
Abstract
Verrucomicrobia is a bacterial phylum that is commonly detected in soil, but little is known about the distribution and diversity of this phylum in the marine environment. To address this, we analyzed the marine microbial community composition in 506 samples from the International Census of Marine Microbes as well as 11 coastal samples taken from the California Current. These samples from both the water column and sediments covered a wide range of environmental conditions. Verrucomicrobia were present in 98% of the analyzed samples, and thus appeared nearly ubiquitous in the ocean. Based on the occurrence of amplified 16S ribosomal RNA sequences, Verrucomicrobia constituted on average 2% of the water column and 1.4% of the sediment bacterial communities. The diversity of Verrucomicrobia displayed a biogeography at multiple taxonomic levels and thus, specific lineages appeared to have clear habitat preference. We found that subdivision 1 and 4 generally dominated marine bacterial communities, whereas subdivision 2 was more frequent in low salinity waters. Within the subdivisions, Verrucomicrobia community composition were significantly different in the water column compared with sediment as well as within the water column along gradients of salinity, temperature, nitrate, depth and overall water column depth. Although we still know little about the ecophysiology of Verrucomicrobia lineages, the ubiquity of this phylum suggests that it may be important for the biogeochemical cycle of carbon in the ocean.
Similar content being viewed by others

Introduction
The phylum Verrucomicrobia is ubiquitous in soil microbial communities, where it sometimes can be detected in high abundance (O’Farrell and Janssen, 1999; Buckley and Schmidt, 2001, 2003; Sangwan et al., 2005; Janssen, 2006; Kielak et al., 2008; Bergmann et al., 2011). In a recent study, Verrucomicrobia was found in >99% of the analyzed soil samples and constituted on average 23% of the ribosomal RNA (rRNA) sequences (Bergmann et al., 2011). The phylum is related to Planctomycetes and Chlamydiales, and cells affiliated with Verrucomicrobia are morphologically diverse including having intracellular compartments (Schlesner, 1987; Hedlund et al., 1997; Schlesner et al., 2006). Nearly all isolates can grow chemoheterotrophically on many organic carbon compounds including simple sugars—although not always the same compounds (Schlesner et al., 2006; Yoon et al., 2007a, 2007b, 2008a, 2008b). Some strains can utilize methane (Pol et al., 2007) whereas others are facultative anaerobes (Choo et al., 2007; Yoon et al., 2008a, 2008b). At least in culture, Verrucomicrobia grow slowly and many isolates from marine environments have a small cell diameter of approximately 1 μm (Yoon et al., 2007a, 2008b).
The phylum has been divided into seven subdivisions on the basis of the phylogeny of 16S rRNA (Hugenholtz et al., 1998; Schlesner et al., 2006). The most common ones include subdivision 1 (Verrucomicrobiae), 2 (Spartobacteria), 3 and 4 (Opitutae). Little is known about the ecological niche of different Verrucomicrobia subdivisions. In most soil communities, subdivision 2 is dominant, whereas 1, 3 and 4 are found at a lower frequency (Sangwan et al., 2005; Kielak et al., 2008; Bergmann et al., 2011). In freshwater environments, subdivision 2 is also abundant along with 4 (Arnds et al., 2010).
Molecular analyses of marine microbial communities have revealed many previously unrecognized groups. It is clear that many bacterial phyla beyond Proteobacteria and Cyanobacteria are present in the ocean (Giovannoni and Stingl, 2005). This includes Bacteriodetes, Actinobacteria and Planctomycetes, which have important biogeochemical roles like degradation of many polymers or anammox. Less is known about the distribution and diversity of Verrucomicrobia in the ocean (Rappe and Giovannoni, 2003). However, this phylum has been detected in some marine samples from the water column (Bano and Hollibaugh, 2002; Jackson and Weeks, 2008; Zaikova et al., 2010) and sediment (Urakawa et al., 1999). In addition, marine strains have been isolated from a variety of marine environments including seawater (Yoon et al., 2007b), sediment (Yoon et al., 2008a) and marine animals (Choo et al., 2007; Yoon et al., 2007a). This suggests that Verrucomicrobia is present in many marine environments but the extent and diversity are currently unknown as well as factors influencing the distribution of different lineages.
As part of the International Census of Marine Microbes (IcoMM), the 16S rRNA diversity of >500 bacterial communities from a range of marine environments was determined using high-throughput sequencing (Zinger et al., 2011). On the basis of this survey and additional samples from the California Current, here we show that Verrucomicrobia are common in the marine environment and then examine the group's biogeographic patterns in detail.
Materials and methods
IcoMM analysis
As described previously, the hypervariable V6 region of the 16S rRNA gene was PCR-amplified using a mixture of five forward and four reverse primers targeting all bacteria, and sequenced using 454 technology as part of the ICoMM project (Zinger et al., 2011) (see Supplementary Table S1 for sample details). Notably, DNA was extracted differently for different samples; for instance, samples from the water column were treated different compared with sediment sample (see also http://icomm.mbl.edu/microbis/project_pages/pp_by_name/ for details). Chimeras and primer sequences, and fragments <50 bp were removed before analysis. To define operational taxonomic units, the sequences were initially pre-clustered to remove sequencing error (denoising) using a modified single-linkage method at 98% sequence similarity, followed by an average neighbor clustering using a 97% sequence similarity cut-off (Huse et al., 2010). Taxonomic assignment was based on a combination of the taxonomic scheme in Silva version 102 (Pruesse et al., 2007) and Bergey's Manual using the GAST pipeline (Huse et al., 2008). The Silva 16S rRNA database was also used to test for primer specificity to Verrucomicrobia.
PCR and sequencing analysis of California Current samples
The California Current data set comprised 11 monthly coastal samples taken from Newport Pier (CA, USA) (location: 33.61°N 117.94°W). In all, 2l samples were prefiltered through a 2.7-μm GF/D (Whatman, Piscataway, NJ, USA) filter and then collected on a 0.22-μm Sterivex (Millipore, Billerica, MA, USA) filter (Supplementary Table S1). DNA was extracted using a combination of lysozyme and proteinase K pretreatment and phenol-chloroform extraction (Bostrom et al., 2004). For PCR, we used Verrucomicrobia-specific primers VER57F and EUB338_3R (Arnds et al., 2010) with at an annealing temperature of 56 °C and 30 cycles. We then removed excess primers with ExoSAP (Affymetrix, Santa Clara, CA, USA) and ran another 10 PCR cycles using primers consisting of a LibL 454 adaptor, a barcode and the Verrucomicrobia primers described above. We used Verrucomicrobium spinosium as positive control. Next, we sequenced the 11 samples using 454-pyrosequencing and analyzed them with QIIME (Caporaso et al., 2010) to denoise and remove chimeras. We only included sequences >200 bp in length (average=329 bp). In parallel to the ICoMM samples, we then clustered the sequence using a 97% 16S rRNA sequence similarity cut-off and assigned taxonomic rankings based on Silva version 102.
Community composition analysis
To identify the difference in community composition between different samples, we did multiple analyses including step-wise linear regression, multidimensional scaling, analysis of similarities (ANOSIM) and partial Canonical Correspondence Analysis. The step-wise linear regression was done in Matlab. For multidimensional scaling, we first calculated the pair-wise sample similarity with square-root-transformed Bray–Curtis similarity indices determined in PRIMER v6 (Primer-E, Lutton, UK). Sample similarity was visualized after multidimensional scaling using Kruskal fit scheme 1 and a minimum stress of 0.01. In order to have a balanced sample set for pair-wise statistical comparisons for ANOSIM, we randomly selected an equal number of samples from each environment (that is, water column versus sediment or water temperature lower or higher than 15 °C—79 and 80 samples, respectively). We only used samples containing >100 Verrucomicrobia sequences for the analysis. This was repeated 100 times. We next randomly picked 100 sequences from each sample to ensure that each sample contained an equal number of sequences. This was also repeated 100 times. Then, we used ANOSIM from the vegan package in R to determine any significant differences (999 permutations) (Oksanen et al., 2011). The variance contribution of multiple environmental factors on community composition was determined with Canonical Correspondence Analysis (forward selection, α=0.05 and 999 perturbations) using Canoco (ver. 4.5, Microcomputer Power, Ithaca, NY, USA) (ter Braak, 1986), and ordination plots were visualized in CanoDraw. It is worth noting that a subset of samples were used for comparisons between environmental variation and community composition, as we were unable to retrieve environmental data for all samples. Therefore, the total number of samples does not match the number of samples used in many comparisons.
Results
Distribution of total Verrucomicrobia
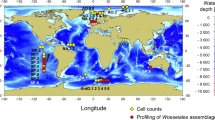
ICoMM (http://icomm.mbl.edu/) covered 506 samples collected from all major ocean basins across a broad range of environmental conditions (Figure 1). This included 391 water column and 115 sediment samples. A total of 9.7 million 16S rRNA sequences (average=19 198) were analyzed with an average of 309 Verrucomicrobia sequences from each ICoMM sample (Table 1 and Supplementary Table S1). In addition to this global data set, we also identified the diversity at a California Current coastal site. Here, we analyzed a total of 103 583 Verrucomicrobia sequences among 11 samples (average=9416) collected over a 1-year period. From the two sample sets, we identified Verrucomicrobia in 98% of the samples and can conclude that this phylum is nearly ubiquitous in the marine environment. Verrucomicrobia constituted on average 1.8% of the sequences and was the sixth most common phylum in the water column (Supplementary Figure S1). The group was more frequent in PCR libraries from the water column (2.0%) compared with the sediment (1.4%) bacterial community (Welch t-test, P<0.0003, n=517). Using a step-wise linear regression model for the water column samples, we also found that the Verrucomicrobia fraction were higher in shallow coastal water versus open ocean sites (n=281, P<0.0074). However, other common oceanographic parameters including salinity, temperature, sample depth or nitrate concentrations were not significant correlated with the frequency of Verrucomicrobia. The highest proportions overall were found in slightly brackish samples (salinity: 30.8–33.4) from a coastal site near a small island (Helgoland) 46 km offshore of Northern Germany. Samples from this area were taken from two filter fractions (0.2–3 μm and 3–10 μm) (Supplementary Table S1). Verrucomicrobia were marginally significantly higher (Student's paired t-test, n=10, P=0.056) in the larger filter fraction (up to 32% of the sequences) compared with the smaller fraction (up to 5.6%). The larger filter fraction likely represented bacteria attached to particles, which suggested that Verrucomicrobia may be more frequent on particles compared with free-living in seawater—at least in this area. However, no other ICoMM samples contained both filter fractions, so we were unable to explore this pattern further. We did not detect the phylum in 10 samples including several samples from deep-sea hydrothermal vents. In summary, the relative occurrence of Verrucomicrobia appears to be highest in coastal ocean water and lowest in sediments—in particular around hydrothermal vents.

Geographical locations of International Census of Marine Microbe (ICoMM) and California Current samples that were analyzed as part of this study.
Distribution and diversity of subdivisions
We observed clear differences in the frequency and distribution of the different Verrucomicrobia subdivisions. Overall, we found that subdivisions 1 and 4 dominated PCR libraries from marine communities—both in the water column (average 73%) and in sediments (85%) (Figure 2). Subdivisions 2 and 3 were only detected at a lower frequency. Around, 22% of the Verrucomicrobia sequences in the water could not be classified below phylum. Many of these sequences originate from samples from the deep ocean. The sequences from the ICoMM data set are too short to accurately build a phylogeny, but the inability to assign those sequences to even a subdivision suggests that a large fraction of the Verrucomicrobia sequences could be associated with unknown lineages.

Average frequency of amplified 16S rRNA sequences affiliated with Verrucomicrobia subdivisions in water column and sediment samples from this study. The frequency of each subdivision was normalized to the frequency of total Verrucomicrobia (nwater=395 and nsediment=112).
To identify environmental factors influencing the distribution of subdivisions, we first found a significant difference in the overall composition between the water column and the sediment (ANOSIM, n=350, P<0.05). For example, subdivision 4 was significantly more frequent in the water column, whereas subdivision 1 was more common in the sediment. A Canonical Correspondence Analysis revealed that salinity, depth, temperature, nitrate concentration and water column depth (which we view as a proxy for coastal influence) all significantly influenced the distribution of sequences associated with different subdivisions in the water column (Figure 3 and Supplementary Table S2). More specifically, we observed that the occurrence of subdivision 4 sequences were highest in the surface photic zone and negatively correlated with depth (Supplementary Figure S2).

Relationship between environmental factors and the occurrence of Verrucomicrobia subdivisions amplified 16S rRNA sequences in the water column (n=281). The ordination plot is based on a partial canonical correspondence analysis with forward selection. See also Supplementary Table S2 for details of ranking and significance values of each environmental parameter.
Subdivision 2 is the most common group in most terrestrial environments, but was generally found at low frequency in the ocean and marine sediments. Within marine samples, the occurrence of subdivision 2 was negatively related to salinity. It was the dominant group in samples from several areas with low salinity including the Baltic Sea, Beaufort Sea and coastal samples from the North Sea. For example, the subdivision constituted on average 50% of the Verrucomicrobia sequences (maximum=69.7%) in the Baltic Sea. However, we did not observe subdivision 2 in two other low salinity areas—the Black Sea and the Hood Canal on the west coast of the United States. These two areas have an unusual seawater chemistry that could influence the presence of subdivision 2. Thus, cells affiliated with subdivision 2 are generally confined to areas with low salinity environments but that other factors may influence the distribution.
Distribution and diversity within subdivisions
We next examined the distribution of Verrucomicrobia diversity within the subdivisions. We first clustered the sequences on the basis of 16S rRNA sequence similarity (97% cut-off) from samples associated with the ICoMM project. This resulted in 2831 operational taxonomic units. Within subdivision 1, the genera Roseibacillus, Persicirhabdus and Rubritalea were the most common (Supplementary Figure S3 and Supplementary Table S1). Roseibacillus constituted 33% of subdivision 1 sequences in the water column and 9% of the sequences from sediment samples. In contrast, Persicirhabdus was more frequent in the sediment (17% versus 10%). We also detected at low frequency in surface water samples the occurrence of operational taxonomic units related to the genus Acidomethylosilex. Several strains from this genus are capable of methanotrophy. The presence of this genus in surface waters suggests that Verrucomicrobia cells might contribute to methane oxidation here. In subdivision 4, the genera Coraliomargarita, Lentimonas, Cerasicoccus, Opitutus and Pelagicoccus were the most common (Supplementary Figure S4). Although many sequences from subdivision 4 were unclassified at the genus level (39.4% in water and 33.1% in sediment), we found that all were associated with the family of Puniceicoccaceae.
A comparison between the water column and sediment communities revealed—with a few exceptions—that communities from these two environments were completely separated (ANOSIM: R=0.52, n=79, P<0.001) (Figure 4). We also found significant difference in community composition within the water column along salinity, depth, water column depth, nitrate and temperature gradients (Supplementary Table S2). For example, communities living below 15 °C were very different from communities living at higher temperature (ANOSIM: R=0.17, n=80, P<0.001) (Figure 4).

Factors influencing Verrucomicrobia community composition using multidimensional scaling. The community composition was based on the distribution of operational taxonomic units defined by at least 97% 16S rRNA sequence similarity. The Verrucomicrobia community composition in sediments and water column (ntreatment=79) and between water column samples above or below 15 °C is significantly different (ntreatment=80).
We observed that most environmental factors influence the distribution of genera within both subdivisions (Supplementary Figure S5 and Supplementary Table S2). However, it was not obvious which environmental factor control the individual distribution of most genera. It appeared that Acidomethylosilex is mostly found in surface waters, whereas we found Opitutus and Fucophilus at highest frequency in brackish waters. Further, Cerasicoccus was almost exclusively found deeper in the water column. Thus, there appeared to be some degree of ecological separation at the genus level of Verrucomicrobia, but further quantitative studies are needed to confirm these patterns.
Discussion
In this global study, we have shown that Verrucomicrobia is nearly ubiquitous in the marine environment and is found in almost all marine environments across a range of environmental conditions. On the basis of the average occurrence of amplified 16S rRNA sequences, Verrucomicrobia may be the sixth most abundant bacterial phylum in ocean water after Proteobacteria, Bacteriodetes, Deferribacteres, Actinobacteria and Cyanobacteria. Overall, the frequency of Verrucomicrobia 16S rRNA sequences appeared to be lower in marine environments compared with what has been observed in soil (Bergmann et al., 2011). We did not find Verrucomicrobia near the hydrothermal vents, although this could be because of the detection limit of the method and deeper sequencing, or the use of specific primers may reveal in Verrucomicrobia in this environment as well. In contrast, Verrucomicrobia constituted a high proportion of bacterial sequences in several samples taken from slightly brackish coastal waters. This included what are likely free-living as well as particle-attached bacterial communities. The coastal sites with the highest frequency of Verrucomicrobia were adjacent to two very small offshore islands (1 km2 and 0.7 km2) with limited river or groundwater outflow. This suggests that terrestrial run-off is not the main cause for the high proportion of Verrucomicrobia here.
It is important to recognize that the data presented here is based on PCR amplification of 16S rRNA followed by pyrosequencing. This can introduce several biases based on variation in DNA extraction, primer specificity, PCR amplification and so on. DNA from individual samples was extracted with different protocols. Thus, specific extraction methods can fail to capture certain sublineages of Verrucomicrobia and otherwise introduce biases in the estimation of the frequency of lineages observed. The primer mixture used for analyzing the ICoMM samples had a perfect match to 97% of the Verrucomicrobia sequences. Primers specifically targeting Verrucomicrobia in the California Current were recently designed by Arnds et al. (2010). They reported that the forward primer (VER47F) captured >78% of all known Verrucomicrobia whereas the reverse primer (EUB338_3R) captured >96%, but the authors noted it is likely the forward primer captured a higher percentage of Verrucomicrobia as many 16S rRNA sequences in this region were of lower quality. These biases could affect the observed biogeographical patterns in unknown ways. However, one should also note that past observed distributions of marine microbial lineages using PCR-amplified 16S rRNA sequence libraries have demonstrated important trends (for example, high abundance of SAR11 and Prochlorococcus or the biogeography of specific ecotypes within these lineages (Giovannoni et al., 1990; Fuhrman et al., 1993; Martiny et al., 2009)) that largely matched later quantitative studies. On the basis of this, we expect that the observed biogeography of Verrucomicrobia using >500 samples generally is robust.
At this point we still have a limited understanding of the biology of Verrucomicrobia in the ocean, but the biogeographical patterns observed here point toward important physiological differences among specific lineages. We observed clear difference in community composition between the water column and sediment, as well as along gradients of common oceanographic environmental variables including salinity, water temperature, nitrate concentration, depth and water column depth (proxy for coastal influence). This was seen at multiple taxonomic levels.
At the subdivision level, previous studies have shown that subdivision 2 dominates many soil communities, but we found that this group was primarily confined to brackish waters. In contrast, subdivision 1 appeared to be very common in the ocean and to some extent in lakes (Allgaier and Grossart, 2006; Arnds et al., 2010), but detected rarely in soil samples (Bergmann et al., 2011). Thus, this lineage may have its primary niche in aquatic environments. Finally, subdivision 4 was more frequent in PCR libraries from the surface ocean. Thus, there appears to be differential distribution patterns of the major phylogenetic lineages within Verrucomicrobia, which suggest that these groups are ecologically distinct. Superimposed on this distinction between subdivisions found on land or in the ocean, we also saw specific Verrucomicrobia communities inhabit sediment and specific water column environments. Thus, our study here demonstrates that Verrucomicrobia is nearly ubiquitous in the marine environment but the phylum appears to consist of several ecologically distinct lineages that occupy unique niches and are differentially distributed along environmental gradients. We hope that this can form the basis of more detailed studies in order to further define the environmental range and biogeochemical role of Verrucomicrobia ecotypes.
References
Allgaier M, Grossart HP . (2006). Seasonal dynamics and phylogenetic diversity of free-living and particle-associated bacterial communities in four lakes in northeastern Germany. Aquat Microb Ecol 45: 115–128.
Arnds J, Knittel K, Buck U, Winkel M, Amann R . (2010). Development of a 16S rRNA-targeted probe set for Verrucomicrobia and its application for fluorescence in situ hybridization in a humic lake. Syst Appl Microbiol 33: 139–148.
Bano N, Hollibaugh JT . (2002). Phylogenetic composition of bacterioplankton assemblages from the Arctic Ocean. Appl Environ Microbiol 68: 505–518.
Bergmann GT, Bates ST, Eilers KG, Lauber CL, Caporaso JG, Walters WA et al. (2011). The under-recognized dominance of Verrucomicrobia in soil bacterial communities. Soil Biol Biochem 43: 1450–1455.
Bostrom KH, Simu K, Hagstrom A, Riemann L . (2004). Optimization of DNA extraction for quantitative marine bacterioplankton community analysis. Limnol Oceanogr Methods 2: 365–373.
Buckley DH, Schmidt TM . (2001). Environmental factors influencing the distribution of rRNA from Verrucomicrobia in soil. FEMS Microbiol Ecol 35: 105–112.
Buckley DH, Schmidt TM . (2003). Diversity and dynamics of microbial communities in soils from agro-ecosystems. Environ Microbiol 5: 441–452.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336.
Choo YJ, Lee K, Song J, Cho JC . (2007). Puniceicoccus vermicola gen. nov., sp. nov., a novel marine bacterium, and description of Puniceicoccaceae fam. nov., Puniceicoccales ord. nov., Opitutaceae fam. nov., Opitutales ord. nov. and Opitutae classis nov. in the phylum ‘Verrucomicrobia’. Int J Syst Evol Microbiol 57: 532–537.
Fuhrman JA, McCallum K, Davis AA . (1993). Phylogenetic diversity of subsurface marine microbial communities from the Atlantic and Pacific Oceans. Appl Environ Microbiol 59: 1294–1302.
Giovannoni SJ, Britschgi TB, Moyer CL, Field KG . (1990). Genetic diversity in Sargasso Sea bacterioplankton. Nature 345: 60–63.
Giovannoni SJ, Stingl U . (2005). Molecular diversity and ecology of microbial plankton. Nature 437: 343–348.
Hedlund BP, Gosink JJ, Staley JT . (1997). Verrucomicrobia div. nov., a new division of the bacteria containing three new species of Prosthecobacter. Antonie Leeuwenhoek 72: 29–38.
Hugenholtz P, Goebel BM, Pace NR . (1998). Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol 180: 6793–6793.
Huse SM, Dethlefsen L, Huber JA, Mark Welch D, Relman DA, Sogin ML . (2008). Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet 4: e1000255.
Huse SM, Mark Welch D, Morrison HG, Sogin ML . (2010). Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ Microbiol 12: 1889–1898.
Jackson CR, Weeks AQ . (2008). Influence of particle size on bacterial community structure in aquatic sediments as revealed by 16S rRNA gene sequence analysis. Appl Environ Microbiol 74: 5237–5240.
Janssen PH . (2006). Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl Environ Microbiol 72: 1719–1728.
Kielak A, Pijl AS, van Veen JA, Kowalchuk GA . (2008). Differences in vegetation composition and plant species identity lead to only minor changes in soil-borne microbial communities in a former arable field. FEMS Microbiol Ecol 63: 372–382.
Martiny AC, Tai APK, Veneziano D, Primeau F, Chisholm SW . (2009). Taxonomic resolution, ecotypes and the biogeography of Prochlorococcus. Environ Microbiol 11: 823–832.
O’Farrell KA, Janssen PH . (1999). Detection of Verrucomicrobia in a pasture soil by PCR-mediated amplification of 16S rRNA genes. Appl Environ Microbiol 65: 4280–4284.
Oksanen J, Blanchet FG, Kindt R, Legendre P, O’Hara RB, Simpson G et al. (2011). Vegan: Community Ecology Package, R package version 1.17-6 edn.
Pol A, Heijmans K, Harhangi HR, Tedesco D, Jetten MSM, den Camp HJMO . (2007). Methanotrophy below pH1 by a new Verrucomicrobia species. Nature 450: 874–U817.
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J et al. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35: 7188–7196.
Rappe MS, Giovannoni SJ . (2003). The uncultured microbial majority. Annu Rev Microbiol 57: 369–394.
Sangwan P, Kovac S, Davis KE, Sait M, Janssen PH . (2005). Detection and cultivation of soil Verrucomicrobia. Appl Environ Microbiol 71: 8402–8410.
Schlesner H . (1987). Verrucomicrobium Spinosum Gen Nov, Sp Nov - a Fimbriated Prosthecate Bacterium. Syst Appl Microbiol 10: 54–56.
Schlesner H, Jenkins C, Staley J . (2006). The Phylum Verrucomicrobia: a phylogenetically heterogeneous bacterial group. In: Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E (eds). The Prokaryotes. Springer: New York, pp 881–896.
ter Braak CJF . (1986). Canonical Correspondence-Analysis - a new eigenvector technique for Multivariate Direct Gradient Analysis. Ecology 67: 1167–1179.
Urakawa H, Kita-Tsukamoto K, Ohwada K . (1999). Microbial diversity in marine sediments from Sagami Bay and Tokyo Bay, Japan, as determined by 16S rRNA gene analysis. Microbiology 145: 3305–3315.
Yoon J, Matsuo Y, Adachi K, Nozawa M, Matsuda S, Kasai H et al. (2008a). Description of Persicirhabdus sediminis gen. nov., sp. nov., Roseibacillus ishigakijimensis gen. nov., sp. nov., Roseibacillus ponti sp. nov., Roseibacillus persicicus sp. nov., Luteolibacter pohnpeiensis gen. nov., sp. nov. and Luteolibacter algae sp. nov., six marine members of the phylum ‘Verrucomicrobia’, and emended descriptions of the class Verrucomicrobiae, the order Verrucomicrobiales and the family Verrucomicrobiaceae. Int J Syst Evol Microbiol 58: 998–1007.
Yoon J, Matsuo Y, Matsuda S, Adachi K, Kasai H, Yokota A . (2007a). Rubritalea spongiae sp. nov. and Rubritalea tangerina sp. nov., two carotenoid- and squalene-producing marine bacteria of the family Verrucomicrobiaceae within the phylum ‘Verrucomicrobia’, isolated from marine animals. Int J Syst Evol Microbiol 57: 2337–2343.
Yoon J, Matsuo Y, Matsuda S, Adachi K, Kasai H, Yokota A . (2008b). Rubritalea sabuli sp. nov., a carotenoid- and squalene-producing member of the family Verrucomicrobiaceae, isolated from marine sediment. Int J Syst Evol Microbiol 58: 992–997.
Yoon J, Yasumoto-Hirose M, Matsuo Y, Nozawa M, Matsuda S, Kasai H et al. (2007b). Pelagicoccus mobilis gen. nov., sp. nov., Pelagicoccus albus sp. nov. and Pelagicoccus litoralis sp. nov., three novel members of subdivision 4 within the phylum ‘Verrucomicrobia’, isolated from seawater by in situ cultivation. Int J Syst Evol Microbiol 57: 1377–1385.
Zaikova E, Walsh DA, Stilwell CP, Mohn WW, Tortell PD, Hallam SJ . (2010). Microbial community dynamics in a seasonally anoxic fjord: Saanich Inlet, British Columbia. Environ Microbiol 12: 172–191.
Zinger L, Amaral-Zettler LA, Fuhrman JA, Horner-Devine MC, Huse SM, Welch DBM et al. (2011). Global patterns of bacterial beta-diversity in seafloor and seawater ecosystems. Plos One 6: e24570.
Acknowledgements
We thank the UCI Undergraduate Research Opportunity Program (SF), the National Science Foundation (OCE-0928544 and OCE-1046297, ACM) and the Alfred P Sloan Foundation (SH, DMW, MS) for supporting the work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
Freitas, S., Hatosy, S., Fuhrman, J. et al. Global distribution and diversity of marine Verrucomicrobia. ISME J 6, 1499–1505 (2012). https://doi.org/10.1038/ismej.2012.3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2012.3
Keywords
This article is cited by
-
Exploring bacterial diversity in Arctic fjord sediments: a 16S rRNA–based metabarcoding portrait
Brazilian Journal of Microbiology (2024)
-
Elimination of antibiotic-resistant bacteria and resistance genes by earthworms during vermifiltration treatment of excess sludge
Environmental Science and Pollution Research (2024)
-
Microbe-driven elemental cycling enables microbial adaptation to deep-sea ferromanganese nodule sediment fields
Microbiome (2023)
-
Rhodopsin-mediated nutrient uptake by cultivated photoheterotrophic Verrucomicrobiota
The ISME Journal (2023)
-
Correlation Between Microbial Community and Hatching Failure in Loggerhead Sea Turtle Caretta caretta
Microbial Ecology (2023)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}