
Abstract
Previous microarray analyses have shown a key role for the two-component system PhoBR (SYNW0947, SYNW0948) in the regulation of P transport and metabolism in the marine cyanobacterium Synechococcus sp. WH8102. However, there is some evidence that another regulator, SYNW1019 (PtrA), probably under the control of PhoBR, is involved in the response to P depletion. PtrA is a member of the cAMP receptor protein transcriptional regulator family that shows homology to NtcA, the global nitrogen regulator in cyanobacteria. To define the role of this regulator, we constructed a mutant by insertional inactivation and compared the physiology of wild-type Synechcococcus sp. WH8102 with the ptrA mutant under P-replete and P-stress conditions. In response to P stress the ptrA mutant failed to upregulate phosphatase activity. Microarrays and quantitative RT-PCR indicate that a subset of the Pho regulon is controlled by PtrA, including two phosphatases, a predicted phytase and a gene of unknown function psip1 (SYNW0165), all of which are highly upregulated during P limitation. Electrophoretic mobility shift assays indicate binding of overexpressed PtrA to promoter sequences upstream of the induced genes. This work suggests a two-tiered response to P depletion in this strain, the first being PhoB-dependent induction of high-affinity PO4 transporters, and the second the PtrA-dependent induction of phosphatases for scavenging organic P. The levels of numerous other transcripts are also directly or indirectly influenced by PtrA, including those involved in cell-surface modification, metal uptake, photosynthesis, stress responses and other metabolic processes, which may indicate a wider role for PtrA in cellular regulation in marine picocyanobacteria.
Similar content being viewed by others

Introduction
Marine cyanobacteria of the genera Prochlorococcus and Synechococcus are globally distributed and ecologically significant (Partensky et al., 1999; Scanlan et al., 2009). These two genera are genetically diverse and numerically abundant across global scales, from the oligotrophic ocean gyres where Prochlorococcus predominate, to subtropical, temperate and coastal systems dominated by Synechococcus (Zwirglmaier et al., 2008). As the base of the food web and as a source of photosynthetically fixed carbon, cyanobacterial limitation by macronutrients such as nitrogen and phosphorus would therefore impact all trophic levels.
Reports of P limitation in some oceanic regimes are accumulating, for example, in the northwestern Atlantic (Sargasso Sea) (Cotner et al., 1997; Ammerman et al., 2003), North Pacific Subtropical Gyre (Karl and Tien, 1997) whereas the Mediterranean Sea shows P limitation during summer stratification (Thingstad et al., 2005). Because dissolved mineral phosphate concentrations fall into the low nanomolar range in oligotrophic regions and the nutrient is rapidly turned over (Karl and Tien, 1997; Thingstad et al., 2005; Zubkov et al., 2007) there is likely an intense competition for bioavailable P.
Marine cyanobacteria have adopted distinct strategies to cope with low and fluctuating levels of P in the environment. Comparative genomics (Palenik et al., 2003, 2006; Dufresne et al., 2008; Scanlan et al., 2009) and metagenomic surveys (Venter et al., 2004) highlight that there are multiple genome-encoded copies of the gene for the high-affinity, periplasmic P-binding protein, PstS, indicating the importance of the affinity capture of inorganic P, even for strains that have undergone significant genome streamlining (Dufresne et al., 2003). It is also evident that distinct isolates, or ecotypes, show different physiological and genetic capabilities to use organic P (Moore et al., 2005; Martiny et al., 2006; Scanlan et al., 2009). Picocyanobacteria that inhabit low P environments show an overall low requirement for P (Bertilsson et al., 2003; Heldal et al., 2003). They are also capable of economizing their use of P in cellular constituents, for example, by substituting phospholipids for sulfolipids (Van Mooy et al., 2006). In addition, contrary to many bacteria, marine cyanobacteria have evolved forms of lipopolysaccharide, a major cellular constituent, that lack phosphate (Snyder et al., 2009).
In model freshwater cyanobacteria a P-regulatory system composed of a two-component response regulator and a sensory kinase, which is homologous to the Escherichia coli PhoBR, has been well characterized (Aiba et al., 1993; Aiba and Mizuno, 1994; Hirani et al., 2001). This system activates the transcription of genes of the P regulon, through activation of the response regulator, SphS (PhoB), by its cognate sensor histidine kinase, SphR (PhoR), presumably in response to a low external concentration of P. In Synechocystis sp. PCC6803, the Pho regulon is composed of at least 12 genes, including two clusters of high-affinity ABC transport systems as well as two colocalized genes encoding an alkaline phosphatase and an extracellular nuclease (Suzuki et al., 2004).
The composition and arrangement of genes in the P regulons of marine cyanobacteria appear to be highly variable (Martiny et al., 2006; Scanlan et al., 2009; Tetu et al., 2009). Among available genome sequences PhoBR homologues are conserved in some, but not all, indicating that P sensing and regulation may have been lost in some lineages. The loss of phoBR in some strains may reflect adaptation to environments with a relatively stable and adequate supply of P such as coastal environments (Palenik et al., 2006). In open ocean Synechococcus sp. WH8102, the P regulon has been characterized by computational predictions (Su et al., 2003, 2007), as well as physiological experiments (Moore et al., 2005) and microarray analyses (Tetu et al., 2009). Comparisons of gene expression in knockout mutants constructed in phoB and phoR confirm a role for P sensing and regulation for these genes in this strain (Tetu et al., 2009). Central elements of the Synechococcus sp. WH8102 P regulon were identified, including four genes encoding paralogues of the periplasmic P-binding proteins, PstS (SYNW1018, SYNW1815, SYNW2507) and SphX (SYNW1286), genes of the ABC transport system for P (SYNW1270, SYNW1271), porins (SYNW2224, SYNW2223), at least three diverse predicted phosphatases (SYNW0196, SYNW2390 and SYNW2391) and a number of genes with weakly associated or no functional predictions (SYNW0165, SYNW0762 and SYNW1333). These experiments also highlight the involvement of another regulator, PtrA, in the P-stress response that was hypothesized to be a potential P regulator in Synechococcus sp. WH7803 (Scanlan et al., 1997a). In Synechococcus sp. WH7803 and WH8102 ptrA is located downstream of the gene for the phosphate-stress-induced periplasmic phosphate-binding protein, PstS. A putative pho box upstream of ptrA suggests that this gene is regulated by PhoB (Su et al., 2007), whereas microarray analysis of Synechococcus sp. WH8102 in response to P depletion shows that ptrA is highly expressed in response to P stress, and confirms that it is most likely under the influence of PhoB (Tetu et al., 2009).
cAMP receptor protein (CRP) family regulators consist of a C-terminal DNA-binding domain that forms a helix–turn–helix motif that slots into the major groove of the DNA and binds to the promoter region of target genes to act either as an activator or an repressor (Korner et al., 2003). The N terminus is composed of a nucleotide-binding domain, which functions as a sensor module that interacts with a signal molecule and is responsible for dimerization and activation. Comparison of amino-acid sequences of putative CRP family regulators from marine cyanobacterial genomes highlights at least four distinct clusters of orthologues (Scanlan et al., 2009). In Synechococcus sp. WH8102, PtrA is one of only two genome-encoded regulators of the CRP family and a homologue of NtcA, the global nitrogen regulator that is conserved in all cyanobacteria (Luque et al., 1994; Lindell et al., 1998; Herrero et al., 2001). The E. coli CRP activates transcription in the presence of cAMP at more than 100 promoters (Salgado et al., 2004) whereas there are a predicted 17–54 NtcA targets in Synechococcus sp. WH8102 (Su et al., 2005).
Given the sequence similarity, PtrA could be the P ‘equivalent’ of NtcA, and therefore a general phosphate regulator in marine cyanobacteria that modulates the expression of part of the P regulon with potentially wider influence on cellular processes. To define the role of this regulator, we constructed a mutant in Synechococcus sp. WH8102 by insertional inactivation and compared the physiology of the ptrA mutant with the isogenic parent strain in response to P stress. Microarrays and quantitative RT-PCR were used to compare patterns of global gene expression in response to P stress. Comparison of the ptrA mutant transcriptome with data from parallel studies (Tai et al., 2009; Tetu et al., 2009) indicates that ptrA is not only an important component of the WH8102 P regulon directly controlling a subset of P-stress genes but also may have a wider influence on the expression of a variety of gene clusters throughout the genome, implicating it in coordinately regulating cellular metabolism during the P stress response.
Materials and methods
Bacterial strains, growth conditions and P-stress experiments
For genetic transformations Synechococcus sp. WH8102 was grown in SN medium (Waterbury and Willey, 1988) prepared with Sargasso Sea water (Sigma-Aldrich, St Louis, MO, USA) with constant illumination at 25 μmol photons m−2 s−1 at 25 °C. For growth experiments Synechococcus sp. WH8102 was grown in synthetic seawater medium with salts based on Aquil (Morel et al., 1979). The final concentration of nutrients in Aquil was as follows: 4.5 mM NaNO3, 90 μM Na2PO4, 100 μM NH4Cl, 13.4 μM Na2EDTA, 10 μM Na2CO3 (N/P=50:1). For P-stress experiments the PO4 concentration was amended to 10 μM, yielding an N/P ratio of 450:1. Cultures were acclimated to 40 μmol photons m−2 s−1 and 10 μM PO4 for a minimum of three serial transfers before the start of each experiment. Growth was monitored on a daily basis by spectrophotometry and flow cytometry, the latter using a FACScan flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). Culture axenicity was monitored regularly by plating an aliquot (200 μl) onto solid Aquil containing 500 mg l−1 yeast extract.
Insertional inactivation of ptrA in Synechococcus sp. WH8102
Directed inactivation of ptrA (SYNW1019) was accomplished according to methods previously described (Brahamsha, 1996). Briefly, a 297 bp internal fragment, corresponding to nucleotides 213–488 of the 684 bp gene, was cloned into the suicide vector pMUT100. The ptrA fragment was amplified with primers PtrAF9 (5′-TGTGCGCGGCATGGTCAAGCTTG-3′) and PtrAR9 (5′-CATTTCCAGAAAACCTCTCACCCG-3′), TA cloned into pCR2.1TOPO (Invitrogen, Carlsbad, CA, USA) and then subcloned into the EcoRI site of pMUT100. Biparental conjugations were conducted exactly as described by Brahamsha (1996) with the exception that SN medium was prepared with Sargasso Sea water. Exconjugants were selected on plates and clonal isolates were propagated in liquid SN with kanamycin (25 and 15 μg ml−1, respectively). Segregation of mutant chromosomes, arising from a single crossover insertion of the plasmid construct, was confirmed by PCR using primer sets targeting sites on the Synechococcus sp. WH8102 genome flanking ptrA and sites internal to the pMUT100 vector (as outlined in Supplementary Figure 1).
Phosphatase and soluble reactive phosphate assays
Phosphatase activity was measured throughout the growth of mutant and wild-type cultures using the para-nitrophenyl phosphate (p-NPP) assay (Bessey et al., 1946) adapted for use in a microplate reader as described in Moore et al. (2005). The concentration of extracellular soluble reactive phosphate was determined spectrophotometrically by the ammonium molybdate assay (Itaya and Ui, 1966) in a quartz cuvette with an optical path length of 5 cm. The limit of detection with this method was 50 nM.
SDS–PAGE analysis of protein expression in response to P stress
Protein expression in low P cultures was monitored by pulse-labeling of a culture aliquot (5.0 ml) with 35S-methionine (1.0 μCi ml−1) for 4 h under identical incubation conditions as the source culture as described previously (Scanlan et al., 1993). At the end of the incubation period the cells were harvested by centrifugation and resuspended in sodium dodecyl sulfate (SDS)–PAGE loading buffer (90 mM Tris-HCl (pH 6.8), 20% (v/v) glycerol, 2% (w/v) SDS, 0.02% (w/v) bromophenol blue, 100 mM DTT). Samples were boiled for 10 min and centrifuged (10 000 g, 5 min) before loading onto a 12% SDS–PAGE. After electrophoresis, the gel was stained with Coomassie blue and the labeled proteins were visualized by exposing the dried gel to a phosphor screen (Fujifilm LifeScience, Bedford, UK) analyzed using a phosphorimager (Fuji FLA5000). Protein bands of interest were excised from a replicate gel prepared with unlabeled cells extracted from the source culture, subjected to tryptic digest and identified by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry using a Micromass MALDI-LR (Waters Corporation, Milford, MA, USA).
RNA isolation and microarray analysis
Total RNA from 1 liter of exponential phase cells was extracted using a Trizol-based method and purified using the Qiagen RNeasy kit following manufacturer's instructions as previously described (Tetu et al., 2009). cDNA was labeled, hybridized to microarrays and the results were captured as previously described (Tetu et al., 2009). RNAs from two wild-type and two ptrA mutant cultures, harvested at approximately 125–150 h after inoculation were used. Six hybridizations were carried out, of which two used different RNA pools (biological replicates) and four were replicates of these two experiments, either dye swapping experiments or direct replicates. Statistical analyses were carried out on the mean of log2-transformed signal ratios of all replicates using the Significance Analysis of Microarrays algorithms (Tusher et al., 2001) with a false discovery rate of less than 1%.
The microarray data presented here are in accordance with the Microarray Gene Expression Data Society's minimum information about a microarray experiment recommendations (Brazma et al., 2001). A description of the experiments, quantitation data and array design has been deposited into the gene expression omnibus database (http://www.ncbi.nlm.nih.gov/geo/) with the assigned accession number GSE18511.
Quantitative RT-PCR
DNA was removed from total RNA using the Turbo-DNase Digestion kit (Ambion, Austin, TX, USA). The absence of DNA was confirmed by PCR and then RNA (200–500 ng) was reverse transcribed with SuperScript II (Invitrogen) in the presence of 200 U SuperaseIN (Ambion) and random hexamers (25 pM). The resulting cDNA was diluted more than 20-fold and used as a template 5–10 ng equivalent of starting RNA. qRT-PCR reactions were carried out in triplicate for biological replicates with Power SYBR Green PCR Master Mix on an ABI Prism 7000 Sequence Detection System (Applied Biosystems, Foster City, CA, USA) according to the manufacturer's recommendations. Primers were designed with ABI Prism Primer Express version 2.0 software for products of 50–65 bp length (Supplementary Table 1). The gene for the class B RNaseP (rnpB) was used as an internal reference. Analysis of RNA transcript abundance was carried out with the ΔΔCT method using ABI Prism SDS 2.1 Software (Applied Biosystems).
Overexpression of PtrA in E. coli
The ptrA gene was PCR amplified with primers 1019pet15bf (5′-AGCCATATGCATGTTGCGCTCCATAC-3′) and 1019pet15br (5′-GGATCCTAGCGACGTGGCAGGTGGGCG-3′) with engineered restriction sites compatible with the in-frame insertion into the NdeI and BamHI sites of the 6 × His tag expression vector pET15b (Novagen, Madison, WI, USA). After the sequence of the construct was confirmed and the plasmid was transformed into the E. coli expression host Rosetta (DE3). Expression cultures were inoculated (1%, 250 ml) from overnight precultures and grown in LB medium supplemented with chloramphenicol (30 μg ml−1) and ampicillin (100 μg ml−1) at 37 °C in an orbital shaker at 200 r.p.m. Upon reaching an OD600 of 0.4, the expression of recombinant protein was induced with IPTG (20 μM) and the culture was transferred to 25 °C with shaking (200 r.p.m.) before the cells were harvested by centrifugation 4–5 h later. These conditions favored the production of soluble recombinant protein despite the decrease in overall yield as a result of using subsaturating concentrations of IPTG. Harvested cell pellets were washed once and then resuspended in lysis buffer (20 mM Tris-HCl (pH 8.0), 500 mM NaCl, 0.1% Triton X-100), frozen for a minimum of 2 h, and then lysed with three passages through a French pressure cell followed by sonication (3 × 30 s). The lysate was fractionated by centrifugation (14 000 g, 30 min, 4 °C). Imidazole was added to the soluble fraction to a final concentration of 50 mM before being applied to a pre-equilibrated Ni(II) immobilized metal affinity column (5 ml, HiTrap Chelating HP; GE Health Sciences; GE Healthcare Life Sciences, Buckinghamshire, UK). The column was washed with 1 volume each of 20 mM Tris-HCl (pH 8.0), 500 mM NaCl (Buffer A) containing 50 and 100 mM imidazole. Bound PtrA was eluted with Buffer A containing 300 mM imidazole and the purity of the fraction was assessed by SDS–PAGE. The purified protein was concentrated and the buffer exchanged for DNA Binding Buffer (20 mM Tris-HCl (pH 7.4), 120 mM KCl, 10 mM MgCl2, 5 mM ZnCl2, 100 μM EDTA) supplemented with 20% (v/v) glycerol by successive concentration and dilution with a Centricon YM-10 centrifugal filter device (Millipore, Billerica, MA, USA). The concentration of recombinant protein was determined with the BCA kit against known quantities of bovine serum albumin (Sigma-Aldrich).
Electrophoretic mobility shift assays
DNA fragments for electrophoretic mobility shift assay (EMSA), encompassing approximately 350 bp upstream and 50 bp downstream of the start codon of target genes, were PCR-amplified from Synechococcus sp. WH8102 using primers listed in Supplementary Table 2 and cloned into pCR2.1TOPO. Each fragment was excised from the plasmid by restriction digest with EcoRI, agarose gel purified and end-labeled with [γ-32P]dATP using polynucleotide kinase. DNA fragments were then incubated with purified proteins at 25 °C for 20 min in DNA-binding buffer supplemented with nonspecific competitor DNA, either 25–50 μg ml−1 polydI/dC or 12.5 μg ml−1 herring sperm DNA. Protein stocks of PtrA were diluted in buffer containing competitor DNA before addition to the binding reaction. Reactions were loaded onto a 5.0% polyacrylamide gel run in 0.5 × Tris-Borate-EDTA buffer for 4–5 h at 180 V. The results were visualized by exposing the dried gel to a phosphor screen (Fujifilm LifeScience) and analyzed using a phosphorimager (Fuji FLA5000).
Results and discussion
Phenotype of the Synechococcus sp. WH8102 ptrA mutant
To define the precise role of ptrA, we constructed a mutant by insertional inactivation in Synechococcus sp. WH8102 and examined the physiology of the mutant relative to the wild-type strain during growth in low-P medium (10 μM PO4). P-stress growth experiments were conducted exactly as described in previous work (Tetu et al., 2009) to enable comparison of gene expression with independent microarrays of phoB and phoR mutants. The maximum rates of growth and yield were similar for wild-type and mutant under P-replete (90 μM PO4) and low-P growth conditions (0.41 and 0.39 per day, respectively; Figure 1). Extracellular P was depleted to undetectable levels (50 nM) within the first 120 h of growth, that is, approximately 100–150 h before the onset of stationary phase (Figure 1), which is similar to previous observations for this strain (Moore et al., 2005; Tetu et al., 2009). The disappearance of P was identical in wild-type and mutant strains, indicating that P uptake was not significantly impaired in the mutant.

(a) Comparison of growth and phosphate (P) uptake of the ptrA mutant compared with the wild-type strain in low-P media in batch cultures and (b) the development of phosphatase activity in mutant and wild-type cultures. (c) The induction of PstS expression in wild-type during early P stress.
Although P uptake was apparently unaffected, the production of phosphatase activity was markedly different in response to P stress (Figure 1b). Wild-type Synechococcus sp. WH8102 shows three distinct levels of phosphatase activity in batch culture, a low constitutive level, followed by an intermediate level approximately 50 h after the disappearance of extracellular P and a maximum level at the point where cell division ceases. The induction of phosphatase activity was significantly affected in the ptrA mutant, where the absolute amount of activity did not increase above a constitutive level. This result suggests that at least one, or more, of the four identified phosphatase genes in Synechococcus sp. WH8102 (SYNW0196, SYNW1799, SYNW2390 and SYNW2391) are directly or indirectly under the control of PtrA, although the p-NPP assay used here may not account for any highly specific nucleotidase activity of phosphatases that possess nucleotidase domains. Insertional inactivation of the ptrA orthologue in Synechococcus sp. WH7803 also resulted in a similar phenotype (Ostrowski and Scanlan, unpublished data) where induction of phosphatase activity was impaired in response to P stress.
Changes in protein expression in low P cultures were monitored using 35S-methionine labeling and SDS–PAGE. It is particularly interesting to note the lag between the induction of PstS (SYNW1018) and porin (SYNW2224) expression after 96 h (Figure 1c), and the time when intermediate and maximum levels of phosphatase activity are observed in the wild-type Synechococcus sp. WH8102 (at ∼172 and 240 h, respectively). There was no significant time difference in the induction of PstS expression between the mutant (not shown) and wild-type Synechococcus sp. WH8102, which produced identical expression profiles, indicating that the initial response to P stress is apparently unaffected by the inactivation of ptrA.
Comparison of expression of P-stress genes in the Synechococcus sp. WH8102 ptrA mutant
To characterize the PtrA regulon in more detail, we used a whole-genome microarray to compare patterns of global gene expression in the mutant and isogenic parent strains. RNA was harvested from mutant and wild-type strains under low-P conditions just after the onset of induction of phosphatase activity in wild-type strain (∼125–150 h after inoculation). The expression levels of 516 genes were negatively affected in the mutant. Genes whose expression level was lower by twofold or more are listed in Table 1 . A total of 645 genes were upregulated in the ptrA mutant and those upregulated more than twofold are shown in Table 2 .
The amount of overlap between sets of genes negatively affected by ptrA and the genes previously reported as upregulated by early P stress or affected by phoB inactivation (Tetu et al., 2009) was surprisingly small (Figure 2). Of the 97 genes that were downregulated by more than twofold, only 7 were shown to be correspondingly upregulated in response to early P stress in wild-type (highlighted in color in Figure 2). Tetu et al. (2009) described nine genes of the PhoB regulon that were highly expressed in response to early P stress and were not upregulated in a phoB knockout. These genes include a possible porin (SYNW2224), three phosphatases (SYNW0196, SYNW2390 and SYNW2391), one PstS gene (SYNW1018) as well as ptrA. Five of those nine genes were also significantly downregulated in the ptrA mutant (Figure 2), encompassing two phosphatase genes (SYNW0196 and SYNW2390), a possible phytase (SYNW0762), a conserved hypothetical gene (SYNW1333) and the gene for a P-starvation inducible polypeptide (psip1, SYNW0165), which is localized to the cell wall (West and Scanlan, unpublished data).

Comparison of the relative expression of phosphate (P) regulon genes from microarray analysis and qRT-PCR that were differentially expressed in the ptrA (SYNW1019) and phoB (SYNW0947) mutants in comparison with early P stress in the wild-type strain. Negative values indicate genes that were downregulated in each mutant. Positive values indicate genes that were upregulated in the wild-type strain during early P stress. Genes with relative expression greater than twofold are highlighted in color (red and green for positive and negative values, respectively).
The direct comparison between the expression profiles of ptrA and phoB knockouts in Figure 2 provides good support for the hypothesis that PtrA may directly regulate a subset of the P-responsive genes in Synechococcus sp. WH8102 (that is, SYNW0165, SYNW0196, SYNW0762, SYNW1333 and SYNW2390) but not others, including the pstS gene directly upstream of ptrA (SYNW1018), a phosphatase/nucleotidase gene (SYWN2391) upstream of SYNW2390 and a porin (SYNW2224). It is highly likely that PtrA is directly regulated by PhoB, either from a predicted pho box promoter in the intergenic region between pstS (SYNW1018) and ptrA (Su et al., 2003, 2007) or by co-transcription directed from the pho box of pstS. Using primers spanning the 3′ end of pstS and the 5′ end of ptrA, we were able to detect reverse transcription–PCR products of expected size indicating that these two genes are, at least partially, co-transcribed (data not shown).
The remaining 92 genes with significantly downregulated expression levels in the mutant are involved in a range of cellular processes. It is interesting to note that many of these potential PtrA-regulated genes are gathered into five distinct genomic clusters (Table 1; Supplementary Table 3; Figure 3). One cluster (cluster 9, SYNW2477–SYNW2485) includes genes for a predicted Zn2+ ABC transport system, a ferredoxin-nitrite reductase, a putative cyanate transporter and four conserved hypothetical genes of unknown function. Zn is found at subnanomolar concentrations in surface waters of the Pacific and Atlantic Oceans yet it is unclear whether this element limits phytoplankton growth (Lohan et al., 2002). In cyanobacteria, Zn is required for the activity of carbonic anhydrase as well as a range of metalloproteins involved in many aspects of metabolism (Blindauer, 2008). Because phosphatases also have a known Zn requirement for activity (Coleman, 2003), the elevated levels of the Zn2+ transport system in wild-type may simply reflect higher Zn requirements to match the increased phosphatase production, rather than direct regulation by PtrA. Synechococcus sp. WH8102 possesses Zur (SYNW2401) and SmtA (SYNW0359) orthologues that have the potential to function as Zn sensors and/or regulators (Blindauer, 2008). Although a direct involvement in Zn regulation has not been experimentally determined, either of these proteins appear to be better candidates to regulate the Zn transport system in response to Zn demand.

Genomic distribution of genes that were differentially expressed in the ptrA mutant relative to wild-type in response to P stress in Synechococcus sp. WH8102. Negative values indicate genes that were downregulated in the mutant. Positive values indicate genes that were upregulated in the mutant. All genes with significant Analysis of Microarrays (SAM) values are plotted. Genes with altered expression levels greater than twofold are highlighted in color (red or green). Single genes and clusters of genes of interest discussed in the text are annotated with cluster numbers.
Cluster 6 (Table 1; Figure 3) is composed of 11 consecutive genes (SYNW0952–SYNW0962) mostly of unknown function. These genes include three that were shown by transposon mutagenesis to have a role in swimming motility in Synechococcus sp. WH8102, including the 34 kb gene for SwmB (SYNW0953) and two associated orfs (SYNW0958 and SYNW0960), that may encode a multicomponent transport apparatus (McCarren and Brahamsha, 2005, 2007). Although SwmB, which is known to be associated with the outer membrane, and surrounding genes in this cluster are required for motility, the genes for other integral components of the swimming apparatus, such as SwmA, do not show a similar pattern of expression in the mutant. It is possible that the genes in this cluster serve an additional role unrelated to motility that involves modifying the cell surface. Indeed, the expression levels of 27 genes (cluster 3, SYNW0424–SYNW0458), which are all related to cell-surface modification and polysaccharide production, are all downregulated in the mutant. As stated previously (Tetu et al., 2009), reorganization and/or strengthening of the cell envelope may be required to accommodate additional P-stress-induced porins (SYNW2224 and SYNW2223). An alternative explanation could be that, as a result of a reduction in cellular growth rate due to limiting P, the cell is capable of diverting nonlimiting resources, for example, C and N, to lipopolysaccharide and carbohydrate modification of the cell wall as a means of avoiding grazers and phage.
A number of genes involved in phycobilisome biosynthesis are also downregulated in the mutant (cluster 7). Other genes of interest with predicted functions that show lower levels of expression in the mutant include genes for cytochrome c oxidase subunits I and II (SYNW1529, SYNW1861 and SYNW1862), a PhoH family protein (SYNW1946), a putative transcriptional regulator (SYNW2105) and the gene for the plastoquinol terminal oxidase (SYNW0887) that may have a role in diverting electrons from PSII out of the photosynthetic electron transport chain under certain conditions (Mackey et al., 2008).
As an independent means of observing trends in gene expression, qRT-PCR was performed on a selection of genes. In each case qRT-PCR resulted in the same directional trend as for the microarray analysis in the mutant relative to wild type, whereas the magnitude of upregulation during P stress in wild type was equivalent to the values reported in previous work (Figure 2; Tetu et al., 2009).
Genes with relatively higher levels of expression in the PtrA mutant
The transcript levels of 63 genes recorded higher levels in the ptrA mutant (Table 2). Among the genes with known functions are the RNA polymerase β-‘subunit (SYNW0615) and 16 ribosomal proteins (SYNW2067–SYNW2091, SYNW2135 and SYNW2136), many of which were significantly repressed in Synechococcus sp. WH8102 (Tetu et al., 2009) and in other organisms in response to P limitation (Martiny et al., 2006) and N limitation (Silberbach et al., 2005). Because the ribosomes themselves are a significant sink for P-rich ribonucleotides, it would be advantageous for a cell to economize the biosynthesis of ribosomes, and associated components, in response to P stress. Given that the mutant and parent strains were grown side by side under identical conditions, the higher levels of expression of ribosomal proteins suggest the mutant fails to downregulate its translation mechanism. It is possible that the expression of central metabolism genes, such as ribosomal proteins, is influenced by PtrA. However, a perhaps more likely explanation is that the growth of the parent and mutant was subtly different and that they were simply in different growth phases, possibly because the mutant used intracellular P at a slower rate.
Ribonucleotide reductase (class II, SYNW1147) also showed a higher level of expression. This enzyme catalyzes the conversion of ribo- to deoxyribonucleotide diphosphates (Elledge et al., 1992) and thus occupies a central role channeling nucleotides to DNA synthesis. The expression of several stress response proteins was also higher, including GroES (SYNW0513), two copies of GroEL (SYNW0514 and SYNW1854), DnaK2 (SYNW2508) and the heat shock protein HtpG (SYNW1278). These stress response genes suggest the ptrA mutant may have been more subtly stressed than wild-type one possibly due to the inability to mount an effective response to early phosphate depletion, and this might explain some gene expression differences not directly related to phosphate. Although a number of genes involved in phycobilisome biosynthesis and cytochrome c oxidase subunits were repressed in the mutant, genes for photosystem II reaction center proteins L, T (SYNW0202 and SYNW1983) and all four D1 forms I and II (SYNW0983, SYNW1470, SYNW1919 and SYNW2151) showed significantly higher levels. This was also the case for NADH dehydrogenase I chain 2 (SYNW1873), ATP synthase subunit c, cytochrome b6f complex subunit 4, apocytochrome b6 and the cytochrome b559 β-chain. However, the reason for these differences is unclear.
PtrA binding to promoters
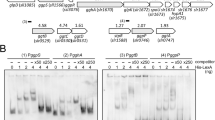
In light of the involvement of additional regulators in the P-stress response (Tetu et al., 2009), we used EMSA as an alternative means of confirming that PtrA is a functional regulator that binds to the upstream regions of regulated genes. Despite attempts to optimize expression of recombinant PtrA in E. coli a soluble product was produced in low quantities. Despite low yield, soluble PtrA was purified using metal affinity chromatography, concentrated and resuspended in DNA-binding buffer supplemented with potential cofactors, 5 mM ZnCl2 and 10 μM cAMP. Figure 4 shows an EMSA experiment to investigate the binding of increasing amounts of purified PtrA to ∼400 bp DNA fragments comprising the upstream regions of SYNW0165, SYNW0196, SYNW1018 and SYNW2390. Up to three distinct PtrA-DNA fragments can be observed for the upstream region of SYNW0165, corresponding to 1, 2 and 3 PtrA target sites in this DNA fragment. Preincubation with nonspecific competitor or 125 × excess of unlabeled competitor DNA shows that PtrA binding to this DNA fragment is sequence specific. Although we were able to detect PtrA binding to specific promoter elements in SYNW0165, SYNW0196 and SYNW2390, no mobility shift was observed for the DNA fragment corresponding to the upstream region of pstS (SYNW1018; Figure 4), which supports the hypothesis that PtrA is a transcription factor that regulates psip1 and these two phosphatases, but not pstS. In general, the amount of DNA specifically bound to recombinant PtrA in each assay was low, which might reflect a low proportion of correctly folded, active protein in the preparation. The relatively weak binding signals observed in vitro also suggest that additional cofactors or cooperative interactions with other transcription factors and possibly RNA polymerase are required for binding in vivo. Indeed, this could even involve cooperative interactions with PhoB at some target sites as a number of PtrA-regulated genes show predicted pho boxes, for example, three operons spanning cluster 3 (SYNW0440–SYNW0458) and a weak prediction for psip1 (Su et al., 2007). However, the majority of genes potentially regulated by PtrA, including the most highly expressed genes, do not possess pho boxes but have regions bound by PtrA, although with motifs we have not been able to determine. Thus despite our hypothesis that PtrA would be a phosphate regulatory factor with a mechanism similar to NtcA for nitrogen it seems to have distinct differences. Further bioinformatic and experimental work is required to identify PtrA-binding motifs in Synechococcus sp. WH8102.

Electrophoretic mobility shift assay showing specific binding of recombinant PtrA (6His-PtrA) to the upstream regions (−350 to +50 bp relative to the start codon) of SYNW0165, SYNW0196 and SYNW2390 but not SYNW1018. The sloping bar indicates increasing concentration of the recombinant protein.
Role of PtrA in response to P limitation in WH8102
Taken together, the respective organization of the PhoB and PtrA regulons suggests a two-tiered response to the level of P limitation in Synechcococcus sp. WH8102. The first level of response is manifest by the affinity scavenging of inorganic P during early P stress involving PhoB-induced expression of pstS genes (SYNW1018, SYNW1286 and SYNW1815) as well as an elevation in the level of PtrA, either from co-transcription of ptrA with SYNW1018 or from a dedicated pho box immediately upstream of ptrA. Elevated levels of PtrA lead to the second level of response characterized by the scavenging of organic P involving PtrA-induced expression of two phosphatases (SYNW0196 and SYNW2390), and a predicted phytase (SYNW0762). Although SYNW2391-SYNW2390 is a predicted transcriptional unit with two tandem pho boxes found upstream of SYNW2391 (Su et al., 2007), the expression of these genes appears to be influenced separately by PtrA and PhoB. The EMSA presented here shows that PtrA binds to the promoter region of SYNW2390. This is further supported by the comparison of ptrA and phoB array data that confirm that SYNW2390 is predominantly regulated by PtrA and SYNW2391 is regulated by PhoB. More complicated regulation could also be occurring for some genes with potentially both PtrA and PhoB-binding sites. Thus, PtrA may be involved in a signal cascade that grades the cellular response initially to P stress, through P limitation and finally to chronic P starvation.
Role of PtrA in adaptation to P limitation in marine Synechococcus and Prochlorococcus
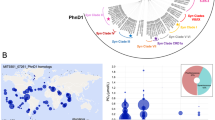
The gene for PtrA was first discovered in Synechococcus sp. WH7803 downstream of the gene for the periplasmic P-binding protein (PstS) which is highly induced in response to P stress in WH7803 (Scanlan et al., 1993; Scanlan et al., 1997b). Since then ptrA homologues have been identified in the genomes of 10 out of 12 oceanic Synechococcus and 5 out of 12 Prochlorococcus. The phylogenetic profile of ptrA and phoBR provides some interesting insights. Out of 12 complete, or nearly complete Synechococcus genomes, ptrA is absent from only two coastal strains isolated from the California Current, CC9311 (clade I) and CC9902 (clade IV) (Palenik et al., 2006). Both strains also lack phoBR and encode a single copy of pstS suggesting that they do not encounter P limitation in their environments. The ptrA gene is represented in fewer Prochlorococcus genomes (5 out of 12) where it is predominantly found in low light-adapted ecotypes. It is also interesting to note the presence of ptrA in at least three strains where phoBR is absent or incomplete, BL107 (clade IV) and Prochlorococcus strains SS120 and MIT9303. This may indicate that PtrA can function in the absence of PhoBR and possibly substitute for some form of P sensing and regulation in these strains.
When present, ptrA is found in a relatively conserved genome context in Synechococcus (Figure 5) where it is almost exclusively found downstream of pstS. Synechococcus sp. RCC307 is the only exception, where the ptrA gene is located in a unique context with no apparent linkage with co-located genes in other strains. Apart from oceanic cyanobacteria, PtrA homologues have not been identified in any other organism, although a potential, yet distant, candidate exists in Cyanobium sp. PCC 7001. PtrA is one of four distinct clusters of CRP regulators found in marine Prochlorococcus and Synechococcus. The similarities in gene context suggest that ptrA has been vertically inherited in Synechococcus, where the surrounding genes, especially the upstream pstS, have been largely conserved. Moreover, ptrA is not located in any predicted island in the Synechococcus genomes sequenced so far (Dufresne et al., 2008). In comparison, in Prochlorococcus the gene order in the vicinity of ptrA is not well conserved. However, the gene is often found in a genomic region that contains all of the P-uptake mechanism (for example, as found in Prochlorococcus MED4, NATL1A, NATL2A and MIT9312; Martiny et al., 2006). The implication here is that the P-uptake machinery, including the regulatory components phoBR and ptrA, resides on a genomic region that may have been horizontally acquired by Prochlorococcus ecotypes that inhabit P-deplete regions of some oceans (Martiny et al., 2006).

The genomic context of ptrA in representative Synechococcus and Prochlorococcus genomes.
As noted previously (Tetu et al., 2009) those few genes that are significantly influenced by PtrA during P stress in Synechococcus sp. WH8102 (SYNW0165, SYNW0196, SYNW0762 and SYNW2390) are represented in a minority of other marine cyanobacterial genomes, suggesting that they have been laterally acquired and their inclusion in the P regulon has been selected by environmental conditions (Palenik et al., 2003; Moore et al., 2005; Dufresne et al., 2008; Scanlan et al., 2009). The clustering of PtrA-regulated genes throughout the genome also has implications for the mechanisms of gene gain (Figure 3). First, these key genes are spread throughout the genome in WH8102 and do not occur in any recognizable island (Palenik et al., 2003; Dufresne et al., 2008), nor are they linked on the chromosome with their cognate regulator. This implies that each gene was independently acquired along with its respective PtrA-binding sequence. Conversely, there are several clusters of genes that appear to be co-regulated, including the swmB cluster (SYNW0952–SYNW0960) and a large region of glycosyltransferases, lipopolysaccharide and cell wall biogenesis-related genes (SYNW0424–SYNW0458) that are clearly genomic islands (Palenik et al., 2003; Dufresne et al., 2008). Whether they are directly regulated by PtrA is not clear but it is clear that the genes within these regions are co-regulated, in this case showing lower levels of expression in the ptrA knockout in comparison to wild-type.
Given that the genomic complement of P-acquisition and regulation genes (Scanlan et al., 2009) and the responses to P stress (Martiny et al., 2006; Tetu et al., 2009) vary quite considerably between marine picocyanobacteria, it will be interesting to determine the role of PtrA in other marine strains. Overall, it is intriguing that PtrA appears to regulate accessory genes in WH8102, providing a potentially novel example of how laterally acquired genes have been recruited into an existing regulon in an environmental isolate. This example shows the importance of regulatory networks in coordinating the expression of the P-stress response, despite relatively low regulatory capacity in this model oligotrophic cyanobacterium.
Accession codes
References
Aiba H, Mizuno T . (1994). A novel gene whose expression is regulated by the response-regulator, SphR, in response to phosphate limitation in Synechococcus species PCC7942. Mol Microbiol 13: 25–34.
Aiba H, Nagaya M, Mizuno T . (1993). Sensor and regulator proteins from the cyanobacterium Synechococcus species PCC7942 that belong to the bacterial signal-transduction protein families—implication in the adaptive response to phosphate limitation. Mol Microbiol 8: 81–91.
Ammerman JW, Hood RR, Case DA, Cotner JB . (2003). Phosphorus deficiency in the Atlantic: an emerging paradigm in oceanography. EOS 84: 165–170.
Bertilsson S, Berglund O, Karl DM, Chisholm SW . (2003). Elemental composition of marine Prochlorococcus and Synechococcus: implications for the ecological stoichiometry of the sea. Limnol Oceanogr 48: 1721–1731.
Bessey OA, Lowry OH, Brock MJ . (1946). A method for the rapid determination of alkaline phosphatase with five cubic millimeters of serum. J Biol Chem 164: 321–329.
Blindauer CA . (2008). Zinc-handling in cyanobacteria: an update. Chem Biodiv 5: 1990–2013.
Brahamsha B . (1996). A genetic manipulation system for oceanic cyanobacteria of the genus Synechococcus. Appl Environ Microbiol 62: 1747–1751.
Brazma A, Hingamp P, Quackenbush J, Sherlock G, Spellman P, Stoeckert C et al. (2001). Minimum information about a microarray experiment (MIAME) toward standards for microarray data. Nat Genet 29: 365–371.
Coleman JE . (2003). Structure and mechanism of alkaline phosphatase. Ann Rev Biophys Biomol Struct 21: 441–483.
Cotner JB, Ammerman JW, Peele ER, Bentzen E . (1997). Phosphorus-limited bacterioplankton growth in the Sargasso Sea. Aquat Microb Ecol 13: 141–149.
Dufresne A, Ostrowski M, Scanlan DJ, Garczarek L, Mazard S, Palenik BP et al. (2008). Unraveling the genomic mosaic of a ubiquitous genus of marine cyanobacteria. Genome Biol 9: R90.
Dufresne A, Salanoubat M, Partensky F, Artiguenave F, Axmann IM, Barbe V et al. (2003). Genome sequence of the cyanobacterium Prochlorococcus marinus SS120, a nearly minimal oxyphototrophic genome. Proc Natl Acad Sci USA 100: 10020–10025.
Elledge SJ, Zhou Z, Allen JB . (1992). Ribonucleotide reductase: regulation, regulation, regulation. Trends Biochem Sci 17: 119–123.
Heldal M, Scanlan DJ, Norland S, Thingstad F, Mann NH . (2003). Elemental composition of single cells of various strains of marine Prochlorococcus and Synechococcus using X-ray microanalysis. Limnol Oceanogr 48: 1732–1743.
Herrero A, Muro-Pastor AM, Flores E . (2001). Nitrogen control in cyanobacteria. J Bacteriol 183: 411–425.
Hirani TA, Suzuki I, Murata N, Hayashi H, Eaton-Rye JJ . (2001). Characterization of a two-component signal transduction system involved in the induction of alkaline phosphatase under phosphate-limiting conditions in Synechocystis sp. PCC 6803. Plant Mol Biol 45: 133–144.
Itaya K, Ui M . (1966). A new micromethod for the colorimetric determination of inorganic phosphate. Clinica Chimica Acta 14: 361–366.
Karl DM, Tien G . (1997). Temporal variability in dissolved phosphorus concentrations in the subtropical North Pacific Ocean. Mar Chem 56: 77–96.
Korner H, Sofia HJ, Zumft WG . (2003). Phylogeny of the bacterial superfamily of Crp-Fnr transcription regulators: exploiting the metabolic spectrum by controlling alternative gene programs. FEMS Microbiol Rev 27: 559–592.
Lindell D, Padan E, Post AF . (1998). Regulation of ntcA expression and nitrite uptake in the marine Synechococcus sp. strain WH7803. J Bacteriol 180: 1878–1886.
Lohan MC, Statham PJ, Crawford DW . (2002). Total dissolved zinc in the upper water column of the subarctic North East Pacific. Deep-Sea Res Part II 49: 5793–5808.
Luque I, Flores E, Herrero A . (1994). Molecular mechanism for the operation of nitrogen control in cyanobacteria. EMBO J 13: 2862–2869.
Mackey KRM, Paytan A, Grossman AR, Bailey S . (2008). A photosynthetic strategy for coping in a high-light, low-nutrient environment. Limnol Oceanogr 53: 900–913.
Martiny AC, Coleman ML, Chisholm SW . (2006). Phosphate acquisition genes in Prochlorococcus ecotypes: evidence for genome-wide adaptation. Proc Natl Acad Sci USA 103: 12552–12557.
McCarren J, Brahamsha B . (2005). Transposon mutagenesis in a marine Synechococcus strain: isolation of swimming motility mutants. J Bacteriol 187: 4457–4462.
McCarren J, Brahamsha B . (2007). SwmB, a 1.12-megadalton protein that is required for nonflagellar swimming motility in Synechococcus. J Bacteriol 189: 1158–1162.
Moore LR, Ostrowski M, Scanlan DJ, Feren K, Sweetsir T . (2005). Ecotypic variation in phosphorus acquisition mechanisms within marine picocyanobacteria. Aquat Microb Ecol 39: 257–269.
Morel FMM, Rueter JG, Anderson DM, Guillard RRL . (1979). Aquil—chemically defined phytoplankton culture-medium for trace-metal studies. J Phycol 15: 135–141.
Palenik B, Brahamsha B, Larimer FW, Land M, Hauser L, Chain P et al. (2003). The genome of a motile marine Synechococcus. Nature 424: 1037–1042.
Palenik B, Ren QH, Dupont CL, Myers GS, Heidelberg JF, Badger JH et al. (2006). Genome sequence of Synechococcus CC9311: insights into adaptation to a coastal environment. Proc Natl Acad Sci USA 103: 13555–13559.
Partensky F, Hess WR, Vaulot D . (1999). Prochlorococcus, a marine photosynthetic prokaryote of global significance. Microbiol Mol Biol Rev 63: 106–127.
Salgado H, Gama-Castro S, Martinez-Antonio A, Diaz-Peredo E, Sanchez-Solano F, Peralta-Gil M et al. (2004). RegulonDB (version 4.0): transcriptional regulation, operon organization and growth conditions in Escherichia coli K-12. Nucleic Acids Res 32: D303–D306.
Scanlan DJ, Bourne JA, Mann NH . (1997a). A putative transcriptional activator of the Crp/Fnr family from the marine cyanobacterium Synechococcus sp WH7803. J Appl Phycol 8: 565–567.
Scanlan DJ, Mann NH, Carr NG . (1993). The response of the picoplanktonic marine cyanobacterium Synechococcus species WH7803 to phosphate starvation involves a protein homologous to the periplasmic phosphate-binding protein of Escherichia coli. Mol Microbiol 10: 181–191.
Scanlan DJ, Silman NJ, Donald KM, Wilson WH, Carr NG, Joint I et al. (1997b). An immunological approach to detect phosphate stress in populations and single cells of photosynthetic picoplankton. Appl Environ Microbiol 63: 2411–2420.
Scanlan DJ, Ostrowski M, Mazard S, Dufresne A, Garczarek L, Hess WR et al. (2009). Ecological genomics of marine picocyanobacteria. Microbiol Mol Biol Rev 73: 249–299.
Snyder DS, Brahamsha B, Azadi P, Palenik B . (2009). Structure of compositionally simple lipopolysaccharide from marine Synechococcus. J Bacteriol 191: 5499–5509.
Silberbach M, Hüser A, Kalinowski J, Pühler A, Walter B, Kramer R et al. (2005). DNA microarray analysis of the nitrogen starvation response of Corynebacterium glutamicum. J Biotech 119: 357–367.
Su ZC, Dam P, Chen X, Olman V, Jiang T, Palenik B et al. (2003). Computational inference of regulatory pathways in microbes. An application to phosphorus assimilation pathways in Synechococcus WH8102. Genome Inform 14: 3–13.
Su ZC, Olman V, Mao FL, Xu Y . (2005). Comparative genomics analysis of NtcA regulons in cyanobacteria: regulation of nitrogen assimilation and its coupling to photosynthesis. Nucleic Acids Res 33: 5156–5171.
Su ZC, Olman V, Xu Y . (2007). Computational prediction of Pho regulons in cyanobacteria. BMC Genomics 8: 156.
Suzuki S, Ferjani A, Suzuki I, Murata N . (2004). The SphS–SphR two component system is the exclusive sensor for the induction of gene expression in response to phosphate limitation in Synechocystis. J Biol Chem 279: 13234–13240.
Tai V, Paulsen IT, Phillippy K, Johnson DA, Palenik B . (2009). Whole-genome microarray analyses of Synechococcus–Vibrio interactions. Environ Microbiol 11: 2698–2709.
Tetu SG, Brahamsha B, Johnson DA, Tai V, Phillippy K, Palenik B et al. (2009). Microarray analysis of phosphate regulation in the marine cyanobacterium Synechococcus sp WH8102. ISME J 3: 835–849.
Thingstad TF, Krom MD, Mantoura RFC, Flaten GAF, Groom S, Herut B et al. (2005). Nature of phosphorus limitation in the ultraoligotrophic eastern Mediterranean. Science 309: 1068–1071.
Tusher VG, Tibshirani R, Chu G . (2001). Significance Analysis of Microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 98: 5116–5121.
Van Mooy BAS, Rocap G, Fredricks HF, Evans CT, Devol AH . (2006). Sulfolipids dramatically decrease phosphorus demand by picocyanobacteria in oligotrophic marine environments. Proc Natl Acad Sci USA 103: 8607–8612.
Venter JC, Remington K, Heidelberg JF, Halpern AL, Rusch D, Eisen JA et al. (2004). Environmental genome shotgun sequencing of the Sargasso Sea. Science 304: 66–74.
Waterbury JB, Willey JM . (1988). Isolation and growth of marine planktonic cyanobacteria. Methods Enzymol 167: 100–105.
Zubkov MV, Mary I, Woodward EMS, Warwick PE, Fuchs BM, Scanlan DJ et al. (2007). Microbial control of phosphate in the nutrient-depleted North Atlantic subtropical gyre. Environ Microbiol 9: 2079–2089.
Zwirglmaier K, Jardillier L, Ostrowski M, Mazard S, Garczarek L, Vaulot D et al. (2008). Global phylogeography of marine Synechococcus and Prochlorococcus reveals a distinct partitioning of lineages among oceanic biomes. Environ Microbiol 10: 147–161.
Acknowledgements
We thank Bianca Brahamsha for assistance and guidance in preparing the ptrA gene knockout and for supplying axenic cultures of Synechococcus sp. WH8102. MO and SM were supported by the EU FP5 Program Margenes (QLRT-2001-0226), the FP6 EU Marine Genomics Network and NERC Grants NE/C0005361/1, NE/F004249/1 and NE/D003385/1 to DJS.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website
Rights and permissions
About this article
Cite this article
Ostrowski, M., Mazard, S., Tetu, S. et al. PtrA is required for coordinate regulation of gene expression during phosphate stress in a marine Synechococcus. ISME J 4, 908–921 (2010). https://doi.org/10.1038/ismej.2010.24
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2010.24
