as interactions between microbial populations themselves that could have a strong influence on the composition of the epipelagic community. Recent studies of surface-ocean communities have shown that biological interactions between taxa can be important in determining community composition (Steele et al., 2011; Gilbert et al., 2012). Thus in the complicated epipelagic environment it is likely that both physical and biogeochemical factors as well as interactions among bacterial populations determine community composition.
In conclusion, this study demonstrates that bacterioplankton communities are consistent and predictable across river to plume environments, with specific taxa defining each season. Epipelagic community composition was less consistent due to seasonal and annual fluctuations in physical factors like mixing and upwelling, and thus correlation with environmental variables was low. In the bottom environments, DO strongly correlated with community composition, and prevalence of taxonomic groups like SUP05 and ARCTIC96BD-19 suggests chemolithoautotrophic processes are important in the carbon cycle of these low oxygen environments. The next step is to further develop a set of key taxa that can be used to model specific conditions in an environment and indicate change in bacterioplankton community composition.
References
Alonso-Saez L, Aristegui J, Pinhassi J, Gomez-Consarnau L, Gonzalez JM, Vaque D et al (2007). Bacterial assemblage structure and carbon metabolism along a productivity gradient in the NE Atlantic Ocean. Aquat Microb Ecol 46: 43–53.
Andersson AF, Riemann L, Bertilsson S . (2010). Pyrosequencing reveals contrasting seasonal dynamics of taxa within Baltic Sea bacterioplankton communities. ISME J 4: 171–181.
Barberan A, Bates ST, Casamayor EO, Fierer N . (2012). Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J 6: 343–351.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336.
Caporaso JG, Paszkiewicz K, Field D, Knight R, Gilbert JA . (2012). The Western English Channel contains a persistent microbial seed bank. ISME J 6: 1089–1093.
Carlson CA, Morris R, Parsons R, Treusch AH, Giovannoni SJ, Vergin K . (2009). Seasonal dynamics of SAR11 populations in the euphotic and mesopelagic zones of the northwestern Sargasso Sea. ISME J 3: 283–295.
Clarke KR, Ainsworth M . (1993). A method of linking multivariate community structure to environmental variables. Mar Ecol Prog Ser 92: 205–219.
Crump BC, Amaral-Zettler LA, Kling GW . (2012). Microbial diversity in arctic freshwaters is structured by inoculation of microbes from soils. ISME J 6: 1629–1639.
Crump BC, Armbrust EV, Baross JA . (1999). Phylogenetic analysis of particle-attached and free-living bacterial communities in the Columbia river, its estuary, and the adjacent coastal ocean. Appl Environ Microbiol 65: 3192–3204.
Crump BC, Kling GW, Bahr M, Hobbie JE . (2003). Bacterioplankton community shifts in an arctic lake correlate with seasonal changes in organic matter source. Appl Environ Microbiol 69: 2253–2268.
Crump BC, Peterson BJ, Raymond PA, Amon RMW, Rinehart A, McClelland JW et al (2009). Circumpolar synchrony in big river bacterioplankton. Proc Natl Acad Sci USA 106: 21208–21212.
Cury JC, Araujo FV, Coelho-Souza SA, Peixoto RS, Oliveira JAL, Santos HF et al (2011). Microbial diversity of a Brazilian coastal region influenced by an upwelling system and anthropogenic activity. PLoS One 6: e16553.
Dufrene M, Legendre P . (1997). Species assemblages and indicator species: the need for a flexible asymmetrical approach. Ecol Monogr 67: 345–366.
Eiler A, Heinrich F, Bertilsson S . (2012). Coherent dynamics and association networks among lake bacterioplankton taxa. ISME J 6: 330–342.
Field KG, Gordon D, Wright T, Rappe M, Urbach E, Vergin K et al (1997). Diversity and depth-specific distribution of SAR11 cluster rRNA genes from marine planktonic bacteria. Appl Environ Microbiol 63: 63–70.
Fortunato CS, Crump BC . (2011). Bacterioplankton community variation across river to ocean environmental gradients. Microb Ecol 62: 374–382.
Fortunato CS, Herfort L, Zuber P, Baptista AM, Crump BC . (2012). Spatial variability overwhelms seasonal patterns in bacterioplankton communities across a river to ocean gradient. ISME J 6: 554–563.
Fuhrman JA, Hewson I, Schwalbach MS, Steele JA, Brown MV, Naeem S . (2006). Annually reoccurring bacterial communities are predictable from ocean conditions. Proceedings of the National Academy of Sciences 103: 13104–13109.
Fuhrman JA, Steele JA, Hewson I, Schwalbach MS, Brown MV, Green JL et al (2008). A latitudinal diversity gradient in planktonic marine bacteria. Proc Natl Acad Sci USA 105: 7774–7778.
Galand PE, Potvin M, Casamayor EO, Lovejoy C . (2010). Hydrography shapes bacterial biogeography of the deep Arctic Ocean. ISME J 4: 564–576.
Ghiglione JF, Palacios C, Marty JC, Mevel G, Labrune C, Conan P et al (2008). Role of environmental factors for the vertical distribution (0–1000 m) of marine bacterial communities in the NW Mediterranean Sea. Biogeosciences 5: 1751–1764.
Gilbert JA, Field D, Swift P, Newbold L, Oliver A, Smyth T et al (2009). The seasonal structure of microbial communities in the Western English Channel. Environ Microbiol 11: 3132–3139.
Gilbert JA, Steele JA, Caporaso JG, Steinbrueck L, Reeder J, Temperton B et al (2012). Defining seasonal marine microbial community dynamics. ISME J 6: 298–308.
Gomez-Pereira PR, Fuchs BM, Alonso C, Oliver MJ, van Beusekom JEE, Amann R . (2010). Distinct flavobacterial communities in contrasting water masses of the North Atlantic Ocean. ISME J 4: 472–487.
Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R . (2008). Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat Methods 5: 235–237.
Hedges JI, Stern JH . (1984). Carbon and nitrogen determination of carbonate-containing solids. Limnol Oceanogr 29: 657–663.
Hickey BM, Banas NS . (2003). Oceanography of the US Pacific Northwest Coastal Ocean and estuaries with application to coastal ecology. Estuaries 26: 1010–1031.
Hickey BM, Banas NS . (2008). Why is the northern end of the California current system so productive? Oceanography 21: 90–107.
Hickey BM, Kudela RM, Nash JD, Bruland KW, Peterson WT, MacCready P et al (2010). River influences on shelf ecosystems: introduction and synthesis. J Geophys Res-Oceans 115: 1–26.
Hickey BM, Pietrafesa LJ, Jay DA, Boicourt WC . (1998). The Columbia River plume study: subtidal variability in the velocity and salinity fields. J Geophys Res-Oceans 103: 10339–10368.
Kent AD, Yannarell AC, Rusak JA, Triplett EW, McMahon KD . (2007). Synchrony in aquatic microbial community dynamics. ISME J 1: 38–47.
Kirchman DL, Cottrell MT, Lovejoy C . (2010). The structure of bacterial communities in the western Arctic Ocean as revealed by pyrosequencing of 16S rRNA genes. Environ Microbiol 12: 1132–1143.
Lauber CL, Hamady M, Knight R, Fierer N . (2009). Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl Environ Microbiol 75: 5111–5120.
Legendre P, Anderson MJ . (1999). Distance-based redundancy analysis: testing multispecies responses in multifactorial ecological experiments. Ecol Monogr 69: 512–512.
Legendre P, Legendre L . (1998) Numerical Ecology. Elsevier Science B.V.: Amsterdam.
Lozupone CA, Knight R . (2007). Global patterns in bacterial diversity. Proceedings of the National Academy of Sciences 104: 11436–11440.
Malmstrom RR, Coe A, Kettler GC, Martiny AC, Frias-Lopez J, Zinser ER et al (2010). Temporal dynamics of Prochlorococcus ecotypes in the Atlantic and Pacific oceans. ISME J 4: 1252–1264.
Martiny JBH, Bohannan BJM, Brown JH, Colwell RK, Fuhrman JA, Green JL et al (2006). Microbial biogeography: putting microorganisms on the map. Nat Rev Microbiol 4: 102–112.
McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A et al (2012). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6: 610–618.
Morris RM, Frazar CD, Carlson CA . (2012). Basin-scale patterns in the abundance of SAR11 subclades, marine Actinobacteria (OM1), members of the Roseobacter clade and OCS116 in the South Atlantic. Environ Microbiol 14: 1133–1144.
Morris RM, Longnecker K, Giovannoni SJ . (2006). Pirellula and OM43 are among the dominant lineages identified in an Oregon coast diatom bloom. Environl Microbiol 8: 1361–1370.
Morris RM, Vergin KL, Cho JC, Rappe MS, Carlson CA, Giovannoni SJ . (2005). Temporal and spatial response of bacterioplankton lineages to annual convective overturn at the Bermuda Atlantic Time-series Study site. Limnol Oceanogr 50: 1687–1696.
Nelson CE . (2009). Phenology of high-elevation pelagic bacteria: the roles of meteorologic variability, catchment inputs and thermal stratification in structuring communities. ISME J 3: 13–30.
Nemergut DR, Costello EK, Hamady M, Lozupone C, Jiang L, Schmidt SK et al (2011). Global patterns in the biogeography of bacterial taxa. Environ Microbiol 13: 135–144.
Pinhassi J, Simo R, Gonzalez JM, Vila M, Alonso-Saez L, Kiene RP et al (2005). Dimethylsulfoniopropionate turnover is linked to the composition and dynamics of the bacterioplankton assemblage during a microcosm phytoplankton bloom. Appl Environ Microbiol 71: 7650–7660.
Pommier T, Canback B, Riemann L, Bostrom KH, Simu K, Lundberg P et al (2007). Global patterns of diversity and community structure in marine bacterioplankton. Mol Ecol 16: 867–880.
Quince C, Lanzen A, Davenport RJ, Turnbaugh PJ . (2011). Removing noise from pyrosequenced amplicons. BMC Bioinformatics 12: 38.
R Development Core Team (2011). R: A language and environment for statistical computing. R Foundation for Statistical Computing: Vienna, Austria. ISBN 3-900051-07-0, available at http://www.R-project.org/.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB et al (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537–7541.
Schwing FB, Mendelssohn R . (1997). Increased coastal upwelling in the California current system. J Geophys Res Oceans 102: 3421–3438.
Shaw AK, Halpern AL, Beeson K, Tran B, Venter JC, Martiny JBH . (2008). It's all relative: ranking the diversity of aquatic bacterial communities. Environ Microbiol 10: 2200–2210.
Simon M, Glockner FO, Amann R . (1999). Different community structure and temperature optima of heterotrophic picoplankton in various regions of the Southern Ocean. Aquat Microb Ecol 18: 275–284.
Steele JA, Countway PD, Xia L, Vigil PD, Beman JM, Kim DY et al (2011). Marine bacterial, archaeal and protistan association networks reveal ecological linkages. ISME J 5: 1414–1425.
Swan BK, Martinez-Garcia M, Preston CM, Sczyrba A, Woyke T, Lamy D et al (2011). Potential for chemolithoautotrophy among ubiquitous bacteria lineages in the dark ocean. Science 333: 1296–1300.
ter Braak CJF . (1986). Canonical correspondence analysis: a new eigenvector technique for multivariate direct gradient analysis. Ecology 67: 1167–1179.
Treusch AH, Vergin KL, Finlay LA, Donatz MG, Burton RM, Carlson CA et al (2009). Seasonality and vertical structure of microbial communities in an ocean gyre. ISME J 3: 1148–1163.
Verardo DJ, Froelich PN, McIntyre A . (1990). Determination of organic carbon and nitrogen in marine sediments using the Carlo Erba NA-1500 analyzer. Deep-Sea Research Part a-Oceanographic Research Papers 37: 157–165.
Walsh DA, Zaikova E, Howes CG, Song YC, Wright JJ, Tringe SG et al (2009). Metagenome of a versatile chemolithoautotroph from expanding oceanic dead zones. Science 326: 578–582.
Acknowledgements
This study was carried out within the context of the Science and Technology Center for Coastal Margin Observation and Prediction (CMOP) supported by the National Science Foundation (grant number OCE-0424602 to Antonio Baptista). We thank the captains and crews of the R/V Wecoma, R/V New Horizon, and R/V Barnes, chief scientists Murray Levine and Fred Prahl, and as well as the many people who helped with sample collection, extraction, and analysis. We want to acknowledge the work of the CMOP Saturn observatory team for compilation and quality control of the environmental data set. We also thank Joe Jones and John Busch for 454-pyrosequencing support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
About this article
Cite this article
Fortunato, C., Eiler, A., Herfort, L. et al. Determining indicator taxa across spatial and seasonal gradients in the Columbia River coastal margin. ISME J 7, 1899–1911 (2013). https://doi.org/10.1038/ismej.2013.79
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2013.79
Keywords
This article is cited by
-
Bacterial community composition of the sediment in Sayram Lake, an alpine lake in the arid northwest of China
BMC Microbiology (2023)
-
Bacterial bioindicators enable biological status classification along the continental Danube river
Communications Biology (2023)
-
Temporal and Spatial Variation Characteristics and Influencing Factors of Bacterial Community in Urban Landscape Lakes
Microbial Ecology (2023)
-
Seasonal and inter-annual variability of bacterioplankton communities in the subtropical Pearl River Estuary, China
Environmental Science and Pollution Research (2022)
-
Sponge holobionts shift their prokaryotic communities and antimicrobial activity from shallow to lower mesophotic depths
Antonie van Leeuwenhoek (2022)
