
Abstract
Little is known about the archaeal diversity of fermented seafood; most of the earlier studies of fermented food have focused on lactic acid bacteria (LAB) in the fermentation process. In this study, the archaeal and bacterial diversity in seven kinds of fermented seafood were culture-independently examined using barcoded pyrosequencing and PCR–denaturing gradient gel electrophoresis (DGGE) methods. The multiplex barcoded pyrosequencing was performed in a single run, with multiple samples tagged uniquely by multiplex identifiers, using different primers for Archaea or Bacteria. Because PCR–DGGE analysis is a conventional molecular ecological approach, this analysis was also performed on the same samples and the results were compared with the results of the barcoded pyrosequencing analysis. A total of 13 372 sequences were retrieved from 15 898 pyrosequencing reads and were analyzed to evaluate the diversity of the archaeal and bacterial populations in seafood. The most predominant types of archaea and bacteria identified in the samples included extremely halophilic archaea related to the family Halobacteriaceae; various uncultured mesophilic Crenarchaeota, including Crenarchaeota Group I.1 (CG I.1a and CG I.1b), Marine Benthic Group B (MBG-B), and Miscellaneous Crenarchaeotic Group (MCG); and LAB affiliated with genus Lactobacillus and Weissella. Interestingly, numerous uncultured mesophilic Crenarchaeota groups were as ubiquitous in the fermented seafood as in terrestrial and aquatic niches; the existence of these Crenarchaeota groups has not been reported in any fermented food. These results indicate that the archaeal populations in the fermented seafood analyzed are diverse and include the halophilic and mesophilic groups, and that barcoded pyrosequencing is a promising and cost-effective method for analyzing microbial diversity compared with conventional approaches.
Similar content being viewed by others

Introduction
Salted and fermented seafood products (‘jeotgal’ in Korean) are widely produced and consumed in Asia (Lee et al., 1993). Jeotgal is made of fish or shellfish, such as shrimp, oysters, fish roes, and fish tripes with lots of salt, followed by a fermentation period so that the jeotgal develops its own distinct taste through the endogenous enzyme activities in the fish or microbes that degrade fish proteins (Cha and Lee, 1985; Lee, 1994). Various kinds of jeotgal were made and consumed; specifically, the 145 different kinds of fermented seafood were classified on the basis of the raw materials available in Korea (Suh and Yoon, 1987). Fermented seafood is often used as a side dish as well as an ingredient to improve the taste of other foods, such as kimchi, a famous fermented vegetable product of Korea. To date, researches aimed at isolating novel strains in the Asian fermented seafood have identified more than 15 novel species in archaea and bacteria (http://www.bacterio.net) (Euzeby, 1997). However, little is known about the microbial diversity and their function in the fermented seafood products, compared with those in fermented dairy products, which have been widely investigated over the past century (Lee et al., 1993; Lee, 1997) even though the use of fermented seafood products, including liquid fish sauce and salt-fermented seafood, is currently widespread in Asia. Most of the existing studies with fermented foods emphasized the lactic acid bacteria (LAB) in the fermented food ecosystem because of their unique physiological and metabolic characteristics (Caplice and Fitzgerald, 1999). Bacterial diversity was partially exploited in fermented seafoods, such as fermented fish sauces or fish products, through culture-based approaches (Sands and Crisan, 1974; Crisan and Sands, 1975; Lee, 1997; Paludan-Muller et al., 1999).
Although the composition and dynamics of the microbial populations in fermented foods were shown through culture-dependent methods, recent molecular ecological investigation showed that the microbial communities of most foods are more diverse and complex than originally thought (Fleet, 1999). Thus, the development of more sensitive, quantitative, and culture-independent tools are needed to explore the microbial ecology of fermented foods (Cheigh and Park, 1994; Giraffa and Neviani, 2001) with their theoretical accuracy detouring ‘the great plate count anomaly’ (Staley and Konopka, 1985). Although molecular methods such as denaturing gradient gel electrophoresis (DGGE), temperature gradient gel electrophoresis, fluorescence in situ hybridization, direct epifluorescence technique, terminal restriction fragment length polymorphism, length heterogeneity–PCR, and microarray (Giraffa, 2004) have been widely applied to study food fermentation and given us a lot of new ecological insights without cultivation bias (ben Omar and Ampe, 2000; Ampe et al., 2001; Cocolin et al., 2001; Randazzo et al., 2002; van Beek and Priest, 2002; Lafarge et al., 2004); these approaches do not allow one to comprehensively analyze all microbial populations within multiple samples (Ercolini, 2004). Thus, new additional in-depth analysis method for quantitatively analyzing the involvement of microbial populations in the process of food fermentation is warranted.
Although most of studies have focused on culture-dependent and culture-independent investigation of bacterial diversity, there is still a scarcity of information regarding the archaeal diversity in fermented seafoods. On the other hand, archaeal diversity was investigated in the various marine and terrestrial environments. Recent studies using cultivation-independent methods have uncovered abundant mesophilic crenarchaeal lineages in marine environments that have not yet been cultivated (Teske and Sorensen, 2008). The crenarchaeal lineages currently consist of several groups: the Marine Group I (MG-I), Marine Benthic Group A, B, D (MBG-A, B, D), Miscellaneous Crenarchaeotic Group (MCG), South African Goldmine Euryarchaeotal Group (SAGMEG), Ancient Archaeal Group (AAG), and Marine Hydrothermal Vent Group (MHVG). Some members of MG-I that were affiliated with the Crenarchaeota Group I.1 (CG I.1) (Ochsenreiter et al., 2003) were known as aerobic, non-extremophilic, ammonia-oxidizing, and autotrophic archaea (Francis et al., 2005; Könneke et al., 2005; Hallam et al., 2006a, 2006b). The ammonia-oxidizing archaea that have ammonia monooxygenase α-subunit (amoA) genes are able to oxidize ammonia into nitrite and are considered to be important to global carbon and nitrogen cycles (Francis et al., 2005; Könneke et al., 2005). Many metagenomic studies have shown that ammonia-oxidizing archaea are abundant and ubiquitous not only in terrestrial ecosystems, such as various soils (Leininger et al., 2006), alkaline sandy loam (Shen et al., 2008), and rhizosphere (Herrmann et al., 2008), but also in aquatic ecosystems, such as marine water columns and sediments (Francis et al., 2005), mucus of corals (Siboni et al., 2008), deep marine sediments (Inagaki et al., 2006), and oceans (Wuchter et al., 2006; Lam et al., 2007; Mincer et al., 2007).
The parallel high throughput pyrosequencing technique, a next-generation DNA sequencing platform based on the detection of pyrophosphate released during nucleotide incorporation, was developed by Margulies et al., (2005) and provides a new way of dramatically reducing the costs associated with sequencing. The pyrosequencing method was applied to microbial ecology, and much more in-depth studies of microbial diversity have been carried out in various ecosystems such as deep mines (Edwards et al., 2006), soil (Roesch et al., 2007), deep marine biospheres (Huber et al., 2007), chronic wounds (Dowd et al., 2008a), tidal flats (Kim et al., 2008), and human oral microflora (Keijser et al., 2008). Although pyrosequencing produces a very large number of reads from a single run, it is not appropriate to analyze multiple samples at a time. Recently, to overcome the limitation in the number of samples that can be sequenced in parallel, sample-specific key sequences called ‘barcode’ or ‘tag’ consisting of smaller sequences of two or four nucleotides (Binladen et al., 2007; Hoffmann et al., 2007; Kasschau et al., 2007) were adapted for use with the pyrosequencing platform. The sample-specific barcode system using 8 or 10 nucleotides was subsequently developed for pyrosequencing analyses so that more samples could be encoded with greater accuracy (Parameswaran et al., 2007; Dowd et al., 2008b; Hamady et al., 2008). The multiplex barcoded pyrosequencing analysis enables us to analyze an increased number of samples at a time, to reduce the cost per sample, and to perform in-depth analyses for studies of comparative microbial ecology (Binladen et al., 2007; Hoffmann et al., 2007; Kasschau et al., 2007; Parameswaran et al., 2007; Dowd et al., 2008b; Hamady et al., 2008). Along with the modified pyrosequencing approaches, efficient and automated bioinformatics pipeline were also used to achieve consistent, rapid, and accurate taxonomic assignments from the 16S rRNA sequence reads (Liu et al., 2008).
As far as we know, no studies have been carried out with the in-depth sequencing approach to evaluate bacterial, and especially archaeal, populations of food ecosystems associated with fermented products. Therefore, we focused our study on the microbial communities of the Archaea and Bacteria domains in fermented seafood products, which is a food ecosystem that is understudied by microbial ecologists. The archaeal and bacterial diversities in seven representative (best selling) kinds of fermented seafood were evaluated with a culture-independent approach. Results of the small subunit ribosomal rRNA (16S rRNA) gene sequencing analysis using multiplex barcoded pyrosequencing (Liu et al., 2008) with uniquely tagged multiple amplicons in a single run were compared with the results obtained with the widely used PCR–DGGE fingerprinting method. The multiplex barcoded pyrosequencing technique enabled us to perform a comparative microbial ecological study by thoroughly analyzing several samples with reduced costs per sample.
Materials and methods
Sampling and DNA extraction
The seven kinds of seafood samples were purchased from the distributors of commercially available brands in Korea. These foods were made of the following raw materials: tiny shrimp (designated J1), shellfish (J2), cuttlefish (J3), oysters (J4), roe of pollack (J5), tripe of pollack (J6), or crab (J7). The bulk community DNAs were extracted separately from 1 ml volume of single samples of each seafood type, obtained just after they were opened, using the bead-beating method (Yeates et al., 1998). All DNA samples were purified and quantified as described earlier (Bae et al., 2005).
PCR amplifications for pyrosequencing
The bulk community DNA sequences were amplified individually with universal primer pairs for the hypervariable V3 region of the 16S rRNA gene (Ashelford et al., 2005). For Archaea, nested PCR amplification was performed using the arch20f-958r primer set (DeLong, 1992; DeLong et al., 1999) for the first round of amplification, followed by the arch344f (Casamayor et al., 2002)-519r (Ovreas et al., 1997) primer set for the second round of amplification because some samples were not successfully amplified with the direct archaeal-specific primer set. For Bacteria, direct PCR amplification was performed using the 341f (Watanabe et al., 2001)-518r (Muyzer et al., 1993) primer set. All PCR amplifications were performed using the PCR mix (Maxime PCR PreMix Kit, iNtRON Biotechnology, Korea), supplemented with 1 μl (50 ng μl−1) template DNA and 20 pmol of each primer, using a PTC-220 DNA Engine Dyad MJ Research thermalcycler (PharmaTech, Seoul, Korea). The PCR conditions were as follows: 94 °C for 1 min; 30 cycles of denaturation (94 °C; 1 min), annealing (60 °C; 0.5 min), and extension (72 °C; 0.5 min); followed by the final elongation (72 °C; 10 min). The only exceptions were that the initial rounds of annealing (60 °C; 1 min) and extension (72 °C; 1.5 min) during the amplification process were modified for the archaeal sequences. After the DNA was quantified using a spectrophotometer (Nanodrop Technologies, Rockland, DE, USA), the archaeal and bacterial DNAs from the same sample were mixed in equivalent proportions.
Pyrosequencing
To create a single-stranded DNA library for the Genome Sequencer FLX system (Roche, Mannheim, Germany), Multiplex Identifiers (MIDs; Roche) were used to label the end fragments of the purified PCR product DNA from the seven samples, according to the manufacturer’s instructions. Specifically, the end fragments were blunted and tagged on both ends with one of seven ligation adaptors that contained a unique 10-bp sequence (Table 1) and a short 4-nucleotide sequence (TCAG) called a sequencing key, which were recognized by the system software and the priming sequences. The quality of the single-stranded DNA libraries generated using GS FLX Standard DNA Library Preparation Kit (Roche) was evaluated using DNA 1000 LabChip software with a Bioanalyzer 2100 (Agilent, Palo Alto, CA, USA). Subsequently, all seven single-stranded DNA libraries with different sample-specific adaptors were pooled in equimolar amounts. The uniquely tagged, pooled DNA samples were immobilized onto DNA capture beads, amplified through emulsion-based clonal amplification (emPCR), and sequenced together in a single region (1 of 16 lanes) of a PicoTiterPlate device on a Genome Sequencer FLX system that has the read length of ∼250 bases, according to the manufacturer's instructions (Roche).
Pyrosequencing data analysis
All sequences were compared against a non-redundant GenBank database constructed in the local BLAST database with BLASTN search (Altschul et al., 1990). Sequences shorter than 100 bp were excluded. The sequences with a greater than 75% identity with the best hit from the BLASTN search and a match length greater than 90% of the query length were selected for further analysis to remove the unspecific or non-rRNA gene sequences. The selected sequences were assigned as Archaea or Bacteria based on the taxonomic annotation of the sequences for the nearest neighbor from the BLASTN search. Directions of the strands were adjusted properly according to the BLASTN results. The sequences were aligned by the NAST program (DeSantis et al., 2006) in the Greengenes (http://greengenes.lbl.gov). The primer sequences were excluded after alignment. The DNA distance matrices were calculated using the DNADIST program in PHYLIP (version 3.68). The matrices were used to define operational taxonomic units with 1% dissimilarity for determination of the coverage percentage by Good’s method (Good, 1953), abundance-based coverage estimator (ACE), bias-corrected Chao1 richness estimator, Jackknife estimator of species richness, and the Shannon–Weaver and Simpson diversity indices by the DOTUR program (Schloss and Handelsman, 2005). Rarefaction analysis was also performed by DOTUR program (Schloss and Handelsman, 2005). Good's coverage percentage was calculated as [1−(n/N)] × 100, where n represents the number of single-member phylotypes and N represents the number of sequences. To determine the value of dissimilarity, we performed in silico modeling using the pre-aligned near full-length 16S rDNA sequence set from the Greengenes database. The 1000 sequences in front of the pre-aligned set were selected as model sequences and the model sequences were trimmed to make two sets of trimmed model sequences simulating the pyrosequencing reads with primers and without primers. The three sets of sequences were analyzed through same procedure as described above. The value of dissimilarity for the trimmed model sequences showing similar results with the 3% dissimilarity for full-length sequences was determined as a criterion to calculate the richness estimators and diversity indices of the jeotgal samples. Clustering analyses of seafood samples and the principal coordinates analysis (PCA) were performed using the UniFrac service (Lozupone et al., 2006). Unique sequences (showing 100% similarity with each other) were selected from the pyrosequencing reads aligned with the NAST program. The neighbor-joining tree was constructed using the MEGA 3.01 (Kumar et al., 2004) program based on the Jukes–Cantor model and used for the UniFrac analysis. The Haloterrigena sp. (AM285297) and Lactobacillus curvatus (EU855223) strains were used as outgroups for Archaea and Bacteria, respectively. The online ribosomal database project classifier (Cole et al., 2007) was used for the taxonomic assignments.
PCR–DGGE and phylogenetic analysis
To amplify the archaeal 16S rRNA genes for the DGGE analysis, PCR amplifications were performed as described above, but with the following exceptions: archaeal sequences were amplified with the archGC340f-519r primer set (Ovreas et al., 1997) for the second round of amplification as described earlier (Ferris et al., 1996) and bacterial sequences were amplified with the GC338f-518r primer set (Muyzer et al., 1993) as described earlier (Henckel et al., 1999). The DGGE analysis and the sequence analysis of the excised DGGE bands were carried out as described earlier (Chang et al., 2008; Nam et al., 2008). The phylogenetic analysis of the DGGE band sequences was performed as described earlier (Roh et al., 2008).
Results
Sequence analysis by pyrosequencing
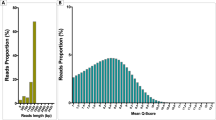
A total of 15 898 reads were obtained from a single lane of a 16-lane PicoTiterPlate on a Genome Sequencer FLX system. Of these, 13 372 sequences (84%) were identified as archaeal or bacterial sequences. Specifically, 12 035 or 1337 sequences from the 13 372 reads were classified as Archaea or Bacteria, respectively. Average read lengths were 147 or 182 bases for Archaea or Bacteria, respectively. The smaller percentage of bacterial sequences (11%) compared with archaeal sequences was not expected because the same amount of archaeal and bacterial DNA was mixed from each sample before preparing the single-stranded DNA library for pyrosequencing. It can be assumed that the biased ratio of the two domains was not only because of an inefficiency in the emPCR technique using different amplicon sizes for Archaea and Bacteria (147 and 182 bp, respectively), but also because of the impurities or unknown compounds disturbing the accurate measurement of DNA amount. For some of the samples (J3–J7), 9.2–14.6% of the sequences were identified as eukaryotic sequences composed mainly of yeast (Pichia fabianii and Candida tropicalis) and some chloroplasts. Detection of chloroplasts was expected because plant materials, such as hot pepper or garlic, are used as ingredients in the fermented seafood products and the chloroplasts contain 16S rRNA genes. Coamplification of non-target eukaryotic 18S rDNA by commonly used universal 16S rDNA-targeting PCR primers has been reported and is probably because of the low specificity of the primer sets used in earlier studies (Ampe et al., 1999; Ampe and Miambi, 2000; Huys et al., 2008; Rivas et al., 2004).
Microflora in food samples observed with pyrosequencing
The phylogenetic classification of sequences from the seafood samples is summarized in Figure 1. Except for sample J3 (cuttlefish jeotgal; 0.4%), the archaeal sequences primarily belonged to the family Halobacteriaceae of the phylum Euryarchaeota (68.6–98.4%). Fermented seafood samples with high NaCl concentration (J1, J2, J4, J7; more than 10% of NaCl) had a larger number of members from the family Halobacteriaceae, and various members in the class Halobacteria were detected in each fermented seafood sample. Genera Halorubum and Halalkalicoccus occurred frequently in most samples. The abundance of genus Halorubum in the archaeal reads was 42.5%, 73.7%, 63.3%, and 19.9% in samples J1, J4, J5, and J7, respectively, and genus Halalkalicoccus comprised 42.0%, 61.2%, 74.8%, and 17.0% of samples J1, J2, J6, and J7, respectively. Genus Halococcus was a minor component of samples J5 (3.3%) and J6 (0.2%). Genera Natrinema (11.8%) and Halobacterium (0.9%) were found only in sample J7, whereas genus Halobiforma (0.1%) was detected in sample J4 only. A significant number of pyrosequencing reads belonged to the unclassified Halobacteriaceae (14.0%, 14.4%, 18.2%, and 34.4% in samples J1, J2, J6, and J7, respectively). On the other hand, archaeal sequences classified as uncultured Crenarchaeota were found in all samples: 1.3%, 24.3%, 99.6%, 22.8%, 31.4%, 2.3%, and 16.0% for samples J1, J2, J3, J4, J5, J6, and J7, respectively. Interestingly, sample J3 (cuttlefish) had the greatest percentage of uncultured Crenarchaeota (99.6%) and the smallest percentage of Halobacteriaceae (0.4%) compared to the other samples.

Phylogenetic classification for the pyrosequencing analysis obtained from a ribosomal database project classifier analyses. The fermented seafood sample in each lane is as follows: J1, shrimp; J2, shellfish; J3, cuttlefish; J4, oyster; J5, roe of pollack; J6, tripe of pollack; and J7, crab. (a) Archaea, (b) Bacteria.
Except for sample J2, most of the pyrosequencing reads (71.1–98.7%) of the bacterial amplicons belonged to the order Lactobacillales, which is related to the LAB. Fermented seafood samples with a low pH (J3, J4, J6, and J7; pH 4.5 or less) contained a larger number of members from the order Lactobacillales. However, only genera Weissella and Lactobacillus, which are LAB, were major components of the seafood samples. The ratio of the two genera varied among the samples. Specifically, samples J1, J3, J5, and J6 contained a similar proportion of each genus, whereas one genus was more predominant than the other genus in samples J4 and J7. Sample J2 had a significantly different bacterial population than the other samples; this sample contained 98.8% Gammaproteobacteria, which consisted of the order Vibrionales (90.1%), which contained the genus Salinivibrio (89.5%), and the order Oceanospirillales (7.6%), which contained the family Halomonadaceae (5.8%). Pyrosequencing reads belonging to Gammaproteobacteria were also found in samples J3 and J4, with the percentage of bacterial reads as follows: 15.9% (family Vibrionaceae was a major component) and 6.7% (the family Pseudomonadaceae), respectively. Only sample J4 contained the orders Rhizobiales (11.1%) and Rhodobacterales (2.2%) in Alphaproteobacteria.
To more clearly confirm that the results of our sequencing study was sufficient for ecosystem analysis, the number of operational taxonomic units and the statistical estimates of species richness were determined (see Table 2). From the in silico modeling, 1% dissimilarity of trimmed model sequences without primers showed most similar results corresponding to 3% dissimilarity of full-length sequences (Supplementary Table 1SM, 2SM, and 3SM). The number and estimated number of operational taxonomic units were less than the values obtained from other ecosystems such as soil (Schloss and Handelsman, 2006) and tidal flats (Kim et al., 2008). The limited microbial diversity of the seafood samples may be because of the processes of fermentation and storage. Comparisons of the cluster analyses with the phylogenetic tree and the PCA performed using the UniFrac service (Lozupone et al., 2006) showed similar results (Figures 2 and 3). Samples J3 (Archaea) and J2 (Bacteria) occupied the most separated positions with the clustering analysis and PCA, as expected from their unique taxonomic classifications. Samples J1, J6, and J7 were clustered each other in both cases of Archaea and Bacteria, whereas samples J2 and J5 showed quite different clustering profiles depending on Archaea and Bacteria. In-depth comparison of seafood samples requires additional analysis of the microbial diversity, which depends on the various kinds of seafood samples.

Clustering analysis for the samples classified as Archaea and Bacteria using the UniFrac service after pyrosequencing. The scale bar indicates the distance between clusters in UniFrac units. (a) Archaea, (b) Bacteria.

PCA of the samples obtained by pyrosequencing, and classified as (a) Archaea and (b) Bacteria using the UniFrac service.
PCR–DGGE and phylogenetic analysis
As the DGGE analysis is now the most widely applied for comparative analysis of parallel samples, results of the barcoded pyrosequencing analysis were compared with the findings with DGGE, for which the bulk community DNAs extracted from the seafood samples were PCR amplified and analyzed and 49 of the 16S rRNA gene sequences were obtained after the bands of interest were excised from the DGGE lanes, reamplified, and sequenced. Archaeal and bacterial DNA DGGE profiles were significantly different from one another and varied with the seafood samples, as shown in Figure 4. However, based on the DGGE analysis, we could not quantitatively compare the bacterial and archaeal populations in the fermented seafood. Only the sequencing results for the 23 archaeal and 26 bacterial DGGE bands (labeled with a number followed by A or B for Archaea or Bacteria, respectively) were summarized in Tables 3 and 4; the results were compared with sequences found in the GenBank and REF 16S databases (Chun et al., 2007). Among the retrieved archaeal sequences (Table 3), 14 of the 23 archaeal DGGE bands (61%) were related to extremely halophilic archaea belonging to the family Halobacteriaceae in the phylum Euryarchaeota; 9 bands (39%) were affiliated with the uncultured Crenarchaeota clones that had ⩽83.3% similarity of their 16S rRNA gene sequence with valid species of the hyperthermophilic Crenarchaeota that belong to genus Sulfolobus, Thermocladium, or Pyrobaculum in class Thermoprotei. Table 4 shows that 13 of the 26 bacterial bands (50%) corresponded to the LAB group affiliated with genus Lactobacillus or Weissella in phylum Firmicutes; 11 bands (42%) belonged to the class Gammaproteobacteria, such as genus Salinivibrio, Vibrio, Cobetia, or Psychrobacter; and 2 bands (8%) were affiliated with the uncultured environmental clones within the domain of Bacteria.

Comparison of pyrosequencing with DGGE analysis
The phylotypes detected in the seven samples with the two molecular analyses, pyrosequencing and PCR–DGGE, generally matched each other with respect to non-detection of genera Halorubrum, Lactobacillus, and Weissella and detection of genera Halalkalicoccus and Salinivibrio in sample J2; and detection of LAB or unclassified bacterial groups in all samples, except sample J2 or sample J4, respectively. However, the archaeal and bacterial communities determined with the pyrosequencing analysis were more diverse than those communities determined with the PCR–DGGE analysis, as there were some phylotypes revealed with the pyrosequencing analysis but not with PCR–DGGE. For example, the sequence reads belonging to uncultured Crenarchaeota, unclassified Halobacteriaceae, several Gammaproteobacteria groups in sample J2 and the minor bacterial groups (excluding LAB) in samples J1, J5, J6, and J7 were shown only with the pyrosequencing analysis. One disadvantage of DGGE fingerprinting is that there is a limit for detecting bands in the DGGE profiles of complex communities (Ercolini, 2004), which may not show minor archaeal and bacterial populations in samples. However, pyrosequencing provides an in-depth analysis of the 16S rRNA gene sequences, which can compensate for the disadvantage with the PCR–DGGE method in detecting minor populations in seafood samples.
Discussion
Application of barcoded pyrosequencing to evaluate food microbial diversity
As the costs are much lower than for conventional methods, such as PCR-cloning, DGGE, so on, and the comprehensive analysis of the microbial diversity of foods is needed, barcoded pyrosequencing could be applied to routine analysis of the microbial diversity of various foods and replace conventional cloning analyses that are conducted to acquire hundreds of sequences. Although the pyrosequencing read lengths of the sequences were significantly short compared with the sequences obtained with traditional Sanger sequencing methods (600–800 bp), these short sequences were long enough to enable classification of the bacterial genera. A systematic evaluation of the accuracy of the classifications, according to the sequence lengths, was carried out earlier and showed that the average read lengths of 80–100 bases provided not only excellent coverage, but also excellent recovery for classification at the genus level (Liu et al., 2008). In addition, 59 base pairs in highly variable region V6 of the 16S rRNA gene sequences were sufficient for phylogenetic affiliation (Andersson et al., 2008). Therefore, the average lengths of the pyrosequencing reads in this study were also thought to be sufficient for assigning the taxa, provided that a 1-bp difference in the 16S rRNA gene sequences differentiated the reads by 0.55% for Bacteria and 0.68% for Archaea. This resolution was at the subspecies level because archaeal and bacterial species are demarcated by Stackebrandt and Goebel's 97% rule (different species share more than 3% of their 16S rRNA identity) (Stackebrandt and Goebel, 1994).
Determination of richness estimators and diversity indices
From the results of in silico modeling, 1% dissimilarity of short (100–140 bp) sequences was corresponding to 3% similarity to full-length sequences, in which diversity indices showed similar values to each other (Supplementary Table 1SM, 2SM, and 3SM). As dissimilarity criterion was changed according to the length of pyrosequencing reads (our data) and the region within the 16S rDNA gene (Andersson et al., 2008), the dissimilarity criterion must be determined before calculation of diversity indices.
Most rarefaction curves of the samples were not saturated. The curves were not saturated even if more 3000 sequences were retrieved (J3 and J6; Supplementary Figure 1SM). Roesch et al. (2007) reported that bacterial rarefaction curves were not saturated up to 5000–10 000 sequences in some soil samples. It needs more sequencing effort to obtain the saturation of rarefaction curves. We performed re-sampling analyses adjusting the sample sizes as same as that of the smallest sample to compare the diversity indices in same condition as described in the next section.
Comparison of diversities of Archaea and Bacteria in seven kinds of fermented seafood
Generally, most of archaeal diversities have been reported to be lower than bacterial diversities. In only 5 libraries among 173 libraries surveyed from published reports, archaeal diversity is higher than bacterial diversity in the same environment (Aller and Kemp, 2008). In the case of this study, archaeal diversity was always higher than bacterial diversity in the same food sample based on the diversity indices and richness estimators except for Good and Simpson index (Table 2). Chao1, ACE, Jackknife richness estimator, and Shannon indices of archaea were always higher than those of bacteria in the same samples. As richness estimators such as Chao1 and ACE were correlated positively with the number of sequences analyzed (Schloss and Handelsman, 2005), we performed a re-sampling analysis. We selected same number of sequences (as same as the number of the bacterial sample J4 that have the smallest number of sequences) randomly 100 times per each jeotgal sample and calculated the diversity indices of the 100 sub-samples. Average and standard deviation values of the estimators and indices according to jeotgal samples were calculated. Although the richness estimators and Shannon index became lower than compared with original samples with large number of sequences, the values of archaeal samples were still higher than those of bacterial samples (Supplementary Figure 2SM).
It is not surprising that the dominant bacterial populations in the seafood samples contained LAB because the food fermentation process is largely dependent on LAB (Caplice and Fitzgerald, 1999). Also expected was the finding that the Archaea in the seafood samples were characterized by extremely halophilic archaea that are phylogenetically affiliated to the family Halobacteriaceae, which inhabit environments saturated or nearly saturated with salt. As seafood samples have high salt concentrations, several extremely halophilic Archaea (GenBank accession number EF077631-EF077641) had already been isolated and two novel species, Halalkalicoccus jeotgali and Natronococcus jeotgali, were validated in earlier studies through culture-dependent methods with another seafood sample (Roh et al., 2007a, 2007b).
Samples J2 and J3 had significantly different bacterial and archaeal communities, respectively. Although raw materials of food samples analyzed were quite different: for example, shellfish for sample 2 and cuttlefish for sample J3, there was nothing peculiar on manufacturing process and fermentation conditions. The significant differences of archaeal and bacterial communities can be considered owing only to the raw materials of food samples.
Unexpected occurrence of uncultivated mesophilic Crenarchaeota groups from fermented seafood
To clearly analyze the archaeal diversity in food samples, phylogenetic trees were constructed with the 16S rRNA gene sequences retrieved from the archaeal DGGE bands and representative pyrosequencing reads selected from each group of operational taxonomic units that were clustered within a DNA distance of 0.03 by the DOTUR program (Figures 5, 6, and 7). It is interesting to note that there are numerous phylogenetic lineages of uncultured crenarchaea in fermented seafoods. The most abundant crenarchaeal sequences obtained from the pyrosequencing and DGGE approaches fell into the clades of the uncultivated mesophilic Crenarchaeota lineages of CG I.1a, CG I.1b, MBG-B, and MCG. This result proved that uncultured mesophilic Crenarchaeota groups are as ubiquitous in fermented seafood ecosystems, as in terrestrial and aquatic niches. Detection of diverse uncultured mesophilic Crenarchaeota groups in food ecosystems was unexpected because the existence of these Crenarchaeota groups has not been reported earlier in any fermented foods. The existence of the Crenarchaeota groups in seafood may be assumed from the fact that the seafood samples were made of fish or shellfish from marine environments in which mesophilic Crenarchaeota groups are enormously abundant. Further studies of the uncultured mesophilic Crenarchaeota groups in fermented food will be necessary to understand the ecological role of Crenarchaeota in the food fermentation process. This new insight into the archaeal community structure in fermented food may provide fundamental information for future investigations.

Archaeal phylogenetic tree showing the relationship among the DGGE bands along with representative reads from the pyrosequencing analysis. DGGE bands and representative pyrosequencing reads are indicated by bold-faced type. Representative pyrosequencing reads were selected from each group of the operational taxonomic units clustered within a DNA distance of 0.03 by the DOTUR program. Sequences that appeared more than 1% in a sample were included as representative sequences. Neighbor-joining tree was constructed with the representative and reference sequences. The Juke–Cantor method was used to calculate the distance matrix. Numbers at the nodes indicate bootstrap values calculated from the neighbor-joining probabilities (expressed as a percentage of 1000 replications). Bootstrap values greater than 50% are shown at the branch points. The scale bar represents 0.05 accumulated changes per nucleotide.

Bacterial phylogenetic tree showing the relationship among the DGGE bands along with representative reads from the pyrosequencing analysis. DGGE bands and representative pyrosequencing reads are indicated by bold-faced type. Representative pyrosequencing reads were selected and neighbor-joining tree was constructed as described in Figure 5. Bootstrap values greater than 50% are shown at the branch points. The scale bar represents 0.05 accumulated changes per nucleotide.

Phylogenetic tree and the quantities of the representative reads derived from the pyrosequencing analysis for each sample. Construction of the phylogenetic tree was based on a neighbor-joining algorithm. Each row denotes the percentage of the representative reads for each sequence. For each sample, the percentage of the representative reads of each sequence was visualized by ArrayColor.exe (http://microbecol.khu.ac.kr), which produces a range of colors, from white to red, to indicate the prevalence of each sequence in each sample. White squares represent low values, whereas red squares represent high values. The scale bar indicates 0.1 accumulated changes per nucleotide.
Conclusions
In this study, the microbial diversity in fermented seafood was evaluated with high-throughput multiplex barcoded pyrosequencing. The results elucidated that archaeal populations are more diverse in food samples than we expected, providing novel insights into the archaeal populations in fermented foods and confirming that the barcoded pyrosequencing approach can be a powerful tool for characterizing the microbiota in fermented food ecosystems compared with classical molecular ecological approaches, such as PCR–DGGE. The barcoded pyrosequencing method will be of value in the area of food microbiology and will allow for a better understanding of food ecosystems, in which fermented food processing can be influenced by diverse archaeal and bacterial microorganisms.
References
Aller JY, Kemp PF . (2008). Are Archaea inherently less diverse than Bacteria in the same environments? FEMS Microbiol Ecol 65: 74–87.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ . (1990). Basic local alignment search tool. J Mol Biol 215: 403–410.
Ampe F, Ben Omar N, Moizan C, Wacher C, Guyot JP . (1999). Polyphasic study of the spatial distribution of microorganisms in Mexican pozol, a fermented maize dough, demonstrates the need for cultivation-independent methods to investigate traditional fermentations. Appl Environ Microbiol 65: 5464–5473.
Ampe F, Miambi E . (2000). Cluster analysis, richness and biodiversity indexes derived from denaturing gradient gel electrophoresis fingerprints of bacterial communities demonstrate that traditional maize fermentations are driven by the transformation process. Int J Food Microbiol 60: 91–97.
Ampe F, Sirvent A, Zakhia N . (2001). Dynamics of the microbial community responsible for traditional sour cassava starch fermentation studied by denaturing gradient gel electrophoresis and quantitative rRNA hybridization. Int J Food Microbiol 65: 45–54.
Andersson AF, Lindberg M, Jakobsson H, Backhed F, Nyren P, Engstrand L . (2008). Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS ONE 3: e2836.
Ashelford KE, Chuzhanova NA, Fry JC, Jones AJ, Weightman AJ . (2005). At least 1 in 20 16S rRNA sequence records currently held in public repositories is estimated to contain substantial anomalies. Appl Environ Microbiol 71: 7724–7736.
Bae JW, Rhee SK, Park JR, Chung WH, Nam YD, Lee I et al (2005). Development and evaluation of genome-probing microarrays for monitoring lactic acid bacteria. Appl Environ Microbiol 71: 8825–8835.
ben Omar N, Ampe F . (2000). Microbial community dynamics during production of the Mexican fermented maize dough pozol. Appl Environ Microbiol 66: 3664–3673.
Binladen J, Gilbert MT, Bollback JP, Panitz F, Bendixen C, Nielsen R et al (2007). The use of coded PCR primers enables high-throughput sequencing of multiple homolog amplification products by 454 parallel sequencing. PLoS ONE 2: e197.
Caplice E, Fitzgerald GF . (1999). Food fermentations: role of microorganisms in food production and preservation. Int J Food Microbiol 50: 131–149.
Casamayor EO, Massana R, Benlloch S, Ovreas L, Diez B, Goddard VJ et al (2002). Changes in archaeal, bacterial and eukaryal assemblages along a salinity gradient by comparison of genetic fingerprinting methods in a multipond solar saltern. Environ Microbiol 4: 338–348.
Cha YJ, Lee EH . (1985). Studies on the processing of low salt fermented sea foods. Bull Korean Fish Soc 18: 206–213.
Chang HW, Kim KH, Nam YD, Roh SW, Kim MS, Jeon CO et al (2008). Analysis of yeast and archaeal population dynamics in kimchi using denaturing gradient gel electrophoresis. Int J Food Microbiol 126: 159–166.
Cheigh HS, Park KY . (1994). Biochemical, microbiological, and nutritional aspects of kimchi (Korean fermented vegetable products). Crit Rev Food Sci Nutr 34: 175–203.
Chun J, Lee JH, Jung Y, Kim M, Kim S, Kim BK et al (2007). EzTaxon: a web-based tool for the identification of prokaryotes based on 16S ribosomal RNA gene sequences. Int J Syst Evol Microbiol 57: 2259–2261.
Cocolin L, Manzano M, Cantoni C, Comi G . (2001). Denaturing gradient gel electrophoresis analysis of the 16S rRNA gene V1 region to monitor dynamic changes in the bacterial population during fermentation of Italian sausages. Appl Environ Microbiol 67: 5113–5121.
Cole JR, Chai B, Farris RJ, Wang Q, Kulam-Syed-Mohideen AS, McGarrell DM et al (2007). The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic Acids Res 35: D169–D172.
Crisan EV, Sands A . (1975). Microflora of four fermented fish sauces. Appl Microbiol 29: 106–108.
DeLong EF . (1992). Archaea in coastal marine environments. Proc Natl Acad Sci USA 89: 5685–5689.
DeLong EF, Taylor LT, Marsh TL, Preston CM . (1999). Visualization and enumeration of marine planktonic archaea and bacteria by using polyribonucleotide probes and fluorescent in situ hybridization. Appl Environ Microbiol 65: 5554–5563.
DeSantis Jr TZ, Hugenholtz P, Keller K, Brodie EL, Larsen N, Piceno YM et al (2006). NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res 34: W394–W399.
Dowd SE, Sun Y, Secor PR, Rhoads DD, Wolcott BM, James GA et al (2008a). Survey of bacterial diversity in chronic wounds using pyrosequencing, DGGE, and full ribosome shotgun sequencing. BMC Microbiol 8: 43.
Dowd SE, Sun Y, Wolcott RD, Domingo A, Carroll JA . (2008b). Bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP) for microbiome studies: bacterial diversity in the ileum of newly weaned Salmonella-infected pigs. Foodborne Pathog Dis 5: 459–472.
Edwards RA, Rodriguez-Brito B, Wegley L, Haynes M, Breitbart M, Peterson DM et al (2006). Using pyrosequencing to shed light on deep mine microbial ecology. BMC Genomics 7: 57.
Ercolini D . (2004). PCR-DGGE fingerprinting: novel strategies for detection of microbes in food. J Microbiol Methods 56: 297–314.
Euzeby JP . (1997). List of bacterial names with standing in nomenclature: a folder available on the Internet. Int J Syst Bacteriol 47: 590–592.
Ferris MJ, Muyzer G, Ward DM . (1996). Denaturing gradient gel electrophoresis profiles of 16S rRNA-defined populations inhabiting a hot spring microbial mat community. Appl Environ Microbiol 62: 340–346.
Fleet GH . (1999). Microorganisms in food ecosystems. Int J Food Microbiol 50: 101–117.
Francis CA, Roberts KJ, Beman JM, Santoro AE, Oakley BB . (2005). Ubiquity and diversity of ammonia-oxidizing archaea in water columns and sediments of the ocean. Proc Natl Acad Sci USA 102: 14683–14688.
Giraffa G . (2004). Studying the dynamics of microbial populations during food fermentation. FEMS Microbiol Rev 28: 251–260.
Giraffa G, Neviani E . (2001). DNA-based, culture-independent strategies for evaluating microbial communities in food-associated ecosystems. Int J Food Microbiol 67: 19–34.
Good IJ . (1953). The population frequencies of species and the estimation of population parameters. Biometrika 40: 237–264.
Hallam SJ, Konstantinidis KT, Putnam N, Schleper C, Watanabe Y, Sugahara J et al (2006a). Genomic analysis of the uncultivated marine crenarchaeote Cenarchaeum symbiosum. Proc Natl Acad Sci USA 103: 18296–18301.
Hallam SJ, Mincer TJ, Schleper C, Preston CM, Roberts K, Richardson PM et al (2006b). Pathways of carbon assimilation and ammonia oxidation suggested by environmental genomic analyses of marine Crenarchaeota. PLoS Biol 4: e95.
Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R . (2008). Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat Methods 5: 235–237.
Henckel T, Friedrich M, Conrad R . (1999). Molecular analyses of the methane-oxidizing microbial community in rice field soil by targeting the genes of the 16S rRNA, particulate methane monooxygenase, and methanol dehydrogenase. Appl Environ Microbiol 65: 1980–1990.
Herrmann M, Saunders AM, Schramm A . (2008). Archaea dominate the ammonia-oxidizing community in the rhizosphere of the freshwater macrophyte Littorella uniflora. Appl Environ Microbiol 74: 3279–3283.
Hoffmann C, Minkah N, Leipzig J, Wang G, Arens MQ, Tebas P et al (2007). DNA bar coding and pyrosequencing to identify rare HIV drug resistance mutations. Nucleic Acids Res 35: e91.
Huber JA, Mark Welch DB, Morrison HG, Huse SM, Neal PR, Butterfield DA et al (2007). Microbial population structures in the deep marine biosphere. Science 318: 97–100.
Huys G, Vanhoutte T, Joossens M, Mahious AS, De Brandt E, Vermeire S et al (2008). Coamplification of eukaryotic DNA with 16S rRNA gene-based PCR primers: possible consequences for population fingerprinting of complex microbial communities. Curr Microbiol 56: 553–557.
Inagaki F, Nunoura T, Nakagawa S, Teske A, Lever M, Lauer A et al (2006). Biogeographical distribution and diversity of microbes in methane hydrate-bearing deep marine sediments on the Pacific Ocean margin. Proc Natl Acad Sci USA 103: 2815–2820.
Könneke M, Bernhard AE, de la Torre JR, Walker CB, Waterbury JB, Stahl DA . (2005). Isolation of an autotrophic ammonia-oxidizing marine archaeon. Nature 437: 543–546.
Kasschau KD, Fahlgren N, Chapman EJ, Sullivan CM, Cumbie JS, Givan SA et al (2007). Genome-wide profiling and analysis of Arabidopsis siRNAs. PLoS Biol 5: e57.
Keijser BJ, Zaura E, Huse SM, van der Vossen JM, Schuren FH, Montijn RC et al (2008). Pyrosequencing analysis of the oral microflora of healthy adults. J Dent Res 87: 1016–1020.
Kim BS, Kim BK, Lee JH, Kim M, Lim YW, Chun J . (2008). Rapid phylogenetic dissection of prokaryotic community structure in tidal flat using pyrosequencing. J Microbiol 46: 357–363.
Kumar S, Tamura K, Nei M . (2004). MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform 5: 150–163.
Lafarge V, Ogier JC, Girard V, Maladen V, Leveau JY, Gruss A et al (2004). Raw cow milk bacterial population shifts attributable to refrigeration. Appl Environ Microbiol 70: 5644–5650.
Lam P, Jensen MM, Lavik G, McGinnis DF, Muller B, Schubert CJ et al (2007). Linking crenarchaeal and bacterial nitrification to anammox in the Black Sea. Proc Natl Acad Sci USA 104: 7104–7109.
Lee CH . (1994). Importance of lactic acid bacteria in non-dairy food fermentation. In: Lee CH, Nisse JA, Barwald G (eds). Lactic Acid Fermentation of Non-Dairy Food and Beverages. Harnrimwon Publishing CoL: Seoul, Korea. pp 17.
Lee CH . (1997). Lactic acid fermented foods and their benefits in Asia. Food Control 8: 259–269.
Lee CH, Steinkraus KH, Reilly PJA . (1993). Fish Fermentation Technology. United Nations University Press: Tokyo.
Leininger S, Urich T, Schloter M, Schwark L, Qi J, Nicol GW et al (2006). Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature 442: 806–809.
Liu Z, DeSantis TZ, Andersen GL, Knight R . (2008). Accurate taxonomy assignments from 16S rRNA sequences produced by highly parallel pyrosequencers. Nucleic Acids Res 36: e120.
Lozupone C, Hamady M, Knight R . (2006). UniFrac—an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics 7: 371.
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA et al (2005). Genome sequencing in microfabricated high-density picolitre reactors. Nature 437: 376–380.
Mincer TJ, Church MJ, Taylor LT, Preston C, Karl DM, DeLong EF . (2007). Quantitative distribution of presumptive archaeal and bacterial nitrifiers in Monterey Bay and the North Pacific Subtropical Gyre. Environ Microbiol 9: 1162–1175.
Muyzer G, de Waal EC, Uitterlinden AG . (1993). Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59: 695–700.
Nam YD, Chang HW, Kim KH, Roh SW, Kim MS, Jung MJ et al (2008). Bacterial, archaeal, and eukaryal diversity in the intestines of Korean people. J Microbiol 46: 491–501.
Ochsenreiter T, Selezi D, Quaiser A, Bonch-Osmolovskaya L, Schleper C . (2003). Diversity and abundance of Crenarchaeota in terrestrial habitats studied by 16S RNA surveys and real time PCR. Environ Microbiol 5: 787–797.
Ovreas L, Forney L, Daae FL, Torsvik V . (1997). Distribution of bacterioplankton in meromictic Lake Saelenvannet, as determined by denaturing gradient gel electrophoresis of PCR-amplified gene fragments coding for 16S rRNA. Appl Environ Microbiol 63: 3367–3373.
Paludan-Muller C, Huss HH, Gram L . (1999). Characterization of lactic acid bacteria isolated from a Thai low-salt fermented fish product and the role of garlic as substrate for fermentation. Int J Food Microbiol 46: 219–229.
Parameswaran P, Jalili R, Tao L, Shokralla S, Gharizadeh B, Ronaghi M et al (2007). A pyrosequencing-tailored nucleotide barcode design unveils opportunities for large-scale sample multiplexing. Nucleic Acids Res 35: e130.
Randazzo CL, Torriani S, Akkermans AD, de Vos WM, Vaughan EE . (2002). Diversity, dynamics, and activity of bacterial communities during production of an artisanal Sicilian cheese as evaluated by 16S rRNA analysis. Appl Environ Microbiol 68: 1882–1892.
Rivas R, Velazquez E, Zurdo-Pineiro JL, Mateos PF, Molina EM . (2004). Identification of microorganisms by PCR amplification and sequencing of a universal amplified ribosomal region present in both prokaryotes and eukaryotes. J Microbiol Methods 56: 413–426.
Roesch LF, Fulthorpe RR, Riva A, Casella G, Hadwin AK, Kent AD et al (2007). Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J 1: 283–290.
Roh SW, Nam YD, Chang HW, Kim KH, Lee HJ, Oh HM et al (2007a). Natronococcus jeotgali sp. nov., a halophilic archaeon isolated from shrimp jeotgal, a traditional fermented seafood from Korea Int. J Syst Evol Microbiol 57: 2129–2131.
Roh SW, Nam YD, Chang HW, Sung Y, Kim KH, Oh HM et al (2007b). Halalkalicoccus jeotgali sp. nov., a halophilic archaeon from shrimp jeotgal, a traditional Korean fermented seafood. Int J Syst Evol Microbiol 57: 2296–2298.
Roh SW, Sung Y, Nam YD, Chang HW, Kim KH, Yoon JH et al (2008). Arthrobacter soli sp. nov., a novel bacterium isolated from wastewater reservoir sediment. J Microbiol 46: 40–44.
Sands A, Crisan EV . (1974). Microflora of fermented Korean seafoods. J Food Sci 39: 1002–1005.
Schloss PD, Handelsman J . (2005). Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol 71: 1501–1506.
Schloss PD, Handelsman J . (2006). Toward a census of bacteria in soil. PLoS Comput Biol 2: e92.
Shen JP, Zhang LM, Zhu YG, Zhang JB, He JZ . (2008). Abundance and composition of ammonia-oxidizing bacteria and ammonia-oxidizing archaea communities of an alkaline sandy loam. Environ Microbiol 10: 1601–1611.
Siboni N, Ben-Dov E, Sivan A, Kushmaro A . (2008). Global distribution and diversity of coral-associated Archaea and their possible role in the coral holobiont nitrogen cycle. Environ Microbiol 10: 2979–2990.
Stackebrandt E, Goebel BM . (1994). Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int J Syst Bacteriol 44: 846–849.
Staley JT, Konopka A . (1985). Measurement of in situ activities of nonphotosynthetic microorganisms in aquatic and terrestrial habitats. Annu Rev Microbiol 39: 321–346.
Suh H-K, Yoon S-S . (1987). A study on the regional characteristics of Korean chotkal. Korean J Dietary Cult 2: 45–54.
Teske A, Sorensen KB . (2008). Uncultured archaea in deep marine subsurface sediments: have we caught them all? ISME J 2: 3–18.
van Beek S, Priest FG . (2002). Evolution of the lactic acid bacterial community during malt whisky fermentation: a polyphasic study. Appl Environ Microbiol 68: 297–305.
Watanabe K, Kodama Y, Harayama S . (2001). Design and evaluation of PCR primers to amplify bacterial 16S ribosomal DNA fragments used for community fingerprinting. J Microbiol Methods 44: 253–262.
Wuchter C, Abbas B, Coolen MJ, Herfort L, van Bleijswijk J, Timmers P et al (2006). Archaeal nitrification in the ocean. Proc Natl Acad Sci USA 103: 12317–12322.
Yeates C, Gillings MR, Davison AD, Altavilla N, Veal DA . (1998). Methods for microbial DNA extraction from soil for PCR amplification. Biol Proced Online 1: 40–47.
Acknowledgements
This work was supported by the Environmental Biotechnology National Core Research Center (KOSEF: R15-2003-012-02002-0) and TDPAF (Technology Development Program for Agriculture and Forestry).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website (http://www.nature.com/ismej)
Rights and permissions
About this article
Cite this article
Roh, S., Kim, KH., Nam, YD. et al. Investigation of archaeal and bacterial diversity in fermented seafood using barcoded pyrosequencing. ISME J 4, 1–16 (2010). https://doi.org/10.1038/ismej.2009.83
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2009.83
Keywords
This article is cited by
-
Terasi, exploring the Indonesian ethnic fermented shrimp paste
Journal of Ethnic Foods (2024)
-
Assessment of diversity of archaeal communities in Algerian chott
Extremophiles (2023)
-
The human gut archaeome: identification of diverse haloarchaea in Korean subjects
Microbiome (2020)
-
Modeling microbial communities from atrazine contaminated soils promotes the development of biostimulation solutions
The ISME Journal (2019)
-
Changes in the metabolic potential of the sponge microbiome under ocean acidification
Nature Communications (2019)
