
Abstract
Deep-sea hydrothermal vent chimneys harbor a high diversity of largely unknown microorganisms. Although the phylogenetic diversity of these microorganisms has been described previously, the adaptation and metabolic potential of the microbial communities is only beginning to be revealed. A pyrosequencing approach was used to directly obtain sequences from a fosmid library constructed from a black smoker chimney 4143-1 in the Mothra hydrothermal vent field at the Juan de Fuca Ridge. A total of 308 034 reads with an average sequence length of 227 bp were generated. Comparative genomic analyses of metagenomes from a variety of environments by two-way clustering of samples and functional gene categories demonstrated that the 4143-1 metagenome clustered most closely with that from a carbonate chimney from Lost City. Both are highly enriched in genes for mismatch repair and homologous recombination, suggesting that the microbial communities have evolved extensive DNA repair systems to cope with the extreme conditions that have potential deleterious effects on the genomes. As previously reported for the Lost City microbiome, the metagenome of chimney 4143-1 exhibited a high proportion of transposases, implying that horizontal gene transfer may be a common occurrence in the deep-sea vent chimney biosphere. In addition, genes for chemotaxis and flagellar assembly were highly enriched in the chimney metagenomes, reflecting the adaptation of the organisms to the highly dynamic conditions present within the chimney walls. Reconstruction of the metabolic pathways revealed that the microbial community in the wall of chimney 4143-1 was mainly fueled by sulfur oxidation, putatively coupled to nitrate reduction to perform inorganic carbon fixation through the Calvin–Benson–Bassham cycle. On the basis of the genomic organization of the key genes of the carbon fixation and sulfur oxidation pathways contained in the large genomic fragments, both obligate and facultative autotrophs appear to be present and contribute to biomass production.
Similar content being viewed by others


Introduction
Deep-sea hydrothermal vent chimneys that form by interactions between hot fluids and cold seawater are regarded as biogeochemical hot spots, with reactive gases, dissolved elements and thermal and chemical gradients operating over spatial scales of millimeters and centimeters up to meters (Schrenk et al., 2003; Kristall et al., 2006). These chemical and thermal gradients along and inside the sulfide chimneys provide a wide range of microhabitats for chemolithoautotrophic microorganisms that fix inorganic carbon using chemical energy obtained through the oxidation of reduced inorganic compounds contained in the hydrothermal fluids, converting the geothermally derived energy into microbial biomass (for example, Reysenbach and Shock, 2002).
Since the discovery of deep-sea hydrothermal vent systems, the microbial diversity in these systems has been the subject of studies using both cultivation and cultivation-independent molecular methods (for example, Nakagawa et al., 2005; Huber et al., 2007). These studies have reported a remarkable phylogenetic diversity of microbes inhabiting the chimney walls, yet the metabolic diversity and physiological potential of these microbial communities are only beginning to be revealed. Progress in understanding this diversity has been made largely because of the recent developments in high-throughput genomic technologies that enable microbial ecologists to address complex evolutionary and ecological hypotheses at a community scale (Tyson et al., 2004; Tringe et al., 2005; DeLong et al., 2006; Grzymski et al., 2008). With advances in sequencing technologies, large-scale genomic surveys of microbial communities (metagenomics) are becoming routine, making deciphering the genetic and functional ‘differences that make a difference’ within and among microbial habitats increasingly feasible. Using metagenomic analysis, the metabolic potential and environmental adaptation strategies of epi- and endosymbionts of deep-sea hydrothermal vent invertebrates have been elucidated (Newton et al., 2007; Grzymski et al., 2008). The metagenome of a Lost City carbonate chimney biofilm, which is so far the only published metagenome from any deep-sea vent chimney, was found to contain a remarkable abundance and diversity of genes potentially involved in lateral gene transfer (Brazelton and Baross, 2009). However, it remains elusive whether lateral gene transfer is a common occurrence in chimney biofilms, or whether it is restricted to certain deep-sea hydrothermal vent environment, such as Lost City. Comparative metagenomic analysis of chimney biofilms from either different locations and/or with different geochemistry would provide valuable information on identifying unique traits of these largely unknown deep-sea microbial communities, in particular in comparison with those from other environments.
Here, we present the first comparative metagenomic analysis of two different deep-sea hydrothermal vent chimney microbiomes: one from a carbonate chimney at the ultramafic hosted Lost City vent site that is characterized by relatively low temperature and high pH (<90 °C, pH 9–11), the other one from sulfide chimney 4143-1 at the basalt-hosted Mothra hydrothermal vent field on the Juan de Fuca Ridge that is characterized by venting of high temperature (>300 °C) and low pH (pH 2–3) fluids. This comparative metagenomic approach is providing very useful information on the adaptation and metabolic potential of the whole microbial community in the chimney wall.
Materials and methods
Samples collection, DNA isolation and fosmid library construction
The sulfide chimney (4143-1) was collected in 2005 by the submersible Alvin, supported by the R/V Atlantis, from the Mothra Field of the Juan de Fuca Ridge, located ∼300 km west of Vancouver Island, Canada. Precautions were taken during sampling and handling of the chimney samples to preserve their integrity for microbiological analyses. The chimneys were stored at −20 °C immediately following collection, kept on dry ice during transportation, and stored at −80 °C until further analysis. The genomic DNA was isolated from the outer portion of the chimney and used for fosmid library construction as previously described (Meng et al., 2009). A total of 2880 fosmid clones were grown overnight in Luria–Bertani broth, extracted separately using the Axyprep-96 Plasmid kit (Axygen, Union City, CA, USA) and pooled in equal quantities of 20 ng/μl for pyrosequencing.
Sequencing, assembly and annotation
Sequencing was performed using pyrosequencing on 454 Life Sciences GS FLX system platforms with a practical limit of 250 bp. The total amount of sequence data obtained for 4143-1 fosmid library are as follows: 578 567 reads, containing 133 MB of raw sequence. SeqClean software (http://compbio.dfci.harvard.edu/tgi/software/) was used to eliminate sequences of host genome (Escherichia coli EPI300) (31 MB) and fosmid sequence (31 MB), this reduced the data set to: 308 034 reads, containing 71 MB of sequence. Those data were assembled with TGICL software (http://compbio.dfci.harvard.edu/tgi/software/) (Zhang et al., 2000). The result is 31 405 reads with average length of 484 bp. And 22 968 singletons with average length of 196 bp cannot be assembled. Initially, those contigs and singletons assembly was mapped to a database of known 16S rDNA sequences (Ribosomal Database Project release 9.3.3) using Blastn algorithm. These 16S rDNA anchors that longer than 50 were classified into respective taxonomic groups using the Ribosomal Database Project Classifier. Open reading frames (ORFs) were predicted by glimmer 3.02 software for those contigs longer than 1000 bp. The predicted ORFs, contigs without any putative ORFs by glimmer 3.02 and singletons were compared against the National Center for Biotechnology Information non-redundant database using Blastx with an expectation value cut-off of <10−5. Those sequences that had reliable hits with the non-redundant database were compared against the Kyoto Encyclopedia of Genes and Genomes (KEGG) sequence database and Clusters of Orthologous Group sequence database by using an expectation value cut-off of <10−5. The range, mean, median of the expectation values were shown in Supplementary Table S1.
Two-way clustering analysis and bootstrap resampling methodologies
For taxonomic binning, all genomic sequences from Lost City hydrothermal field, acid mine drainage, gutless worm consortium, Peru Margin sub-seafloor sediments, whale falls and North Pacific Gyre water were analyzed with the method described in the previous paragraph. Blast results were tabulated and the percentage of sequences within each KEGG pathway or Clusters of Orthologous Group category was calculated. Dendrograms were generated by using CLUSTER (http://rana.lbl.gov/EisenSoftware.htm) and visualized with TREEVIEW (http://rana.lbl.gov/EisenSoftware.htm). To test whether significant difference exists between any two protein sequence bins for a particular KEGG subsystem, a bootstrap sampling method (DeLong et al., 2006) was used and is summarized in the following. For any two protein sequence bins to be compared, the process includes three steps: (1) 10 000 sequences were sampled from each bin and the difference in the number of KEGG subsystems was calculated. We repeated this process 1000 times and recorded the median difference for each subsystem; (2) two sets with 10 000 sequences were sampled from either bin with equal probability, and the difference in the number of KEGG subsystems between these two sets of 10 000 was calculated. This process was also repeated 1000 times and (3) the P-value for a particular subsystem was calculated as the number of differences in step (2) larger than the median difference from step (1), divided by the number of replicates in step 2 (that is, 1000 here).
Screening for target fosmid clones
Eight fosmids carrying genes coding for Calvin–Benson–Bassham cycle (CBB), reductive tricarboxylic acid (rTCA), sulfur oxidation or denitrification functions were identified by PCR with specific primers designed from target contigs or singletons. Information is shown in Supplementary Table S2. Positive clones were grown overnight in Luria–Bertani broth, extracted using the Axyprep plasmid miniprep kit and sequenced by primer walking with ABI 3730 XL from Applied Biosystems (Foster City, CA, USA). Those sequences were also assembled with TGICL software found with the glimmer 3.02 software. Then, Blastx was used to compare all the predicted ORFs the National Center for Biotechnology Information non-redundant database, using an expectation value cut-off of <10−5.
Reconstructions of the microbiome's metabolism
Identified genes were assigned KEGG Orthology numbers using the latest release of KEGG (v. 47), which allowed us to assign identified genes to KEGG maps. The odds ratio was then used to define if a gene was enriched in the environment. The odds ratio can be thought of as the likelihood of observing a given gene in the sample relative to the comparison data set. We calculated the odds ratios using (A/B)/(C/D) where A is the number of hits to a given gene in the deep-sea hydrothermal vent metagenome, B is the number of hits to all other genes in the deep-sea hydrothermal vent metagenome, C is the number of hits to a given gene in the comparison data set and D is the number of hits to all other genes in the comparison data set (Gill et al., 2006). To minimize the error caused by the database size, both of the chimneys metagenomes were resampled to 3 000 000 to approximate the size of the KEGG database.
Accession numbers
The GenBank Sequence Read Archive accession number for the source sequences of 4143-1 Fosmid library is SRA009990.1. The accession numbers of eight fosmids are GU191796-803.
Results and discussion
Fosmid library construction and sequencing
The sample 4143-1 represented the outer-layer of an actively venting (316 °C) black-smoker chimney from the Mothra hydrothermal vent field on the Endeavour segment of the Juan de Fuca Ridge. From 4143-1, a fosmid library was constructed that contained 2880 fosmid clones without amplification. The sizes of the inserted fragments of the clones in the library were checked by enzyme digestion and gel electrophoresis. It was calculated that the average insert size was about 20 kb. Each of the fosmid clones was extracted separately and mixed in equal quantity for direct pyrosequencing. A total of 308 034 reads with an average sequence length of 227 bp were generated after removing the pCC1FOS vector sequence and the chromosome sequence contamination from the host strain. Sequence data were assembled with TGICL software. The clustering is performed by a slightly modified version of National Center for Biotechnology Information's megablast, and the resulting clusters are then assembled using CAP3 assembly program (Details see the Materials and methods). The sequenced microbiome of the hydrothermal vent sample consisted of 31 405 contigs and 22 968 singletons, a total of 15.3 MB. The average G-C% of the sequences was determined to be 49%, and a total of 21 836 ORFs were predicted from the data set, more than 70% of which could be classified into function sets in the Clusters of Orthologous Group category (Table 1).
The taxonomic diversity of the metagenomic sequence was assessed by SSU rRNA gene sequence analysis. All 26 contigs and 11 singletons that could be assigned to SSU rRNA gene sequences were identified as bacterial 16S rRNA gene sequences. These bacterial 16S rRNA gene sequences were found to be mainly affiliated with Gammaproteobacteria (25.5%); Bacteroidetes (15.7%); Alphaproteobacteria (13.7%); Betaproteobacteria (7.8%); and Planctomycetes and Deltaproteobacteria (5.9% each) (Supplementary Figure S1), supporting our previous 16S rRNA gene library analysis that also had predominantly Gammaproteobacteria (Wang et al., 2009).
Comparative metagenome analysis
The predicted proteins from the 4143-1 chimney metagenome were classified into functional categories at higher order cellular processes from the KEGG database. Comparative metagenomic analyses using the two-way clustering method (DeLong et al., 2006) were conducted. The metagenomes being compared were obtained from diverse environments, including a biofilm of a carbonate chimney from the Lost City hydrothermal vent field (90°C, pH 9–11 fluids); two biofilm samples from acid mine drainage (Tyson et al., 2004), a gutless worm consortium (Woyke et al., 2006); a whale fall microbial community (which also represents a deep-sea reducing environment with similarities to hydrothermal vents (Tringe et al., 2005)); deeply buried marine sediments (Biddle et al., 2008), and finally pelagic microbial communities from the North Pacific Subtropical Gyre (DeLong et al., 2006).
The two-way clustering of samples and KEGG functional pathway, in which categorized sequences percentage is indicated by yellow shading, is displayed in Figure 1. The Hawaii Ocean water samples clustered together and predicted protein sequences were differentially distributed in photic zone and deep water as has been shown before (DeLong et al., 2006). The sub-seafloor sediment samples formed one group, and also had a depth-related clustering tendency. The clustering pattern of the three whale fall samples is also consistent with the one that has been reported before (Tringe et al., 2005). The two samples from chimneys clustered most closely and they formed a group with the acid mine drainage biofilm, whereas the Gutless worm consortium grouped with the whale fall samples (Figure 1). A very similar grouping pattern was obtained by clustering of samples and Clusters of Orthologous Group functional gene categories (Supplementary Figure S2). Although the physicochemical conditions of the two chimney environments are strikingly different (for example, the venting fluid of 4143-1 is acidic, around 310 °C, whereas that of Lost City sample is <90 °C, pH 9–11), they both represent chimney wall samples that are continuously exposed to highly dynamic conditions characterized by vigorous mixing of reduced, high-temperature fluids of extreme pH with oxygenated, cold sea water of pH 8. The two samples from chimneys are enriched in genes associated with mismatch repair and homologous recombination (Figure 1 and Table 2), suggesting that the microbial communities therein have evolved extensive DNA repair systems to cope with the extreme environment that subjects the genomes to damaging effects by physical and toxic chemical agents (such as high concentrations of hydrogen sulfide, trace metals, high temperature and radionuclides) in the deep-sea hydrothermal vent environment (Pruski and Dixon, 2003).

Two-way Clustering analysis of samples and Kyoto Encyclopedia of Genes and Genomes (KEGG) categories on the basis of the percentage of KEGG-annotated sequences found in each category. The yellow shading is proportional to the percentage of identified sequences falling into each sample. KEGG categories with a standard deviation greater than 0.2 of observed values, having >0.2 and <8.0 of the total KEGG-categorized genes are shown. KEGG categories mainly discussed in the text are boxed. Hot_10 to Hot_4000: sea water of Hawaii Ocean Time Series station; Peru_Margin: Peru Margin subseafloor biosphere project (D04684S01, D04684S02, D3EISHW01, D3EISHW02, EC6HCKB01); Whale_Fall_1 to Whale_Fall_3: a rib bone, a microbial mat isolated from gray whale carcass in the Santa Cruz Basin, and a whale bone in the Southern Ocean, respectively; Lost City: Lost City carbonate chimneys sample; Vent 4143-1: hydrothermal vent sample 4143-1 (this study); Acid mine drainage: Biofilm microbial community from acid mine drainage site at Iron Mountain, CA; Gutless worm consortium: endosymbionts of gutless worm, Mediterranean Sea.
Beside the abundant transposases in the Lost City carbonate chimneys metagenome as described by Brazelton and Baross (2009), the percentage transposases in the metagenomes of the chimney_4143-1, acid mine drainage biofilm and gutless worm consortium are 1.10, 2.27 and 2.8%, respectively, which are higher than other samples with little or no biomass contribution from biofilms. Brazelton and Baross (2009) have shown that the possible carriers of transposases in the Lost City sample are small extracellular DNA fragments (Brazelton and Baross, 2009), similar to our findings for chimney 4143-1 (Supplementary Figure S3).
The two samples from chimneys have a high percentage of genes for chemotaxis and flagellar assembly, commonly used by prokaryotes to sense and respond to environmental cues (Figure 1 and Table 2). The relatively high abundance of methyl-accepting chemotaxis proteins in the two metagenomes from vent chimneys could be seen as an adapting strategy of the microbial communities in response to the steep chemical and thermal gradients observed at hydrothermal vents.
Besides the similarities between the two metagenomes from chimneys, there are also distinct differences. For example, the sulfide chimney 4143-1 appeared more enriched in genes in nitrogen metabolism (Figure 1 and Table 2). Nevertheless, the comparative metagenome analysis clearly demonstrates that functional gene profiling is a useful and reliable proxy to delineate biological communities from different environments.
Autotrophic carbon metabolism
The assimilation of carbon dioxide (CO2) into organic material is globally the most important biosynthetic process. There are six pathways for CO2 fixation, that is, the CBB cycle, rTCA cycle (Arnon cycle), the 3-hydroxypropionate (3-HP) cycle, the reductive acetyl coenzyme A (acetyl-CoA) pathway (Wood–Ljungdahl pathway), the 3-hydroxypropionate/4-hydroxybutyrate (3-HP/4-HB) cycle and the dicarboxylate/4-hydroxybutyrate cycle (Nakagawa and Takai, 2008), the latter two of which were only recently characterized in a thermoacidophilic archaeon, Metallosphaera sedula and Ignicoccus hospitalis, respectively (Huber et al., 2008).
Odds ratios for cbbLS and/or cbbM encoding RubisCO and cbbP encoding phosphoribulokinase in chimney 4143-1 and Lost City carbonate chimney metagenome are highly enriched relative to the KEGG database (Figure 2a). To obtain the genomic context of the CBB cluster in the chimney 4143-1 metagenome, four fosmids were positively identified by PCR with specific primers and subsequently sequenced as described in Materials and methods. The two unique genes for the CBB cycle, cbbLS and/or cbbM encoding RubisCO and cbbP encoding phosphoribulokinase, were detected at high frequencies (Figure 2a). One fosmid clone containing cbbL and three clones containing cbbM genes were selected, and the adjacent regions were partially sequenced. The obtained cbbL sequence had 91% identity with that from the gammaproteobacterial endosymbiont of the clam Solemya velum, which expresses a form IA RuBisCO. The fosmid cbbLS genes were followed by genes encoding CbbQ and CbbO, which are important proteins implied in the assembly of RubisCO. Upstream of cbbLS, no genes involved in the CBB cycle were identified (Figure 3). A similar cbb gene cluster with conserved cbbLSQO gene organization has been found in the chemoautotrophic Gammaproteobacteria Thiomirospira crunogena XCL-2, Hydrogenovibrio marinus and the endosymbiont of Solemya velum (Badger and Bek, 2008). It has been observed that the CBB cycle genes of obligate autotrophs apparently do not form CBB operons that would facilitate coordinated regulation, presumably because these genes are constitutively expressed and, therefore, do not require regulation in these obligate autotrophs (Scott et al., 2006). In the obligate autotroph T. crunogena XCL-2, as well as in H. marinus, the genes encoding other enzymes of the CBB cycle are scattered throughout their respective genomes. Similar to T. crunogena XCL-2, the fosmid Rubisco genes cbbLS do not form a CBB operon with cbbP, suggesting that the fosmid cbbL gene may come from an obligate chemoautotrophic organism (Figure 3). The three obtained cbbM genes exhibit 60–70% sequence identity to one another. A DNA fragment of approximately 8.6 kb containing the cbbM1 was analyzed in greater detail, and it was found that the cbbM1 gene was likely part of a CBB operon containing other CBB genes, as well as carboxysome structural genes, similar to those of Rhodopseudomonas palustris BisB5, a facultative chemoautotroph (Figure 3). Although only short DNA fragments were sequenced (around 3.4 kb each) around the remaining two cbbM genes (Fosmid_aI, Fosmid_contig), they are not likely to constitute CBB operons with other CBB genes (opposite gene direction of cbbR and cbbM, see Figure 3), and these two cbbM genes may come from obligate autotrophs. Form I and form II RuBisCOs have different affinities to CO2, with form I RuBisCOs generally found to be adapted to higher CO2 concentrations. Some autotrophs contain both the form I and form II Rubiscos, indicating the adaptation of these organisms to varying CO2 concentrations in the environment. Similarly, both form I and form II Rubiscos from putative obligate and facultative autotrophs were found in the chimney wall, suggesting that members of the microbial community reflect adaptations to the highly dynamic physicochemical conditions in the chimney wall.

Enrichment of genes for major carbon fixation pathways. (a) CBB cycle; (b) rTCA cycle; (c) 3-HP cycle; (d) reductive acetyl-CoA pathway. Diagrams are based on KEGG pathway maps. When available, enzyme classification numbers for each step were included in boxes. Box color indicated the odds ratio of each enzyme with darker red and green color representing higher odds ratio of respective enzyme in the chimney 4143-1 and lost city chimney metagenome, respectively.

Genomic organization of the CBB cluster. The open reading frame (ORF) Finder program was used to perform ORF analysis. RubisCO genes (cbbLS and cbbM) are green, cbbR genes are blue, other genes encoding CBB cycle enzymes are black, carboxysome structural genes are gray. hp, hypothetical protein; ribC, friboflavin synthase subunit alpha; tkt, transketolase; fbaC2, fructose-1,6-bisphosphate aldolase, class II; fbp2, fructose-1,6-bisphosphatase, class II; cbbR, transcriptional regulator LysR family; cbbP, phosphoribulokinase genes; cbbQ, P-loop containing nucleoside triphosphate hydrolases; cbbO, Nitric oxide reductase activation protein; pgcM, phosphatase/phosphohexomutase HAD superfamily; natB, ABC-type transporter permease protein.
The rTCA cycle has recently been considered as an important CO2 fixation pathway in the deep-sea vent environments (Campbell and Cary, 2004; Nakagawa and Takai, 2008). The three key enzymes of rTCA cycle, ATP citrate lyase, 2-oxoglutarate: ferredoxin oxidoreductase and pyruvate:ferredoxin oxidoreductase, were identifiable in the 4143-1 metagenome (Figure 2b), suggesting the presence of the complete rTCA cycle for inorganic carbon fixation by autotrophic organisms in the chimney 4143-1 wall. For the Lost City carbonate chimney sequences, fewer putative orthologs involved in rTCA cycle were detected and ATP citrate lyase was missing. Two gene fragments annotated as putative ATP citrate lyase genes were identified in the 4143-1 metagenome through KEGG pathway analysis, and the fosmid clone named fosmid_aF containing the putative aclB-like gene was positively identified and sequenced. Blast analysis of this gene showed that it had high sequence identity (70%) with a putative succinyl-CoA synthetase β-subunit gene (Mmc13639) from the magnetotactic coccus strain MC-1. Strain MC-1 has been demonstrated to use the rTCA cycle for carbon fixation, but initially no bona fide ATP citrate lyase could be identified (Williams et al., 2006). However, recently, the genome sequence of MC-1 has been completed, which has led to the suggestion that the genes Mmc13638 and Mmc13639 encode the large (aclA) and small (aclB) subunits of an ATP-dependent citrate lyase (Schubbe et al., 2009). The aclA gene of strain MC-1 contains an inserted portion that prevented its previous identification as an ATP citrate lyase. Phylogenetic analysis suggests that the putative aclB contained on the fosmid forms a new aclB cluster together with the putative aclB from MC-1 (Supplementary Figure S4A). The genomic region surrounding the putative aclB on the fosmid was sequenced (approximately 18 kb, Supplementary Figure S4B). A putative malate dehydrogense was found upstream of the putative aclB, whereas no other genes that may be involved in the rTCA cycle were found throughout the 18 kb DNA sequence fragment. In particular, no homolog to Mmc13638, which represents the large subunit of the ATP citrate lyase, could be identified on the fosmid. Up till now, there have been no reports indicating that the large and the small subunit of the ATP citrate lyase may be separately located on the chromosome, making it questionable if the identified aclB-like gene on the fosmid is functional. Our results suggest that some microorganisms in the chimney environment may use the rTCA cycle for carbon fixation, utilizing the novel ATP citrate lyase for citrate cleavage. The possible presence of an ATP citrate lyase most closely related to that of the Gammaproteobacterium Endoriftia persephone and the magnetotactic Alphaproteobacterium strain MC-1 further argues for the importance of Gamma- and Alphaproteobacteria for carbon fixation in the chimney wall. It is possible that organisms use both rTCA and CBB, similar to what has been hypothesized for Endoriftia persephone (Markert et al., 2007).
There are two evolutionarily distinct forms of carbon monoxide dehydrogenase (CODH). The aerobic CODH is used by CO oxidizers to couple the oxidation of CO to oxygen reduction, whereas the anaerobic CODH is used by anaerobic microorganisms to couple CO oxidation to sulfate reduction or to reduce CO2 to either acetate (acetogenesis) or methane (methanogenesis) (Wu et al., 2005; King and Weber, 2007). Comparing with 910 sequences coding for CODH in about 61 M of Peru Margin subseafloor biosphere sequences, only seven and four sequences were found in the 4143-1 and the Lost City carbonate chimney metagenome, respectively (Figure 2d). There were four gene fragments annotated as carbon monoxide dehydrogenase, large subunit or catalytic subunit in the 4143-1 metagenome. These genes are most closely related to those of Jannaschia sp. CCS1 (Figure not shown). The fourth gene is closely related to the CODH gene from Methanosaeta thermophila, suggesting its origin from a methanogen. At deep-sea hydrothermal vents, the majority of microorganisms using the reductive acetyl-CoA pathway for carbon fixation are likely to be methanogens. The existence of putative methanogens in the 4143-1 sample was also revealed by mcrA gene analysis, indicating that methanogens affiliated with Methanomicrobiales, Methanosarcinales and Methanobacteriales exist in the chimney wall (Wang et al., 2009).
The key enzymes for the 3-HP cycle are acetyl-CoA/propionyl-CoA carboxylase, malonyl-CoA reductase and propionyl-CoA synthase. Not all of these enzymes were detected, and the detection frequencies of 3-HP genes were not high in the two chimney metagenomes (Figure 2c). Furthermore, 4-hydroxybutyryl-CoA dehydratase, the key enzyme of the 3-HP/4-hydroxybutyrate cycle was not found, although it occurs at high frequencies in the Global Ocean Sampling database (Berg et al., 2007). Our data further support the previous observations that the 3-HP cycle of the 3-HP/4-hydroxybutyrate may not be an important carbon fixation pathway at deep-sea hydrothermal vents.
Sulfur oxidation
The key enzymes involved in both the sulfur-compound oxidation (Sox)-dependent pathway and the adenosine-5′-phosphosulfate-dependent pathway were found to be abundant in the metagenome, whereas sulfite oxidase (EC1.8.3.1) and/or sulfite dehydrogenase (EC 1.8.2.1), which are involved in the sulfite:cytochrome c oxidoreductase pathway, were not present in the chimney 4143-1 metagenome (Figure 4). A similar sulfur oxidation pathway was found in the Lost City carbonate chimney, but in lower abundance compared to chimney 4143-1.

Sox-dependent and Sox-independent sulfur oxidation pathways identified in this study. SO42−, sulfate; SO32−, sulfite; S0, sulfur; S2O32−, thiosulfate; S2−, sulfide; APS, adenylylsulfate; sqr, sulfide:quinone oxidoreductase; fccAB, flavocytochrome c/sulfide dehydrogenase; sox, sulfur oxidation multienzyme complex. Box color indicated the odds ratio of each enzyme with darker red and green color representing higher odds ratio of respective enzyme in the chimney 4143-1 and lost city chimney metagenome, respectively. The color scales are the same as Figure 2a.
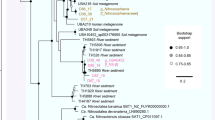
In the metagenome, the discovered sox genes include soxA, soxB, soxD, soxX, soxY and soxZ, indicating the presence of chemoautotrophs capable of oxidizing various reduced sulfur compounds to sulfate. To obtain more information about these lithotrophs, two fosmid clones containing soxB genes were selected and sequenced. The two retrieved soxB genes are closely related to each other and clustered with the corresponding genes from Halothiobacillus hydrothermalis and H. neapolitanus, two halophilic obligate sulfur-oxidizing chemolithoautotrophic Gammaproteobacteria (Sievert et al., 2000; Figure 5). The soxB gene from fosmid_soxB1 was separated from other sox genes, as has been observed in T. crunogena (Scott et al., 2006). The soxB from fosmid_soxB2 was linked with other sox genes to form a soxXYZAB cluster on the chromosome, but the single sox operon that has been described in facultative autotrophic sulfur-oxidizers, such as Paracoccus versutus and Rhodopseudomonas palustris (Schutz et al., 1999; Friedrich et al., 2000; Figure 5) was not found. Obligate autotrophic sulfur-oxidizers appear to not have the complete set of the sox genes organized in a single operon as observed in facultative autotrophs, possibly because the sox genes are constitutively expressed and, therefore, the sox gene organization into a single operon may not be strongly evolutionarily selected (Scott et al., 2006). On the basis of these observations, as well as the phylogeny of soxB, the two fosmid fragments containing the sox genes are most likely derived from obligate chemoautotrophic sulfur-oxidizing bacteria related to the sulfur-oxidizing Halothiobacillus. In the metagenome, the genes encoding the putative sulfide quinone oxidoreductase and the flavocytochrome c/sulfide dehydrogenase (FccAB) were both identified (Figure 4). Both these enzymes are known to catalyze the oxidation of sulfide to sulfur, whereas FccAB was hypothesized to be adapted to low sulfide concentrations (Mussmann et al., 2007). The detection of these enzymes may be a reflection of different niches corresponding to varying sulfide concentrations in the chimney wall. The elemental sulfur formed by sulfide quinone oxidoreductase or FccAB could be further oxidized. The genes encoding the dissimilatory sulfate reductase, adenosine-5′-phosphosulfate reductase and ATP sulfurylase were enriched in the metagenome, suggesting the presence of the so-called ‘reverse dissimilatory sulfate reductase’ pathway for sulfur oxidization (Hipp et al., 1997). From the microbiome, a total of 17 gene fragments annotated as partial dissimilatory sulfite reductase subunit A were retrieved. Around 16 of them could be potentially placed into the dsrA clusters from sulfide oxidizers (data not shown). The fact that the majority of the dissimilatory sulfite reductases cluster with those from sulfide-oxidizers further confirms the presence of a functional reverse dissimilatory sulfate reductase pathway for sulfur oxidization in the chimney microbial system.

Sox gene cluster organization in proteobacteria. Sox genes are gray, the gene between the sox genes are white. The letters in the boxes are abbreviated from corresponding sulfur oxidizing enzymes. Fosmid_soxB1 and fosmid_soxB2 in this study were marked with black diamonds. The cladogram was based on an alignment of 820 amino acids of the soxB genes. Sequences were aligned using ClustalW and the dendrogram was constructed using the neighbor-joining method by Mega 4.1.
In general, the oxidation of reduced sulfur compounds can be coupled to the reduction of electron acceptors, including oxygen and nitrate. In the metagenome, genes encoding the cbb3-type cytochrome c oxidase were identified, whereas genes encoding the widespread aa3-type cytochrome c oxidase were not found. The cbb3-type cytochrome c oxidase has a higher affinity for oxygen than the regular aa3-type cytochrome c oxidase, suggesting that members of the microbial community in the chimney wall are adapted to microoxic to anoxic conditions, similar to what has been described for the sulfur-oxidizing bacterium T. crunogena (Scott et al., 2006).
Nitrogen metabolism
The nitrogen cycle at deep-sea hydrothermal vent environments is less understood than the carbon and sulfur cycles. Intense nitrification and denitrification have been detected in some hydrothermal environments (Mehta et al., 2003), and biological nitrogen fixation has been identified as an important process contributing to the nitrogen cycle at deep-sea vents (Mehta et al., 2003; Rau, 1981). However, most of the microorganisms that are involved in the nitrogen cycle at vents are still largely unknown. High ammonium and nitrate concentration can be found in sediment-hosted hydrothermal fields, such as the Guaymas Basin and Okinawa Trough Backarc Basin, as well as the Endeavor segment of the Juan de Fuca Ridge (Ishibashi et al., 1995; Mehta et al., 2003). It has been demonstrated that ammonium is rapidly consumed by chemoautotrophs in the hydrothermal plume of vents on the Juan de Fuca Ridge (Lam et al., 2004). However, we did not find genes encoding for ammonia monooxygenase (amoA), a key enzyme for ammonia oxidization, in the metagenome.
On the other hand, the chimney 4143-1 metagenome is highly enriched in genes required for the complete denitrification pathway (Figures 1 and 6), suggesting that in addition to oxygen, nitrate could be an important electron acceptor, possibly coupled to sulfur-oxidation. All the genes, including nar (nitrate reductase), nos (nitrous oxide reductase), nir (nitrite reductase) and nor (nitric oxide reductase), for denitrification could be identified in the metagenome (Figure 6). The majority of the genes involved in denitrification are closely related to those from Beta and Alphaproteobacteria (data not shown). A fosmid clone (Fosmid_X) containing a narG fragment was selected and sequenced. The obtained narG has the highest identity with narG from Thiobacillus denitrificans (76%), an obligate chemolithoautotrophic bacterium capable of gaining energy by coupling the oxidation of reduced sulfur compounds to denitrification, as well as to aerobic respiration (Figure 7). The DNA fragment further showed synteny to Thiobacillus denitrificans, as the genes narK, narH, narJ and narI were found in identical order adjacent to narG. These data imply that sulfur oxidation coupled to denitrification is likely to be an important energy-generating pathway fueling the microbial community in the outer wall of the chimney. However, most of the key enzymes of denitrification pathway were not found in the Lost City carbonate chimney sequences (Figure 6), indicating that denitrification is not a universal pathway utilized by microbial communities inhabiting deep-sea vent chimneys, but is rather specific for particular vent communities, such as those from the Juan de Fuca Ridge. More analyses of chimneys from a variety of environments and locations should shed more light on this interesting observation.

Components of the nitrogen metabolism pathways identified in this study. Box color indicated the odds ratio of each enzyme with darker red and green color representing higher odds ratio of respective enzyme in the chimney 4143-1 and lost city chimney metagenome, respectively. The color scales are the same as Figure 2a.

Nitrate reductase gene cluster organization in the fosmid library. narL, nitrate/nitrite response regulator; narX, nitrate/nitrite sensor kinase; nark1, nitrate/proton symporter; nark2, nitrate/nitrite antiporter; narG, nitrate reductase, α-chain; narH, nitrate reductase, β-subunit; narJ, chaperone; narI, nitrate reductase γ-subunit.
In summary, comparative metagenomic analyses demonstrated that functional gene profiling could be a useful and reliable proxy to reflect the specific environment in which the biological communities reside. The metagenomes from the sulfide chimney 4143-1 and the carbonate chimney from Lost City clustered most closely. They are especially enriched in genes for mismatch repair and homologous recombination, suggesting that the microorganisms in the chimney walls have to cope with a higher rate of DNA damage caused by the extreme conditions of their habitat. The high percentage of transposases in the two samples from chimneys suggests that lateral gene transfer may be a common occurrence in the high temperature chimney biospheres. The metagenome further reveals that autotrophic carbon fixation in the sulfide chimney 4143-1 community was mainly a product of the CBB cycle, which appeared to be primarily driven by the oxidation of reduced sulfur compounds through both Sox-dependent and adenosine-5′-phosphosulfate-dependent pathways, using either oxygen or nitrate as terminal electron acceptors. Thus, the availability of reduced sulfur compounds, as well as oxygen and nitrate, appear to be key parameters structuring the microbial community. On the basis of the genomic organization of the key genes of the carbon fixation and sulfur oxidation pathways contained in the large genomic fragments, both obligate and facultative autotrophs appear to be present and contributing to biomass production. In addition, high abundance of chemotaxis genes in the metagenome reflected the adaptation of the organisms to a highly dynamic environment.
Accession codes
References
Badger MR, Bek EJ . (2008). Multiple Rubisco forms in proteobacteria: their functional significance in relation to CO2 acquisition by the CBB cycle. J Exp Bot 59: 1525–1541.
Berg IA, Kockelkorn D, Buckel W, Fuchs G . (2007). A 3-hydroxypropionate/4-hydroxybutyrate autotrophic carbon dioxide assimilation pathway in Archaea. Science 318: 1782–1786.
Biddle JF, Fitz-Gibbon S, Schuster SC, Brenchley JE, House CH . (2008). Metagenomic signatures of the Peru Margin subseafloor biosphere show a genetically distinct environment. Proc Natl Acad Sci USA 105: 10583–10588.
Brazelton WJ, Baross JA . (2009). Abundant transposases encoded by the metagenome of a hydrothermal chimney biofilm. ISME J 3: 1420–1424.
Campbell BJ, Cary SC . (2004). Abundance of reverse tricarboxylic acid cycle genes in free-living microorganisms at deep-sea hydrothermal vents. Appl Environ Microbiol 70: 6282–6289.
DeLong EF, Preston CM, Mincer T, Rich V, Hallam SJ, Frigaard NU et al. (2006). Community genomics among stratified microbial assemblages in the ocean's interior. Science 311: 496–503.
Friedrich CG, Quentmeier A, Bardischewsky F, Rother D, Kraft R, Kostka S et al. (2000). Novel genes coding for lithotrophic sulfur oxidation of Paracoccus pantotrophus GB17. J Bacteriol 182: 4677–4687.
Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS et al. (2006). Metagenomic analysis of the human distal gut microbiome. Science 312: 1355–1359.
Grzymski JJ, Murray AE, Campbell BJ, Kaplarevic M, Gao GR, Lee C et al. (2008). Metagenome analysis of an extreme microbial symbiosis reveals eurythermal adaptation and metabolic flexibility. Proc Natl Acad Sci USA 105: 17516–17521.
Hipp WM, Pott AS, Thum-Schmitz N, Faath I, Dahl C, Truper HG . (1997). Towards the phylogeny of APS reductases and sirohaem sulfite reductases in sulfate-reducing and sulfur-oxidizing prokaryotes. Microbiology 143: 2891–2902.
Huber H, Gallenberger M, Jahn U, Eylert E, Berg IA, Kockelkorn D et al. (2008). A dicarboxylate/4-hydroxybutyrate autotrophic carbon assimilation cycle in the hyperthermophilic Archaeum Ignicoccus hospitalis. Proc Natl Acad Sci USA 105: 7851–7856.
Huber JA, Butterfield DA, Baross JA . (2002). Temporal changes in archaeal diversity and chemistry in a mid-ocean ridge subseafloor habitat. Appl Environ Microbiol 68: 1585–1594.
Huber JA, Mark Welch DB, Morrison HG, Huse SM, Neal PR, Butterfield DA et al. (2007). Microbial population structures in the deep marine biosphere. Science 318: 97–100.
Ishibashi J, Sano Y, Wakita H, Gamo T, Tsutsumi M, Sakai H . (1995). Helium and carbon geochemistry of hydrothermal fluids from the Mid-Okinawa Trough Back Arc Basin, southwest of Japan. Chem Geol 123: 1–15.
King GM, Weber CF . (2007). Distribution, diversity and ecology of aerobic CO-oxidizing bacteria. Nat Rev Microbiol 5: 107–118.
Kristall B, Kelly D, Hannington M, Delaney J . (2006). Growth history of a diffusely venting sulfide structure from the Juan de Fuca Ridge: a petrological and geochemical study. Geochem Geophys Geosyst 7: 30.
Lam P, Cowen JP, Jones RD . (2004). Autotrophic ammonia oxidation in a deep-sea hydrothermal plume. FEMS Microbiol Ecol 47: 191–206.
Markert S, Arndt C, Felbeck H, Becher D, Sievert SM, Hugler M et al. (2007). Physiological proteomics of the uncultured endosymbiont of Riftia pachyptila. Science 315: 247–250.
Mehta MP, Butterfield DA, Baross JA . (2003). Phylogenetic diversity of nitrogenase (nifH) genes in deep-sea and hydrothermal vent environments of the Juan de Fuca Ridge. Appl Environ Microbiol 69: 960–970.
Meng J, Wang F, Zheng Y, Peng X, Zhou H, Xiao X . (2009). An uncultivated crenarchaeota contains functional bacteriochlorophyll a synthase. ISME J 3: 106–116.
Mussmann M, Hu FZ, Richter M, de Beer D, Preisler A, Jorgensen BB et al. (2007). Insights into the genome of large sulfur bacteria revealed by analysis of single filaments. PLoS Biol 5: e230.
Nakagawa S, Takai K . (2008). Deep-sea vent chemoautotrophs: diversity, biochemistry and ecological significance. FEMS Microbiol Ecol 65: 1–14.
Nakagawa S, Takai K, Inagaki F, Chiba H, Ishibashi J, Kataoka S et al. (2005). Variability in microbial community and venting chemistry in a sediment-hosted backarc hydrothermal system: Impacts of subseafloor phase-separation. FEMS Microbiol Ecol 54: 141–155.
Newton IL, Woyke T, Auchtung TA, Dilly GF, Dutton RJ, Fisher MC et al. (2007). The Calyptogena magnifica chemoautotrophic symbiont genome. Science 315: 998–1000.
Pruski AM, Dixon DR . (2003). Toxic vents and DNA damage: first evidence from a naturally contaminated deep-sea environment. Aquat Toxicol 64: 1–13.
Rau GH . (1981). Low 15N/14N in hydrothermal vent animals: ecological implications. Nature 289: 484–485.
Reysenbach AL, Shock E . (2002). Merging genomes with geochemistry in hydrothermal ecosystems. Science 296: 1077–1082.
Schrenk MO, Kelley DS, Delaney JR, Baross JA . (2003). Incidence and diversity of microorganisms within the walls of an active deep-sea sulfide chimney. Appl Environ Microbiol 69: 3580–3592.
Schubbe S, Williams TJ, Xie G, Kiss HE, Brettin TS, Martinez D et al. (2009). Complete genome sequence of the chemolithoautotrophic marine magnetotactic coccus strain MC-1. Appl Environ Microbiol 75: 4835–4852.
Schutz M, Maldener I, Griesbeck C, Hauska G . (1999). Sulfide-quinone reductase from Rhodobacter capsulatus: requirement for growth, periplasmic localization, and extension of gene sequence analysis. J Bacteriol 181: 6516–6523.
Scott KM, Sievert SM, Abril FN, Ball LA, Barrett CJ, Blake RA et al. (2006). The genome of deep-sea vent chemolithoautotroph Thiomicrospira crunogena XCL-2. PLoS Biol 4: e383.
Sievert SM, Heidorn T, Kuever J . (2000). Halothiobacillus kellyi sp. nov., a mesophilic, obligately chemolithoautotrophic, sulfur-oxidizing bacterium isolated from a shallow-water hydrothermal vent in the Aegean Sea, and emended description of the genus Halothiobacillus. Int J Syst Evol Microbiol 50: 1229–1237.
Tringe SG, von Mering C, Kobayashi A, Salamov AA, Chen K, Chang HW et al. (2005). Comparative metagenomics of microbial communities. Science 308: 554–557.
Tyson GW, Chapman J, Hugenholtz P, Allen EE, Ram RJ, Richardson PM et al. (2004). Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 428: 37–43.
Wang F, Zhou H, Meng J, Peng X, Jiang L, Sun P et al. (2009). GeoChip-based analysis of metabolic diversity of microbial communities at the Juan de Fuca Ridge hydrothermal vent. Proc Natl Acad Sci USA 106: 4840–4845.
Williams TJ, Zhang CL, Scott JH, Bazylinski DA . (2006). Evidence for autotrophy via the reverse tricarboxylic acid cycle in the marine magnetotactic coccus strain MC-1. Appl Environ Microbiol 72: 1322–1329.
Woyke T, Teeling H, Ivanova NN, Huntemann M, Richter M, Gloeckner FO et al. (2006). Symbiosis insights through metagenomic analysis of a microbial consortium. Nature 443: 950–955.
Wu M, Ren Q, Durkin AS, Daugherty SC, Brinkac LM, Dodson RJ et al. (2005). Life in hot carbon monoxide: the complete genome sequence of Carboxydothermus hydrogenoformans Z-2901. PLoS Genet 1: e65.
Zhang Z, Schwartz S, Wagner L, Miller W . (2000). A greedy algorithm for aligning DNA sequences. J Comput Biol 7: 203–214.
Acknowledgements
This work was supported by the China Ocean Mineral Resources R&D Association (COMRA DYXM115-05), the National Natural Science Foundation of China (40625016), the State High-Tech Development Project (2006AA09Z433, 2007AA091904 and 2008AA092603), project 2007DFA30840 of International S&T Cooperation Program from the Ministry of Science and Technology of China, key project of the Commission of Science and Technology of Guangdong Province and Guangzhou City and project of Sun Yet-sen University Science Foundation. SMS was supported by National Science Foundation grant OCE-0452333 and a fellowship awarded by the Alfried Krupp Wissenschaftskolleg Greifswald, Germany. We thank Huaiyang Zhou (Tongji University, Shanghai, China), Maurice A. Tivey (Woods Hole Oceanographic Institution, Woods Hole, MA, USA), Marvin D Lilley (University of Washington, Seattle, WA, USA), Deborah Kelly (University of Washington, Seattle, WA, USA), Kang Ding (University of Minnesota, Minneapolis, MN, USA) and all of the crew members from R/V Atlantis/DSV Alvin for their efforts and help in sample collection.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary Information accompanies the paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
Xie, W., Wang, F., Guo, L. et al. Comparative metagenomics of microbial communities inhabiting deep-sea hydrothermal vent chimneys with contrasting chemistries. ISME J 5, 414–426 (2011). https://doi.org/10.1038/ismej.2010.144
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2010.144
Keywords
This article is cited by
-
Global patterns of diversity and metabolism of microbial communities in deep-sea hydrothermal vent deposits
Microbiome (2022)
-
Bacterial diversity of geochemically distinct hot springs located in Maharashtra, India
Archives of Microbiology (2022)
-
Recent Advances of Marine Sponge-Associated Microorganisms as a Source of Commercially Viable Natural Products
Marine Biotechnology (2022)
-
Indus and Nubra Valley hot springs affirm the geomicrobiological specialties of Trans-Himalayan hydrothermal systems
Journal of Earth System Science (2022)
-
Biomass-degrading glycoside hydrolases of archaeal origin
Biotechnology for Biofuels (2020)
