
Abstract
Bacteriovorax marinus SJ is a predatory delta-proteobacterium isolated from a marine environment. The genome sequence of this strain provides an interesting contrast to that of the terrestrial predatory bacterium Bdellovibrio bacteriovorus HD100. Based on their predatory lifestyle, Bacteriovorax were originally designated as members of the genus Bdellovibrio but subsequently were re-assigned to a new genus and family based on genetic and phenotypic differences. B. marinus attaches to Gram-negative bacteria, penetrates through the cell wall to form a bdelloplast, in which it replicates, as shown using microscopy. Bacteriovorax is distinct, as it shares only 30% of its gene products with its closest sequenced relatives. Remarkably, 34% of predicted genes over 500 nt in length were completely unique with no significant matches in the databases. As expected, Bacteriovorax shares several characteristic loci with the other delta-proteobacteria. A geneset shared between Bacteriovorax and Bdellovibrio that is not conserved among other delta-proteobacteria such as Myxobacteria (which destroy prey bacteria externally via lysis), or the non-predatory Desulfo-bacteria and Geobacter species was identified. These 291 gene orthologues common to both Bacteriovorax and Bdellovibrio may be the key indicators of host-interaction predatory-specific processes required for prey entry. The locus from Bdellovibrio bacteriovorus is implicated in the switch from predatory to prey/host-independent growth. Although the locus is conserved in B. marinus, the sequence has only limited similarity. The results of this study advance understanding of both the similarities and differences between Bdellovibrio and Bacteriovorax and confirm the distant relationship between the two and their separation into different families.
Similar content being viewed by others

Introduction
Bacteriovorax is a genus of bacteria within the unique group of predatory prokaryotes known as the Bdellovibrio and like organisms (BALOs) that prey upon Gram-negative bacteria. These predatory bacteria share a unique lifecycle consisting of two distinct phases, the predatory attack phase that is highly motile, which facilitates predation and an intraperiplasmic growth phase. On colliding with their prey, the BALOs penetrate the cell wall and lodge in the periplasm where they alter the cell wall to form a bdelloplast, grow, multiply and finally free themselves by lysing the prey cell wall.
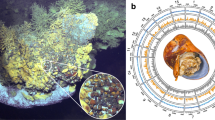
Although Bacteriovorax share some attributes with Bd. bacteriovorus, they are distinct in several important features including the requirement of the former for sodium chloride (>0.5%), for growth and multiplication and a lower % G+C ratio, in the mid 30%s (Marbach et al., 1976), compared with about 50% for the freshwater group (Seidler et al., 1972). Salt tolerant or marine Bacteriovorax organisms also differ from their terrestrial cousins by being generally smaller, Bdellovibrio are typically 0.2–0.5 μm in width and 0.5–2.5 μm in length (Burnham et al., 1968; Burnham and Conti, 1984), whereas Bacteriovorax ranges from 0.6 to 1.0 μm in length in our collection of electron micrographs (Figures 1a–c), thereby passing through a 0.2 μm filter. Bacteriovorax also thrive at lower temperatures than freshwater BALOs (Uematsu and Wakimoto, 1971; Williams, 1979) and have a preference for prey native to estuarine or other saltwater environments (Marbach et al., 1976; Sutton and Besant, 1994). Formerly, Bacteriovorax was in the genus Bdellovibrio (Conti and Burnham, 1984) but was not given a species name. The designation of marine Bdellovibrio sp., persisted for more than a decade.

Electron micrographs of B. marinus SJ (a) single attack phase flagellate B. marinus cell (b) inhabiting the intraperiplasmic space of Vibrio parahaemolyticus prey cells, and (c) septating into progeny.
Recent advances in the taxonomy and systematics of BALOs have proceeded with knowledge of 16S ribosomal DNA sequences. This approach was most useful as wild type obligate predators have not been grown in pure culture, a necessary step for many metabolic and biochemical methods traditionally used to characterize and classify bacteria. These predators can be grown in co-culture solely with prey bacteria. The 16S ribosomal DNA sequence data from isolates of the saltwater and freshwater species revealed major differences and have resulted in establishment of new genera, Bacteriovorax, and family, Bacteriovoraxaceae, for the marine groups (Baer et al., 2000; Snyder et al., 2002; Baer et al., 2004; Davidov and Jurkevitch, 2004) and a second family, Peredibacteraceae that includes some of the freshwater strains (Pineiro et al., 2007). Subsequently, further differentiation based on sequence comparisons of the rpoB gene revealed that there were at least eight operational taxonomic units within the marine group (Pineiro et al., 2007). Prey or host-independent (HI) mutants of marine (Bacteriovorax marinus and Bacteriovorax litoralis) and freshwater (Bd. bacteriovorus 109J) species have been isolated in the laboratory, and tested for metabolic characteristics (Shoeffield, 1990; Seidler and Starr, 1969).
Both Bd. bacteriovorus and B. marinus are members of the delta-proteobacteria. This phylum includes organisms highly diverse in terms of niche, metabolic strategies and adaptations (Karlin et al., 2006). Members of the delta-proteobacteria share a few particular genomic features but use distinct ecological strategies such as using iron as electron acceptor (Geobacter metallireducens), swarming with self-organizing behavior, (Myxococcus xanthus), and as with B. marinus, exhibiting a predatory lifestyle (Coppi et al., 2004; Goldman et al., 2006).
B. marinus strains are unusual and potentially useful (Sockett and Lambert, 2004), but have not been studied extensively. To date, only a terrestrial strain Bd. bacteriovorus HD100, has been sequenced (Rendulic et al., 2004). Here, we report the complete sequencing and analyses of the first halophilic Bacteriovorax genome. B. marinus SJT (=ATCC BAA-682T=DSM 15412T) was isolated from coastal waters near St John’s Island, US Virgin Islands (Baer et al., 2004). Bacteriovorax have a ubiquitous distribution in saltwater environments. However, not all operational taxonomic units are similarly distributed. Some have a widespread distribution and others are restricted to certain ecosystems. B. marinus is placed in operational taxonomic units phylogenetic Cluster III based upon similarity of the 16S ribosomal RNA gene sequences. In a global survey this group was found to be the most widely distributed in oceans and seas among the eight phylotype clusters identified (Pineiro et al., 2007). It is typically found in higher salinity marine environments than in lower salinity brackish waters and estuarine systems. Differences between ocean and estuarine isolates were revealed by 16S rDNA sequence analysis.
Comparisons between the genomes of Bdellovibrio and Bacteriovorax will advance knowledge of these unique predators, their predatory behavior and other common and distinct features. The results of this study will also expand interest in sequencing Bacteriovorax and other BALO phylotypes that vary in their ecology and genomic profile.
Materials and methods
For DNA isolation, B. marinus SJ was grown in prey seawater broth using Vibrio parahemolyticus as the prey bacterium as previously described (Williams and Baer, 2005). Briefly, prey (108 to 109) was inoculated into sterile seawater to which a 24–48 h culture (filtered through 0.2 μm filter to remove prey cells) of SJ was added and the turbid suspension was incubated on a shaker at 25° C until the turbidity cleared. The culture was filtered to remove prey cells and the filtrate centrifuged to pellet the SJ cells from which DNA was extracted.
DNA sequencing was carried out using Sanger sequencing technology. DNA was sonicated, size selected and libraries were constructed in pUC19, pMAQ1b and pBACe3.6. The genome assembly was based on paired end-reads from pUC19 and pMAQ1b libraries to give 10-fold sequence coverage of the genome. Directed finishing reads were used to produce scaffolds from large-insert libraries in pBACe3.6. Repeats were bridged using read-pairs or end-sequenced PCR products. Gene prediction was carried out on the complete finished genome sequence using the programs Glimmer3 and Orpheus, and manually curated (Frishman et al., 1998; Delcher et al., 1999). Annotation was carried out using the Artemis (Rutherford et al., 2000) platform, with marked InterPro (Hunter et al., 2009), TMHMM (Sonnhammer et al., 1998) and SigP (Bendtsen et al., 2004) domains. The transfer RNAs (tRNAs) were determined using tRNAscan-SE (Lowe and Eddy, 1997) and ribosomal RNAs were determined using BLASTN and manual markup.
Phylogenetic Tree analysis was carried out with 1000 replicates using the RAxML-VI-HPC (Stamatakis, 2006), Phylip (Felsenstein, 2005) and NJ Plot (Perriere and Gouy, 1996) programs. This was independently verified using neighbor joining and whole-genome composition vector Tree (Xu and Hao, 2009a) approaches.
Gene orthologues were calculated by reciprocal best match (bidirectional best hit analysis using Fasta global alignments with sequence identity cutoffs as described in the text. Orthologues were removed from the B. marinus annotation using Artemis Select Function to leave genes shared only with Bdellovibrio that are not found in other members of the delta-proteobacteria. This geneset was additionally verified using a unidirectional (BLASTP) best hit analysis and visualized manually. Accession numbers of the strains used in the study are as follows: Bd. bacteriovorus HD100 (NC_005363.1), Lawsonia intracellularis PHE/MN1-00 (NC_008011.1), Myxococcus xanthus DK1622 (NC_008095.1), Anaeromyxobacter dehalogenans 2CP-1 (NC_011891.1), Pelobacter carbinolicus DSM238 0 (NC_007498.2), Geobacter metallireducens GS-15 (NC_007517.1), Haliangium ochraceum DSM14365 (NC_013440.1), Desulfatibacillum alkenivorans AK-01 (CP001322.1), Desulfovibrio desulfuricans subsp. desulfuricans strain ATCC27774 (NC_011883.1), Desulfovibrio vulgaris str. Hildenborough (NC_002937), Desulfococcus oleovorans Hxd3 (NC_009943.1), Desulfohalobium retbaense DSM 5692 (NC_13223.1), Sorangium cellulosum ‘Soce56’ (NC_010162.1), Syntrophus aciditrophicus SB (NC_007759.1), Syntrophobacter fumaroxidans MPO (NC_008554.1).
Electron microscopy was carried out at University of Florida, USA (UF) and University of Nottingham, UK. A 50 ml sample of 4 and 24 h culture of the predators and prey was fixed for electron microscopy examination according to Koval and Bayer (1997) with modification. Briefly, samples were centrifuged for 30 min at 27 485 g, resuspended in 1 ml of 0.1 M sodium phosphate buffer (pH 7) and centrifuged for 20 min at 20 142 × g. The pellets were resuspended in 2 ml of 0.1 M cacodylate buffer containing 2% glutaraldehyde and 1% formaldehyde, both diluted from 25% (v/v) and 16% (v/v) stock solutions, respectively. After 1 h at 4 °C and centrifugation at 20 142 g for 20 min, the pellets were overlaid with cacodylate buffer. Samples were embedded in epoxy for further study of thin sectons. Cells were post-fixed in OsO4, washed, dehydrated and embedded in resin. Ultrathin sections were counterstained with uranyl acetate and lead citrate and examined with a Hitachi H-7600 transmission electron microscope Hitachi high technologies America Inc., Pleasanton, CA, USA.
Results
Electron microscopy studies show B. marinus SJ as a predatory species
To determine whether B. marinus carries out the same predatory strategy as the terrestrial Bd. bacteriovorus, or other halophilic Bacteriovorax, we examined cells co-cultured with Vibrio parahaemolyticus. The results from the laboratories of two independent groups show conclusively the invasion and replication tactic employed by B. marinus is the same as Bd. bacteriovorus. Typically, the first observable interaction of BALOs with its prey is attachment (Figure 1a). Following attachment is penetration (not shown) and invasion into the periplasmic space of the prey (Figure 1b) where a predator cell alters the prey cell wall, consumes the cytoplasmic contents of the prey and grows to fully occupy the area within the prey cell. The final steps in the intracellular cycle are multiplication (Figure 1c shows predator cells septating to form new cells) and lysis of the prey cell releasing predator progeny. The predatory growth of Bx. marinus had not previously been demonstrated unequivocally.
Comparison of B. marinus to the features of delta-proteobacteria
The genome of B. marinus strain SJ comprises a single circular chromosome and a small plasmid. The chromosome is moderately short at 3 435 933 bp with 3230 predicted coding sequences. Eighteen pseudogenes and two ribosomal RNA operons were identified in the sequence. The general features of the chromosome are shown in Table 1. Remarkably, 1116 (34.5%) of the predicted genes of over 500 nt in length were found to have no significant hit in the databases (including the GenBank marine metagenomics samples), nor did they possess any Pfam domains. Overall, 4496 hits to Bacteriovorax were found in the MG-RAST databases.
The delta-proteobacteria are a highly diverse class of the phylum Proteobacteria. A phylogenetic tree shows the positioning of B. marinus SJ within the delta-proteobacteria (Figure 2). While the closest sequenced relative to B. marinus SJ is Bd. bacteriovorus, no genomic synteny is observed by dotplot or by Artemis Comparison Tool (Carver et al., 2005) BLASTN analysis. Using Artemis Comparison Tool with a more sensitive TBLASTX indicates only very short match regions. MUMmer (Kurtz et al., 2004) alignments indicated only one short region showing direct synteny that corresponds to the ribosomal proteins (Supplementary Figure 1).

Phylogenetic tree based on small subunit ribosomal RNA to show the placing of B. marinus SJ among the delta-proteobacteria. Numbers on branches correspond to bootstrap values based on 1000 replicates. This phylogenetic tree was produced with maximum likelihood with bootstrapping by RAxML (Stamatakis, 2006) and Phylip (Felsenstein, 2005). Species included in this analysis are as listed on the tree and in the methods. The topology of the tree was conserved (excluding B.marinus SJ) using the neighbor-joining method in the Integrated Microbial browser and with the composition vector Tree (Xu and Hao, 2009b) approach (excluding Desulfatibacillum alkenivorans and Myxococcus xanthus). Escherichia coli was used to root the tree.
From investigations by Karlin et al. (2006), the major characteristics of delta-proteobacteria are as follows:
-
1
Delta-proteobacteria have two giant S1 ribosomal proteins genes compared with one or none in other bacteria.
-
2
In most delta-proteobacteria, the major ribosomal protein gene cluster near the origin.
-
3
Delta-proteobacteria have a rare combination of discriminating asparagine and glutamine tRNA synthetases to modify Asp-tRNA into Asn-tRNA and Glu-tRNA into Gln-tRNA.
-
4
Delta-proteobacteria have numerous TonB receptors and ferric siderophore receptors.
-
5
Anaerobic deltas have mutiple copies of reuberythrin.
-
6
Sigma 54 has a more important role in delta-proteobacteria than other bacteria.
-
7
There are multiple beta lactamases.
The genome of B. marinus appears to conform to some, but not all of these findings.
Surprisingly, there is only one giant S1 protein in B. marinus, only five TonB Pfam domains and only three large putative TonB receptor proteins encoded in the genome. However, in keeping with other delta-proteobacteria, the major ribosomal protein gene cluster is located near the ori. Discriminating asparagine and glutamine tRNA synthetases are found in the genome, and there are 11 Pfam domains classified as beta lactamases or beta-lactamase-like.
Plasmid, general structure and function
B. marinus possesses a unique plasmid of 1973, bp and of 36.2% GC content, designated pBMS1. The % GC content is in accordance with that of the chromosome. There is only one strongly predicted coding sequence on the plasmid, repA. Throughout the remainder of this short plasmid, there are also a number of short direct and palindromic DNA repeats and a potential stem-loop structure. This plasmid may represent a truly selfish piece of DNA, in that it may only carry one gene specifying its replication. A selfish plasmid, pKL1, has been described from E. coli that carries only a repA gene (Burian et al., 1997). In addition, the German outbreak E. coli O104:H4 also carries a selfish plasmid (BGI, 2011).
Although the repA gene remains the only strongly predicted coding sequence, BLAST analysis indicates a predicted 38-amino-acid polypeptide with N-terminal matches to conserved hypothetical genes. Recently, two plasmids have been characterized from Bd. bacteriovorus (Roschanski and Strauch, 2011), however, these plasmids do not resemble pBMS1.
Orthologous genes shared with delta-proteobacteria
To date, B. marinus SJ remains divergent from all other sequenced prokaryotes. Table 2 shows a list of important genes with their June 2012 top BLASTP hit against the non-redundant databases shown by sequence identity, while Figure 3 indicates the number of orthologous genes shared with other delta-proteobacteria. Only four coding sequences have an 80% or higher identity shared with its closest sequenced relative, Bd. bacteriovorus. Investigations indicate a common delta core solely consisting of 891 genes in bidirectional best hit analyses with the genome sequences of the species described in the Materials and methods section that represent different clades of the delta-proteobacteria.

Orthologous genes shared between delta-proteobacteria (by percentage identity) as determined by bidirectional best hit analysis. Bar graph to show the percentage identities of B. marinus SJ gene orthologues with other delta-proteobacteria. Orthologues were calculated by bidirectional best hit analysis, altering the percentage identity cutoff between 30–90%. The species of delta-proteobacteria used in the investigation were: Anaeromyxobacter dehalogenans, Desulfotalea psychrophila, Bdellovibrio bacteriovorus, Desulfovibrio vulgaris, Geobacter metallireducens, Lawsonia intracellularis, Myxococcus xanthus, Pelobacter carbinolicus and Syntrophus aciditrophicus.
Genomic islands of B. marinus
Regions of difference were determined. Some of these genomic islands of B. marinus were (i) unique to B. marinus among delta-proteobacteria or (ii) shared only with Bd. bacteriovorus (Figure 4). Each of these genetic regions has been systematically numbered, and the key coding sequences (CDS) carried on these islands are listed in Supplementary Table 1.

Orthologous genes shared with delta-proteobacteria as determined by bidirectional best hit analysis. The circles on the diagram are as follows (outermost to innermost): B. marinus SJ DNA coordinates. Regions of major differences, known as BMRD for B. marinus regions of difference (marked in black). Orthologous genes from the following genera, with all predicted orthologues shown on the forward strand only, for clarity: Bdellovibrio, Myxococcus, Anaeromyxobacter, Desulfovibrio, Desulfococcus, Pelobacter, Lawsonia, Sorangium, GC content, GC skew.
Following alien hunter analysis (Vernikos and Parkhill, 2006), approximately half of the genomic islands detected above were potentially acquired by horizontal gene transfer. These regions have been numbered systematically and are listed in and summarized in Table 3. The phylogenetic origin of the potentially horizontally transferred regions was accessed by dividing the regions into 150 bp sequences and compared with Uniprot using BLAST. The output was entered into MEGAN (Huson et al., 2007). Approximately 1/3 of the sequences had no hit, 1/3 matched unassigned sequences and 1/4 matched proteo-bacterial sequences (Supplementary Figure 2). In the main, the regions of difference carry large amounts of conserved and hypothetical proteins, many of which are predicted by the SigP program to be exported (Emanuelsson et al., 2007). For example, BMRD (B. marinus major regions of difference)8 and 14 carry secondary efflux transporters of the RND family. BMRD27 carries a predicted ABC transporter and a putative sigma factor, while BMRD28 and 29 carry pili and glycosyl transferases and a methyl-accepting chemotaxis protein, respectively. BMRD1 carries some amino acid biosynthetic enzymes and BMRD9 carries CDS of bacteriophage origin in addition to a putative type I restriction-modification system.
In addition to the larger islands described above, there are potentially laterally transferred regions that comprise few or single genes. One such gene, BMS1619, shares high sequence similarity with a hemaggluntinin/hemolysin of Staphylococcus epidermidis, a surface protein from the planctomycete Rhodopirellula baltica, and two separate CDS from Bd. bacteriovorus. This potential island has a very high alien hunter score (>54) consistent with horizontal gene transfer. A second gene, BMS_3202, has another high alien hunter score (>32) and shares sequence similarity with a cell surface/hemagglutinin protein of the plant pathogenic bacterium Xanthomonas campestris.
Larger genomic islands present in Bd. bacteriovorus and B. marinus and not in free-living delta-proteobacteria are of interest in that they may represent islands involved or required for predation and/or host-interaction. Most of these islands carry high quantities of transmembrane and exported proteins. BMRD11 carries glycosyl transferases in addition to other transmembrane/exported proteins. BMRD26 carries a gene with similarity to microcin immunity (BMS2821), together with xanthine dehydrogenase, chemotaxis and efflux transporter proteins.
Genes important in predation
A small common core of 891 genes is shared between Bacteriovorax and Bd. bacteriovorus genomes. Using an in silico subtractive hybridization using both bidirectional and unidirectional best hit analysis revealed 291 genes were shared between Bacteriovorax and Bdellovibrio but not shared with the common core gene data set of delta-proteobacteria. The orthologous genes from the in silico subtractive hybridization may represent the core set required for bacterial invasion, macromolecular consumption and escape from the (eventually) degraded prey cell or bdelloplast. Identifying them by comparative genomics is a potential insight into the machinery of intracellular bacterial predation.
Gene expression in Bdellovibrio at the beginning of the predatory cycle has been studied by a transcriptomic analysis at only 30 min of predator–prey interaction (Lambert et al., 2010). At this time-point the Bdellovibrio are attaching to the outer-membrane of the prey, entering the prey periplasm, resealing the prey outer-membrane and inducing the genes whose products will commence the systematic digestion of the prey cell. Remarkably, only 15 of the 291 genes were observed as upregulated in Bdellovibrio at this time-point. Ten of these genes (BMS_1308, BMS_0406, CMS_2372, BMS_2940, BMS_2521, BMS_1074, BMS_0590, BMS_1798, BMS_0407) and BMS_2266 are homologous to Bdellovibrio genes (Bd0301, Bd0370, Bd0965, Bd1176, Bd2033, Bd2288, Bd2480, Bd3094, Bd3134, Bd3748 and Bd3823) in the conserved hypothetical category and as such may represent unexplored novel pathways contributing to predator–prey interactions. The genes are scattered throughout the genomes Bacteriovorax and Bdellovibrio, reflecting the variable synteny that is evident between the genomes; however, they are enriched particularly in BMRD26, which also shares genes in common with the predator-lysis bacterium Myxococcus xanthus and are not found in non-predatory delta-proteobacteria. Present in this region are transmembrane proteins and regulators (Supplementary Table 1).
The remaining five orthologues with a predicted encoded function include BMS_2143, the homolog of Bd0087. This resembles a guanosine triphosphate cyclohydrolase that encodes the de novo synthesis pathway for tetrahydrofolate, a coenzyme involved in transfer of one-carbon groups. A second orthologue, BMS_0990 (a Bd1990 homolog), is annotated as a gene encoding a putative auxin regulated protein as it has a Pfam domain denoted as plant auxin responding. Third, BMS3383 (Bd3359) has a RAS-like guanosine-5′-triphosphatase motif and may be involved in signal transduction as is the case in Myxobacteria (Leonardy et al., 2010). Fourth, BMS_1097 (Bd3533) has serine esterase similarity and finally, BMS_2214 (Bd1370) is annotated as an RNA helicase.
Motility/chemotaxis
Motility and chemotaxis is important for predation in Bd. bacteriovorus (Lambert et al., 2003, 2006). In B. marinus, there are eight predicted methyl-accepting chemotaxis proteins (MCPs), with one extra MCP found as a pseudogene with a central stop codon. The MCPs are important in predation in Bd. bacteriovorus where chemotaxis aids detection of high concentrations of prey bacteria (Lambert et al., 2003, 2006) B. marinus has an apparent full complement of motility and chemotaxis genes, although the number of fliCs encoding the flagellar filament is five rather than the six found in Bdellovibrio genes (Lambert et al., 2006). A key fliC3 gene, required for motility in Bd. bacteriovorus and important in predation has a close homolog in B. marinus. In contrast to the three pairs of motAB genes found in Bd. bacteriovorus (Morehouse et al., 2011), a sole pair of flagellar motor protein genes is present in B. marinus.
Type IV pili are encoded by both of the above predatory species. Type IV pili are generally implicated in gliding/twitching motility, biofilm formation and pathogenicity/host interaction (Evans et al., 2007). Type IV pili are essential for predation in Bdellovibrio (Evans et al., 2007; Mahmoud and Koval, 2010). This homology may reflect the common prey invasion mechanisms of these bacteria.
Host interaction (hit) locus
Spontaneous mutants of Bd. bacteriovorus that did not require host cells for growth on rich media (Cotter and Thomashow, 1992) were referred to as HI mutants and shown to have lesions in the hit locus. A short open reading frame within a gene cluster encoding structural elements of a type IV pilus (tadAB) and a cellulose binding protein (wapA) was mutated in these mutants and termed hit locus (Cotter and Thomashow, 1992; Rendulic et al., 2004). Predatory growth can be restored on introduction of a wild-type hit gene to saprophytic mutants (Roschanski et al., 2011). A subsequent study has shown that Flp pilus-like proteins encoded at this locus show increased transcription in synchronized cultures with prey bacteria by reverse transcription-PCR, whereas transcriptional activity was reduced in intracellular replication phase (Schwudke et al., 2005). More recently, mariner transposon mutants were isolated in this locus and implicated in predation of E.coli biofilms (Medina et al., 2008). Thus, the hit locus is thought to be involved in host invasion and penetration.
Synteny in the core region of the hit locus (Figure 5) is conserved between Bd. bacteriovorus and B. marinus where genes upstream of cpaBC in B. marinus also show limited similarity with pilus assembly genes, including tadV and pilA. The position of the tad genes for Flp pilus assembly being conserved in the hit locus of B. marinus and Bd. bacteriovorus suggests these genes are important. This region is syntenic except that in Bacteriovorax the region is inverted with respect to the Bdellovibrio sequence. It has been suggested that the hit locus bears some resemblance to the widespread colonization island (Tomich et al., 2007). Although there is one putative Flp pilus coding sequences in Myxococcus xanthus, and Desulfovibrio desulfuricans G20 has two potential coding sequences involved in flp pili production, the hit locus is not intact in other delta-proteobacteria. This is as expected should the hit locus have an important involvement in predation, as the other sequenced delta-proteobacteria are free-living and non-parasitic.

tBLASTx analysis of the hit locus region comparing conservation and genome synteny. Artemis comparison tool (ACT (Carver et al., 2005)) tBLASTx investigation of the hit locus. The top of the figure shows the hit locus in B. marinus SJ. The middle line constitutes the hit locus of Bd. bacteriovorus HD100. The extent of the described hit locus in Bd. bacteriovorus (Schwudke et al., 2005) is shown. The base of the figure shows a partial match equivalent to the hit locus region in Desulfovibrio desulfuricans G20. This organism, as all other sequenced delta-proteobacteria, does not have a hit orthologue present.
Recent findings highlighted the importance of a single gene, hit, in Bd. bacteriovorus within the hit locus in the conversion from HI growth to predatory growth (Wurtzel et al., 2010; Roschanski et al., 2011). Deletion of hit abolished plaque formation on prey lawns, which was restored by complementation with just this gene. Additionally, mutations that genuinely affected HI growth resided solely within hit, implying that hit is required, in a direct or regulatory role, for predatory entry to other bacteria. Given this importance it is striking that at the same locus in Bacteriovorax, a gene duplication event has given rise to two gene products, BMS0183 and BMS0184, each with limited similarity to Bd. bacteriovorus hit. The 18-N-terminal amino-acids of all three genes are highly conserved. Both B. marinus genes are 34-amino-acids shorter than 101 amino acids of hit and have limited similarity in the 60-C-terminal amino-acids of hit. This low similarity was unexpected because C-terminal deletions and point mutations causing truncations or frameshifts after codon 30 in hit were common in Bdellovibrio HI strains (Barel and Jurkevitch, 2001).
Other aspects
Analysis of the B. marinus genome was undertaken using the program Pathway Tools (Karp et al., 2002), including the pathway hole filler (Green and Karp, 2004), and KEGG analysis (Ogata et al., 1999). The results indicate that B. marinus is able to synthesize many but not all amino acids, for example, complete pathways are lacking for the biosynthesis of arginine, histidine, isoleucine, phenylalanine, threonine, tryptophan, tyrosine and valine (or selenocysteine). Bd. bacteriovorus is similarly predicatively unable to synthesize the following amino acids: arginine, histidine, isoleucine, methionine, phenylalanine, tryptophan and valine (Rendulic et al., 2004). These findings are suggestive that the organism can gain essential amino acids from its prey.
It remains possible that these metabolic pathways are unusual in these organisms, and that the method of synthesizing these amino acids cannot be recognized. Interestingly, asparagine synthetase along with a divergently transcribed AsnC family transcriptional regulator is found in a region unique to B. marinus among deltas. A tRNA-Arg is also located here (BMRD1; see Supplementary Table 1). A full complement of tRNA synthetases is present in the genome. In terms of nucleotide synthesis, the ready-made DNA supplied by the prey is sufficient for one extra progeny predator cell to form, yet to form several progeny the predator must convert other prey cellular materials de novo to DNA and RNA.
Discussion
The delta-proteobacteria are a highly diverse phylogenetic group and as expected, the sequenced B. marinus SJ strain conforms to most, but not all of the expected characteristics of phylum. B. marinus SJ is most closely related to Bd. bacteriovorus, however, they are very distinct genomically. B. marinus SJ has a unique gene content that comprises 66% of the genome when bidirectional best hit analyses are performed against other sequenced delta-proteobacteria at a 30% level of identity across most of the sequence. In addition, hypothetical genes of larger than 500 nucleotides predicted by ab-initio and manual curation methods with no significant BLASTP matches in the non-redundant sequence databases as of July 2011 constitute 35% of the genome. These regions did not appear to be repetitive when searched using the Reputer (Kurtz and Schleiermacher, 1999) and MUMmer (Kurtz et al., 2004) packages. Several of these predicted hypothetical coding sequences are found in pairs in the genome. Of these pairs, five were related by sequence albeit slightly to other pairs in the genome, and a further, separate two pairs were related. The size and organization of these gene pairs is reminiscent of insertion sequence mobile elements, but there is no indication that these could represent such elements by domain or sequence similarity.
The high level of sequence diversity is rarely seen in current sequencing projects (Young et al., 2006; Crossman et al. 2008a, 2008b; Holden et al., 2009; Crossman et al., 2010; Carter et al., 2011; Crossman, 2011; Heavens et al., 2011; Seipke et al., 2011). It raises the question of the origin of such unique DNA. Bacteriovorax marinus SJ appears to be related to Bd. bacteriovorus by phylogenetic analysis with long branch length and may represent an ancient delta-proteobacterium. The unusual lifestyle of this predatory bacterium lends weight to a suggestion that some of the unique DNA could represent sequences that have been gained from prey consistent with our analysis that 25% of the sequences are most closely related to bacterial sequences (Supplementary Figure 2). We have observed in our laboratory that SJ is capable of preying on several other bacteria including V. vulnificus, V. cholera, a Vibrio sp. and E. coli and may prey on the wealth of bacteria in oceans that have never before been cultured or sequenced. Effectively this organism may have been carrying out its own DNA sampling of prey bacteria.
Unique hypothetical genes are sprinkled throughout the genome rather than being confined to predicted islands, although BMRD9 carries the highest concentration of these genes. In addition to the BMRD, there are also short islands that carry a few or single genes such as one with similarity to haemagglutinins implicated in adhesion in Xanthomonas campestris that may be involved in attachment/recognition. Hemagglutinins have been implicated in adhesion and may be involved in plant–parasite relationships in plant pathogenic bacteria (Alavi et al., 2008).
Orthologous genes shared between Bacteriovorax and Bdellovibrio from an in silico subtractive hybridization may represent the core set required for the invasive predatory processes. Little is known about the mechanisms employed by the invasive Bdellovibrio-like predatory bacteria (also known as BALOs) during invasion and intraperiplasmic growth phase when they utilize prey macromolecules for biosynthesis and energy metabolism. However, it is known that motility/chemotaxis and pili are important factors in predation in Bd. bacteriovorus.
Few studies have addressed the metabolic and energy pathways for the BALOs and much remains to be done in this area. Of the studies that have been carried out, nearly all have been on strains of Bd. bacteriovorus or other freshwater/soil BALOs. None have focussed on the saltwater species, Bacteriovorax, other than to study the utilization patterns of some nutrient substrates. It is obvious that the prey cell can provide all the nutrients necessary for the intraperiplasmic growth of the predators in the case of both the freshwater and saltwater species. Beyond that, results of studies on Bd. bacteriovorus have shown that the organisms are capable of utilizing prey cell-derived amino acids as a major energy source for growth within the prey’s intraperiplasmic space (Hespell et al., 1973). Hespell later reported that Bd. bacteriovorus has many of the enzymes of the tricarboxylic acid cycle and that during intraperiplasmic growth these enzymes increase (Hespell, 1976). A number of proteolytic enzymes associated with BALO predation cycle have been reported from studies on the freshwater BALOs. A recent report describes the twin-arginine translocation system in Bdellovibrio, which transports folded proteins across the cytoplasmic membrane as it does in other bacteria (Chang et al., 2011). Recent observations in our laboratory have revealed that the Bacteriovorax and Bdellovibrio and other freshwater BALOs, with few exceptions, are not capable of predation on the same genera of bacteria perhaps because a different set of enzymes are required. Additional study on the metabolic machinery and growth requirements of the saltwater Bacteriovorax is needed to clarify the similarities or differences between this group and the freshwater BALOs.
Specific flagellar gene differences between Bacteriovorax and Bdellovibrio genomes can reflect the marine versus terrestrial lifestyles of the two bacteria but reinforce the importance of motility in encountering prey in both environments. Previous investigators (Lambert et al., 2006; Iida et al., 2009) showed that in Bd. bacteriovorus of six fliC genes only, fliC3, is essential for flagellar motility and hence efficient location of/collision with prey. The highest similarity among the fli orthologues is with the fliC3 gene product at 70%. Thus, the importance of flagellar motility for prey encounters is strong in Bacteriovorax to hunt prey-rich regions in the oceans. Related to that marine lifestyle, a sole pair of flagellar ion-channel flagellar motor protein genes are present in the Bacteriovorax genome rather than the three pairs, which were all shown to contribute to the single flagellar motor of Bd. bacteriovorus (Morehouse et al., 2011).
A host-interaction (hit) locus identified in Bd. bacteriovorus is only partially conserved in B. marinus. A hit gene is tandemly duplicated giving rise to the BMS0183 and BMS0184 genes of divergent proteins. We found this surprising given that this protein has been implicated in Bdellovibrio, by two separate research groups, as being uniquely important to strains switching between predatory and prey-independent HI growth (Wurtzel et al., 2010; Roschanski et al., 2011) and suggesting that the conserved genes may be the only ones necessary for a successful predator–prey interaction.
There is a theory that these predatory bacteria or certain of their enzymes could be used as therapeutic agents in a biological control type scenario. Furthermore, predators could be designed to invade specific cell types should the molecular machinery of predation be fully understood. The halophilic nature of B. marinus could be beneficial in its use for treating infections in humans and certain animals whose body environment is at 0.85 percent salts, tolerable for many of the halophilic predators. This theory will be interesting to explore and the genome sequence of Bacteriovorax, together with the comparison with Bd. bacteriovorus, provides us with a major stepping-stone to achieve the application of the predators as antibacterial agents. Understanding the machinery of predation may advance consideration of the possible use of these predatory bacteria or their enzymes as therapeutic agents or alternative applications in biological control.
References
Alavi SM, Sanjari S, Durand F, Brin C, Manceau C, Poussier S . (2008). Assessment of the genetic diversity of Xanthomonas axonopodis pv. phaseoli and Xanthomonas fuscans subsp. fuscans as a basis to identify putative pathogenicity genes and a type III secretion system of the SPI-1 family by multiple suppression subtractive hybridizations. Appl Environ Microbiol 74: 3295–3301.
Baer ML, Ravel J, Chun J, Hill RT, Williams HN . (2000). A proposal for the reclassification of Bdellovibrio stolpii and Bdellovibrio starrii into a new genus, Bacteriovorax gen. nov. as Bacteriovorax stolpii comb. nov. and Bacteriovorax starrii comb. nov., respectively. Int J Syst Evol Microbiol 50 (Part 1): 219–224.
Baer ML, Ravel J, Pineiro SA, Guether-Borg D, Williams HN . (2004). Reclassification of salt-water Bdellovibrio sp. as Bacteriovorax marinus sp. nov. and Bacteriovorax litoralis sp. nov. Int J Syst Evol Microbiol 54: 1011–1016.
Barel G, Jurkevitch E . (2001). Analysis of phenotypic diversity among host-independent mutants of Bdellovibrio bacteriovorus 109J. Arch Microbiol 176: 211–216.
Bendtsen JD, Nielsen H, von Heijne G, Brunak S . (2004). Improved prediction of signal peptides: signalp 3.0. J Mol Biol 340: 783–795.
BGI (2011). The complete map of the Germany E.coli O104 genome released. EurekAlert! AAAS.
Burian J, Guller L, Macor M, Kay WW . (1997). Small cryptic plasmids of multiplasmid, clinical Escherichia coli. Plasmid 37: 2–14.
Burnham J, Conti S . (1984) Bergey’s Manual of Systematic Bacteriology. Springer: New York, p 11824.
Burnham JC, Hashimoto T, Conti SF . (1968). Electron microscopic observations on the penetration of Bdellovibrio bacteriovorus into gram-negative bacterial hosts. J Bacteriol 96: 1366–1381.
Carter AT, Pearson BM, Crossman LC, Drou N, Heavens D, Baker D et al. (2011). Complete genome sequence of the proteolytic Clostridium botulinum type A5 (B3′) strain H04402 065. J Bacteriol 193: 2351–2352.
Carver TJ, Rutherford KM, Berriman M, Rajandream MA, Barrell BG, Parkhill J . (2005). ACT: the Artemis Comparison Tool. Bioinformatics 21: 3422–3423.
Chang CY, Hobley L, Till R, Capeness M, Kanna M, Burtt W et al. (2011). The Bdellovibrio bacteriovorus twin-arginine transport system has roles in predatory and prey-independent growth. Microbiology 157: 3079–3093.
Conti S, Burnham J . (1984). Genus Bdellovibrio. In: Holt JG (ed) Bergey’s Manual of Systematic Bacteriology. Williams & Wilkins: Baltimore, MD.
Coppi MV, O′Neil RA, Lovley DR . (2004). Identification of an uptake hydrogenase required for hydrogen-dependent reduction of Fe(III) and other electron acceptors by Geobacter sulfurreducens. J Bacteriol 186: 3022–3028.
Cotter TW, Thomashow MF . (1992). Identification of a Bdellovibrio bacteriovorus genetic locus, hit, associated with the host-independent phenotype. J Bacteriol 174: 6018–6024.
Crossman L . (2011). Large Scale expansion of mobile elements in specific hotspot regions of the German outbreak Escherichia coli O104:H4. Nature Precedings Available at http://hdl.handle.net/10101/npre.2011.6466.1.
Crossman LC, Castillo-Ramirez S, McAnnula C, Lozano L, Vernikos GS, Acosta JL et al. (2008a). A common genomic framework for a diverse assembly of plasmids in the symbiotic nitrogen fixing bacteria. PLoS One 3: e2567.
Crossman LC, Gould VC, Dow JM, Vernikos GS, Okazaki A, Sebaihia M et al. (2008b). The complete genome, comparative and functional analysis of Stenotrophomonas maltophilia reveals an organism heavily shielded by drug resistance determinants. Genome Biol 9: R74.
Crossman LC, Chaudhuri RR, Beatson SA, Wells TJ, Desvaux M, Cunningham AF et al. (2010). A commensal gone bad: complete genome sequence of the prototypical enterotoxigenic Escherichia coli strain H10407. J Bacteriol 192: 5822–5831.
Davidov Y, Jurkevitch E . (2004). Diversity and evolution of Bdellovibrio-and-like organisms (BALOs), reclassification of Bacteriovorax starrii as Peredibacter starrii gen. nov., comb. nov., and description of the Bacteriovorax-Peredibacter clade as Bacteriovoracaceae fam. nov. Int J Syst Evol Microbiol 54: 1439–1452.
Delcher AL, Harmon D, Kasif S, White O, Salzberg SL . (1999). Improved microbial gene identification with GLIMMER. Nucleic Acids Res 27: 4636–4641.
Emanuelsson O, Brunak S, von Heijne G, Nielsen H . (2007). Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc 2: 953–971.
Evans KJ, Lambert C, Sockett RE . (2007). Predation by Bdellovibrio bacteriovorus HD100 requires type IV pili. J Bacteriol 189: 4850–4859.
Felsenstein J . (2005), PHYLIP (phylogeny inference package) version 3.6. Distributed by the author, Department of Genome Sciences, University of Washington, Seattle.
Frishman D, Mironov A, Mewes HW, Gelfand M . (1998). Combining diverse evidence for gene recognition in completely sequenced bacterial genomes. Nucleic Acids Res 26: 2941–2947.
Goldman BS, Nierman WC, Kaiser D, Slater SC, Durkin AS, Eisen JA et al. (2006). Evolution of sensory complexity recorded in a myxobacterial genome. Proc Natl Acad Sci USA 103: 15200–15205.
Green M, Karp P . (2004). A Bayesian method for identifying missing enzymes in predicted metabolic pathway databases. BMC Bioinformatics 9: 76.
Heavens D, Tailford LE, Crossman L, Jeffers F, Mackenzie DA, Caccamo M et al. (2011). Genome sequence of the vertebrate gut symbiont Lactobacillus reuteri ATCC 53608. J Bacteriol 193: 4015–4016.
Hespell RB . (1976). Glycolytic and tricarboxylic acid cycle enzyme activities during intraperiplasmic growth of Bdellovibrio bacteriovorus on Escherichia coli. J Bacteriol 128: 677–680.
Hespell RB, Rosson RA, Thomashow MF, Rittenberg SC . (1973). Respiration of Bdellovibrio bacteriovorus strain 109J and its energy substrates for intraperiplasmic growth. J Bacteriol 113: 1280–1288.
Holden MT, Seth-Smith HM, Crossman LC, Sebaihia M, Bentley SD, Cerdeno-Tarraga AM et al. (2009). The genome of Burkholderia cenocepacia J2315, an epidemic pathogen of cystic fibrosis patients. J Bacteriol 191: 261–277.
Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D et al. (2009). InterPro: the integrative protein signature database. Nucleic Acids Res 37: D211–D215.
Huson DH, Auch AF, Qi J, Schuster SC . (2007). MEGAN analysis of metagenomic data. Genome Res 17: 377–386.
Iida Y, Hobley L, Lambert C, Fenton AK, Sockett RE, Aizawa S . (2009). Roles of multiple flagellins in flagellar formation and flagellar growth post bdelloplast lysis in Bdellovibrio bacteriovorus. J Mol Biol 394: 1011–1021.
Karlin S, Brocchieri L, Mrazek J, Kaiser D . (2006). Distinguishing features of delta-proteobacterial genomes. Proc Natl Acad Sci USA 103: 11352–11357.
Karp PD, Paley S, Romero P . (2002). The pathway tools software. Bioinformatics 18 (Suppl 1): S225–S232.
Koval SF, Bayer ME . (1997). Bacterial capsules: no barrier against Bdellovibrio. Microbiology 143: 749–753.
Kurtz S, Schleiermacher C . (1999). REPuter: fast computation of maximal repeats in complete genomes. Bioinformatics 15: 426–427.
Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C et al. (2004). Versatile and open software for comparing large genomes. Genome Biol 5: R12.
Lambert C, Chang CY, Capeness MJ, Sockett RE . (2010). The first bite--profiling the predatosome in the bacterial pathogen. Bdellovibrio. PLoS One 5: e8599.
Lambert C, Evans KJ, Till R, Hobley L, Capeness M, Rendulic S et al. (2006). Characterizing the flagellar filament and the role of motility in bacterial prey-penetration by Bdellovibrio bacteriovorus. Mol Microbiol 60: 274–286.
Lambert C, Smith MC, Sockett RE . (2003). A novel assay to monitor predator-prey interactions for Bdellovibrio bacteriovorus 109J reveals a role for methyl-accepting chemotaxis proteins in predation. Environ Microbiol 5: 127–132.
Leonardy S, Miertzschke M, Bulyha I, Sperling E, Wittinghofer A, Sogaard-Andersen L . (2010). Regulation of dynamic polarity switching in bacteria by a Ras-like G-protein and its cognate GAP. Embo J 29: 2276–2289.
Lowe TM, Eddy SR . (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25: 955–964.
Mahmoud KK, Koval SF . (2010). Characterization of type IV pili in the life cycle of the predator bacterium Bdellovibrio. Microbiology 156: 1040–1051.
Marbach A, Varon M, Shilo M . (1976). Properties of marine bdellovibrios. Microb Ecol 2: 284–295.
Medina AA, Shanks RM, Kadouri DE . (2008). Development of a novel system for isolating genes involved in predator-prey interactions using host independent derivatives of Bdellovibrio bacteriovorus 109J. BMC Microbiol 8: 33.
Morehouse KA, Hobley L, Capeness M, Sockett RE . (2011). Three motAB stator gene products in Bdellovibrio bacteriovorus contribute to motility of a single flagellum during predatory and prey-independent growth. J Bacteriol 193: 932–943.
Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M . (1999). KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 27: 29–34.
Perriere G, Gouy M . (1996). WWW-query: an on-line retrieval system for biological sequence banks. Biochimie 78: 364–369.
Pineiro SA, Stine OC, Chauhan A, Steyert SR, Smith R, Williams HN . (2007). Global survey of diversity among environmental saltwater Bacteriovoracaceae. Environ Microbiol 9: 2441–2450.
Rendulic S, Jagtap P, Rosinus A, Eppinger M, Baar C, Lanz C et al. (2004). A predator unmasked: life cycle of Bdellovibrio bacteriovorus from a genomic perspective. Science 303: 689–692.
Roschanski N, Klages S, Reinhardt R, Linscheid M, Strauch E . (2011). Identification of genes essential for prey-independent growth of Bdellovibrio bacteriovorus HD100. J Bacteriol 193: 1745–1756.
Roschanski N, Strauch E . (2011). Assessment of the mobilizable vector plasmids pSUP202 and pSUP404.2 as genetic tools for the predatory bacterium Bdellovibrio bacteriovorus. Curr Microbiol 62: 589–596.
Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA et al. (2000). Artemis: sequence visualization and annotation. Bioinformatics 16: 944–945.
Shoeffield A . (1990). Ecological, serological and molecular characterization of halophilic bdellovibrios. PhD Thesis, University of Maryland, Baltimore, MD, USA.
Schwudke D, Bernhardt A, Beck S, Madela K, Linscheid MW, Appel B et al. (2005). Transcriptional activity of the host-interaction locus and a putative pilin gene of Bdellovibrio bacteriovorus in the predatory life cycle. Curr Microbiol 51: 310–316.
Seidler R, Mandel M, Baptist J . (1972). Molecular heterogeneity of the bdellovibrios: evidence of two new species. J Bacteriol 109: 209.
Seidler RJ, Starr MP . (1969). Isolation and characterization of host independent bdellovibrios. J Bacteriol 100: 769–785.
Seipke RF, Crossman L, Drou N, Heavens D, Bibb MJ, Caccamo M et al. (2011). Draft genome sequence of Streptomyces strain S4, a symbiont of the leaf-cutting ant Acromyrmex octospinosus. J Bacteriol 193: 4270–4271.
Snyder AR, Williams HN, Baer ML, Walker KE, Stine OC . (2002). 16S rDNA sequence analysis of environmental Bdellovibrio-and-like organisms (BALO) reveals extensive diversity. Int J Syst Evol Microbiol 52: 2089–2094.
Sockett RE, Lambert C . (2004). Bdellovibrio as therapeutic agents: a predatory renaissance? Nat Rev Microbiol 2: 669–675.
Sonnhammer EL, von Heijne G, Krogh A . (1998). A hidden Markov model for predicting transmembrane helices in protein sequences. Proc Int Conf Intell Syst Mol Biol 6: 175–182.
Stamatakis A . (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22: 2688–2690.
Sutton D, Besant P . (1994). Ecology and characteristics of bdellovibrios from three tropical marine habitats. Mar Biol 119: 313–320.
Tomich M, Planet PJ, Figurski DH . (2007). The tad locus: postcards from the widespread colonization island. Nat Rev Microbiol 5: 363–375.
Uematsu T, Wakimoto S . (1971). Biological and ecological studies on Bdellovibrio. Ann Phytopath Soc Japan 37: 52–57.
Vernikos GS, Parkhill J . (2006). Interpolated variable order motifs for identification of horizontally acquired DNA: revisiting the Salmonella pathogenicity islands. Bioinformatics 22: 2196–2203.
Williams H, Baer M . (2005). Bacteriovorax. In: Brenner KStaley and Garrity (ed) The Proteobacteria, Bergey’s 2nd Manual of Systematic Bacteriology. Springer-Verlag: New York, p 1053.
Williams HN . (1979) Cultural, Immunologic and Ecologic Studies of Marine Bdellovibrios Isolated from the Atlantic Ocean and the Chesapeake Bay. PhD Thesis, University of Maryland, Baltimore, MD, USA.
Wurtzel O, Dori-Bachash M, Pietrokovski S, Jurkevitch E, Sorek R . (2010). Mutation detection with next-generation resequencing through a mediator genome. PLoS One 5: e15628.
Xu Z, Hao B . (2009a). CVTree update: a newly designed phylogenetic study platform using composition vectors and whole genomes. Nucleic Acids Res 37: W174–W178.
Xu Z, Hao B . (2009b). CVTree update: a newly designed phylogenetic study platform using composition vectors and whole genomes. Nucleic Acids Res 37: W174–W178.
Young JP, Crossman LC, Johnston AW, Thomson NR, Ghazoui ZF, Hull KH et al. (2006). The genome of Rhizobium leguminosarum has recognizable core and accessory components. Genome Biol 7: R34.
Acknowledgements
Sequencing and annotation of Bacteriovorax marinus SJ was funded by the Wellcome Trust. Microscopy was supported by National Science Foundation award HRD 0932137 to HW and by BBSRC UK G003092/1 to RES. LC is funded by BBSRC in partnership with East of England Development Agency, Norfolk County, Norwich City and South Norfolk Councils and the Greater Norwich Development Partnership, UK. The complete manually annotated chromosome of Bacteriovorax marinus SJ is available at FQ312005; plasmid pBMS1 has been assigned accession number HE610506.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Crossman, L., Chen, H., Cerdeño-Tárraga, AM. et al. A small predatory core genome in the divergent marine Bacteriovorax marinus SJ and the terrestrial Bdellovibrio bacteriovorus. ISME J 7, 148–160 (2013). https://doi.org/10.1038/ismej.2012.90
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2012.90
Keywords
This article is cited by
-
Phagocytosis-like cell engulfment by a planctomycete bacterium
Nature Communications (2019)
-
Identification and Characterization of Differentially-Regulated Type IVb Pilin Genes Necessary for Predation in Obligate Bacterial Predators
Scientific Reports (2017)
-
Correlating microbial community compositions with environmental factors in activated sludge from four full-scale municipal wastewater treatment plants in Shanghai, China
Applied Microbiology and Biotechnology (2016)
-
Draft genome sequences for the obligate bacterial predators Bacteriovorax spp. of four phylogenetic clusters
Standards in Genomic Sciences (2015)
-
Ankyrin-mediated self-protection during cell invasion by the bacterial predator Bdellovibrio bacteriovorus
Nature Communications (2015)

{kind=link}
{kind=link}