
Abstract
Work on soils suppressive to Thielaviopsis basicola-mediated tobacco black root rot has focused on antagonistic pseudomonads to date. The role of non-Pseudomonas rhizosphere populations has been neglected, and whether they differ in black root rot-suppressive versus -conducive soils is unknown. To assess this possibility, tobacco was grown in a suppressive and a conducive soil of similar physicochemical properties, and rhizobacterial community composition was compared using a 16S rRNA taxonomic microarray. The microarray contains 1033 probes and targets 19 bacterial phyla. Among them, 398 probes were designed for Proteobacteria, Firmicutes, Actinomycetes, Cyanobacteria and Bacteroidetes genera/species known to include strains relevant for plant protection or plant growth promotion. Hierarchical clustering as well as principal component analysis of microarray data discriminated clearly between black root rot-suppressive and -conducive soils. In contrast, T. basicola inoculation had no impact on rhizobacterial community composition. In addition to fluorescent Pseudomonas, the taxa Azospirillum, Gluconacetobacter, Burkholderia, Comamonas and Sphingomonadaceae, which are known to comprise strains with plant-beneficial properties, were more prevalent in the suppressive soil. Mycobacterium, Bradyrhizobium, Rhodobacteraceae, Rhodospirillum and others were more prevalent in the conducive soil. For selected taxa, microarray results were largely corroborated by quantitative PCR and cloning/sequencing. In conclusion, this work identified novel bacterial taxa that could serve as indicators of disease suppressiveness in soil-quality assessments, and it extends the range of bacterial taxa hypothesized to participate in black root rot suppression.
Similar content being viewed by others

Introduction
Soil microorganisms have a major role in soil functioning and health, including the ability to control pathogens of plants (van Elsas et al., 2007). In disease-suppressive soils, some of the indigenous microorganisms protect susceptible crops from certain phytopathogens, whereas disease-conducive soils do not provide protection and permit spread of the pathogen (Alabouvette et al., 1996; Weller et al., 2002; Garbeva et al., 2004). Despite being an important component of soil quality (van Bruggen and Semenov, 2000), disease suppressiveness is not well understood. In certain cases, suppression is induced by crop monoculture and materializes as a decline of the disease, which follows earlier outbreaks. Induced disease suppression is well documented with wheat take-all, a disease mediated by Gaeumannomyces graminis var. tritici (Weller et al., 2002; Lebreton et al., 2004), and take-all decline involves enrichment effects of antagonistic root-colonizing Pseudomonas bacteria (Sarniguet and Lucas, 1992; Weller, 2007). In other cases, disease suppression is a natural property of the soil and is compatible with crop rotation (Stutz et al., 1986), even though the extent of disease suppression may change during crop rotation (Ramette et al., 2003). Natural/long-standing suppression is known for several diseases, such as Fusarium oxysporum-mediated Fusarium wilt (Alabouvette et al., 1996) and black root rot caused by the fungus Thielaviopsis basicola (Stutz et al., 1986).
Soils naturally suppressive to T. basicola-mediated black root rot of tobacco have been described at Morens (Switzerland), on sandstone overlaid by shallow morainic material (Stutz et al., 1985, 1989). Disease-conducive soils occur nearby on sandstone sediments. The comparison of up to 96 Morens fields indicated that both types of soils are cambisols, with a largely similar soil chemistry (Stutz et al., 1985, 1986; Ramette et al., 2003). Indeed, the only consistent difference is clay mineralogy, as vermiculite predominates in suppressive soils and illite in conducive soils (Stutz et al., 1989). None of the other characteristics correlates with soil suppressiveness (Stutz et al., 1985; Ramette et al., 2003).
Disease suppressiveness is a property conferred by soil microbial community. This is indicated by the facts that (i) soil pasteurization or sterilization abolished disease-control capacity in Morens suppressive soil, and (ii) the transfer of suppressiveness occurred when conducive soil received a small amount of suppressive soil (Stutz et al., 1985, 1986). Soil pasteurization/sterilization data (and results from artificial vermiculitic soils; Ramette et al., 2006) also showed that the presence of vermiculite did not impede the ability of T. basicola to infect tobacco. This is consistent with previous observations that the suppressive status of the soils did not influence the number of indigenous, virulent T. basicola propagules (Gasser and Défago, 1981) or the survival of inoculated T. basicola (Berling et al., 1984).
The initial studies attributed disease suppression to fluorescent pseudomonads (Stutz et al., 1986). Indeed, many Pseudomonas isolates from these soils (including the well-established model strain CHA0) (i) produced the antifungal compounds 2,4-diacetylphloroglucinol (Phl) and/or hydrogen cyanide (HCN), (ii) were antagonistic to T. basicola in vitro and (iii) protected tobacco from black root rot (Stutz et al., 1986; Ramette et al., 2003). However, in contrast to the situation of the take-all decline soils, HCN+ Phl+ Pseudomonas were also isolated in high numbers from black root rot-conducive soils in the same Morens area (Ramette et al., 2003; Frapolli et al., 2008). Furthermore, they could also protect tobacco from T. basicola when used as inoculants (Ramette et al., 2006). Therefore, disease suppressiveness of Morens soils to black root rot is unlikely to result only from enrichment effects of HCN+ Phl+ pseudomonads.
This study is based on the rationale that natural disease suppressiveness of soil may involve far more than the extensively studied biocontrol contribution of antagonistic fluorescent pseudomonads (Ramette et al., 2006). Indeed, the rhizosphere microbial community is highly diverse (van Elsas et al., 2007), and it is conceivable that several other microbial taxa could have an important part in disease suppression at Morens. They may act by protecting the plant directly, for example, through the release of pathogen inhibitors (Raaijmakers et al., 2008), or indirectly by promoting plant growth (Bally and Elmerich, 2005) or enhancing rhizosphere functioning of antagonistic pseudomonads (Lemanceau and Alabouvette, 1991). Community-level assessment of taxa more prevalent in suppressive soils was advocated to identify candidate microorganisms that may contribute to disease suppressiveness (Borneman and Becker, 2007), but this has rarely been performed with naturally suppressive soils (Rimé et al., 2003; Hjort et al., 2007) and never at Morens.
Therefore, the objective of this work was to compare the composition of the tobacco rhizobacterial community in black root rot-suppressive versus -conducive soils from Morens, and to identify non-Pseudomonas taxa more prevalent in black root rot-suppressive soil. To this end, a taxonomic microarray based on the 16S rRNA gene rrs and validated for rhizosphere assessments was further developed from earlier prototypes (Sanguin et al., 2006a, 2006b, 2008; Demanèche et al., 2008; Kyselková et al., 2008) and was used to assess tobacco at an early stage of plant development that is, at the time when biocontrol interactions are of particular relevance. In addition, the possible effect of the pathogen on rhizobacterial community composition was addressed by studying both non-inoculated tobacco as well as tobacco seedlings inoculated with T. basicola.
Materials and methods
Soil harvesting and growth chamber experiment
Soils were collected (5–30 cm depth) from fields MS8 (disease-suppressive cambisol; sandy loam, pH 7.8, CEC 5.7 cmol kg−1 with ammonium acetate method, 1.3% organic matter, 0.13% total N) and MC112 (disease-conducive cambisol; sandy loam, pH 6.8, CEC 8.5 cmol kg−1, 2.2% organic matter, 0.13% total N) near Morens, Switzerland, in June 2006 (Frapolli et al., 2008). Preparation of soil, tobacco seedlings (Nicotiana glutinosa L.) and endoconidia of T. basicola Ferraris strain ETH D127 was performed as described by Ramette et al. (2003). Soil received 103 endoconidia per cm3 soil (or sterile water in the controls) when transplanting 4-week-old tobacco plants in soil. The number of pots was eight per treatment. Soil water content was adjusted to 70% of water retention capacity. Plants were cultivated in a growth chamber at 22 °C (day, 16 h) and 18 °C (night, 8 h) at 70% relative humidity. Disease severity was recorded for each plant at 3 weeks after inoculation, as the percentage of root surface covered by T. basicola chlamydospores (Stutz et al., 1986), confirming that MS8 was suppressive (disease index 9.2%±1.8% after T. basicola inoculation) and MC112 conducive (41.7%±2.8% after inoculation). No disease was observed without inoculation. Rhizosphere soil, that is soil tightly adherent to roots (Frapolli et al., 2008), was used (250 mg) for DNA extraction with PowerSoil DNA Isolation Kit (MoBio Laboratories, Carlsbad, CA, USA).
PCR amplification of rrs genes and transcription labeling
The universal eubacterial primers T7-pA (forward: TAATACGACTCACTATAGAGAGTTTGATCCTGGCTCAG) and pH (reverse: AAGGAGGTGATCCAGCCGCA) (Bruce et al., 1992) were used to amplify rrs from DNA extracts obtained from pure strains (Supplementary Table 1) or rhizosphere soil. Primer T7-pA includes at the 5′-end of the sequence of T7 promoter (in italics above), which enabled a subsequent T7 RNA polymerase-mediated in vitro transcription. The PCR conditions were as described by Sanguin et al. (2008). PCR products were purified with MinElute PCR purification kit (Qiagen, Courtaboeuf, France). DNA concentration was determined spectrophotometrically and was adjusted to 50 ng μl−1. Fluorescence labeling by in vitro transcription (Stralis-Pavese et al., 2004), RNA purification and fragmentation was carried out as described by Sanguin et al. (2008).
Design of probes and microarray manufacturing
Probes were designed using ARB (Technical University Munich, Munich, Germany) and its rrs database (ssu_jan04_corr_opt.arb; http://www.arb-home.de; Ludwig et al., 2004), and were verified using the Silva-94 database (http://www.arb-silva.de; Pruesse et al., 2007). The parameters of the Probe Design function were chosen according to Sanguin et al. (2006a, 2006b), especially a weighted mismatch (WMM) value below 2 with the targeted taxa and more than 2 with non-targets. The WMM value is computed using ARB (Ludwig et al., 2004), and is 0 in the absence of any mismatch. The probes were further tested in silico, according to Sanguin et al. (2006a, 2006b). Probes were custom synthesized (Eurogentec, Seraing, Belgium) with a 5′-NH2-C6 group for covalent attachment onto aldehyde slides AL (Schott Nexterion AG, Mainz, Germany). Spotting and treatment of slides were performed as described previously (Sanguin et al., 2008). Each probe was repeated four times per slide.
Hybridization protocol
Two slides were hybridized per sample. Hybridization (overnight at 57 °C) was carried out in a custom-tailored aluminum block used as an insert for a temperature-controlled Belly Dancer (Stovall Life Sciences, Greensboro, NC, USA) set at maximum bending (Bodrossy et al., 2003), as described (Sanguin et al., 2008). Slide washing and handling were carried out as described by Sanguin et al. (2008).
Scanning, image analysis, filtration and normalization of microarray data
The slides were scanned at 532 nm with 10 μm resolution, using a GeneTac LS IV scanner (GenomicSolutions, Huntingdon, UK). Images were analyzed with the GenePix 4.01 software (Axon, Union City, CA, USA). Spot quality was always visually checked, and spots of poor quality (presence of dust) were excluded from further analyses, as described previously (Sanguin et al., 2006b).
Data filtration was conducted with the R 2.2.0 statistical computing environment (http://www.r-project.org). A given spot was considered hybridized when 80% of the spot pixels had an intensity higher than the median local background pixel intensity plus twice the s.d. of the local background. The intensity signals (median of signal minus background) were replaced by their square root value and the intensity of each spot was then expressed as a fraction of the total intensity signal of the basic pattern it belongs to (Sanguin et al., 2006b). Finally, a given feature probe was considered positive when (i) hybridization signals were superior to the mean signal of the negative controls and (ii) at least three of four replicate spots were hybridized.
Microarray validation by cloning/sequencing
Validation of microarray data was sought for important probes by cloning/sequencing, after amplification with specific primer pairs (Supplementary Table 2). When possible, the probe was used as one of the two primers (with if necessary a modification in 3′ to improve primer specificity). The new primers were designed using the ARB software (Ludwig et al., 2004). For each PCR, 20 ng of purified rrs PCR product (see above) obtained from one non-inoculated plant in soil MC112 or one non-inoculated plant in soil MS8 was used as template (see above for PCR conditions and purification of PCR products). Annealing temperature for each primer pair is indicated in Supplementary Table 2.
Purified PCR products were cloned into the plasmid vector pGEM-T (pGEMs-T Easy Vector System kit; Promega, Charbonnieres, France) according to the manufacturer's protocol. Five to six clones were sequenced on both strands (CoGenics, Meylan, France). Sequences were checked and edited with BioEdit version 5.0.9 (Ibis Therapeutics, Carlsbad, CA, USA; http://www.mbio.ncsu.edu/BioEdit/BioEdit.html). Chimeric 16S rRNA gene sequences were identified using the chimera detection program Pintail version 0.33 from Bioinformatics Toolkit (University of Cardiff, Cardiff, UK; http://www.bioinformatics-toolkit.org/; Ashelford et al., 2002), and putative chimeric clones were discarded. Sequence affiliation of non-chimeric sequences was performed using algorithm BlastN with default parameters at NCBI Blast (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi). The sequences are available at GenBank (accession numbers FJ447917–FJ447984).
Quantitative PCR
Relative amounts of rrs sequences of selected bacterial taxa (that is Sphingobacteriaceae, Mycobacterium and Bradyrhizobium) in soil MC112 versus MS8 were assessed with a quantitative PCR (qPCR) approach. The primers from the cloning/sequencing approach were used for Sphingomonadaceae and Mycobacterium, whereas a new primer was needed for Bradyrhizobium (Supplementary Table 2). PCRs (20 μl) were carried out in 96-well microplates. Each reaction contained 5 μl of PCR grade water, 2 μl of each primer (a final concentration of 0.5 μM), 10 μl of LightCycler-DNA Master SYBER Green I master mix (Roche Applied Science, Meylan, France) and 30 pg of soil DNA template. Thermal cycling was carried out with Light Cycler 480 (Roche Applied Science), with an initial denaturation step at 95 °C for 10 min, followed by 55 cycles with 30 s denaturation at 94 °C, annealing (Sphingobacteriaceae 30 s, Mycobacterium 20 s, Bradyrhizobium 20 s; annealing temperatures in Supplementary Table 2) and 30 s of elongation at 72 °C. Each sample was amplified in three replicates. The presence of a single specific PCR product was checked by assessing occurrence of (i) a single band after DNA migration in a 1.5% (w/v) agarose gel and (ii) a single peak in product melting curves provided by Light Cycler 480.
Three non-inoculated rhizosphere samples from each soil were studied. Relative quantities of rrs genes were assessed according to Pfaffl (2001), based on the ratio R(MS8/MC112) of rrs copies in MS8 versus MC112, which was computed as R(MS8/MC112)=EΔCp(MC112−MS8), where E means PCR efficiency and ΔCp (MC112-MS8) is computed for each MS8 sample as the difference between the mean crossing point for the three MC112 samples and the crossing point for the MS8 sample. PCR efficiency was calculated from dilution series curves (5, 10, 20, 40 and 80 pg of input DNA, each in three replicates) according to the equation E=10(−1/slope), the slope being determined by plotting of crossing points to the log value of DNA quantity input.
For the validation of qPCR data by cloning sequencing, qPCR products obtained for each primer pair, using one plant from each soil, were purified with MinElute kit (Qiagen) and reamplified by PCR, as described above. The resulting amplicons were purified (MinElute kit; Qiagen), cloned and eight clones from each were sequenced, as described above. The sequences are available at GenBank (accession numbers FJ890751-FJ890797).
Statistical analysis
Microarray data were treated by hierarchical clustering as well as principal component analysis. Hierarchical clustering was performed with CLUSFAVOR version 6.0 (Baylor College of Medicine, Houston, TX, USA; Peterson, 2002) using the Unweighted Pair Group with Mathematical Average method, based on Euclidean distance matrix calculated for all 199 positive probes. The output is displayed along with a Heatmap image (obtained using CLUSFAVOR 6.0) showing hybridization levels for the 76 positive probes that differed significantly in their signal intensities between treatments based on Fisher's least significant difference tests (see below). Principal component analysis of hybridization data was performed with ADE-4 (Thioulouse et al., 1997) in R environment (http://www.r-project.org), based on the correlation matrix. This was followed by a comparison of the treatments by two-factor (that is, soil × T. basicola inoculation) analysis of variance and Fisher's least significant difference tests (using DSAASTAT for Excel, version 1.0192; available at http://www.unipg.it/~onofri/DSAASTAT/DSAASTAT.htm; Onofri, 2006) along each of the first two axes, that is based on PC1 coordinates as well as PC2 coordinates (P<0.05). Two-factor analysis of variance was also used to compare treatments probe by probe.
Results
Extension of probe set and validation with pure strains
In this study, 468 new 16S rRNA probes were designed based on the criteria established for the previous 684 rrs probes of Sanguin et al. (2006a, 2006b, 2008), Kyselková et al. (2008) and Demanèche et al. (2008). When the whole probe set (1152 probes) was hybridized to rrs genes amplified from 23 bacterial strains (Supplementary Table 1) and one Mycobacterium environmental clone, aberrant results (that is, high signal levels despite high WMM values) were found for 70 of the 468 new probes (that is, about 15%), 1 of the 113 probes from Kyselková et al. (2008) and 48 of the 571 probes (that is, about 7%) from Demanèche et al. (2008). Rarefaction analysis of excluded probe number with increasing number of hybridized strains showed that the number of strains used was sufficient to reveal aberrant probes (not shown).
Once the 119 aberrant probes were excluded, the signals obtained for the 1033 probes left (398 new probes and 634 published probes; Table 1, Supplementary Table 3) decreased with the WMM values, and a steep dropout was observed at WMM=1.5 (Supplementary Figure 1). Above a WMM of 1.6, the median signal was close to zero and WMM 1.5 was taken as a threshold for expected hybridization. For WMM=0, the signal intensity was always above 0.025 normalized intensity units (Supplementary Figure 2, indicated in orange or red). On the basis of the WMM threshold of 1.5, only 2.3% of all 24 792 hybridizations (1033 probes × 24 strains) were unexpected (that is, false positive). In addition, 1.7% of the expected hybridizations (corresponding to less than 0.02% of all hybridizations) were false negative, that is a probe did not give any signal despite expectation (and it was always at WMM between 1.2 and 1.5).
The final list of probes with probe characteristics will be available at the ProbeBase site (http://www.microbial-ecology.net/probebase/). Generally, the probes are 17–26-mer oligonucleotides, the majority being 20-mers, with a G+C content between 35% and 70% (average 53%). The melting temperature (Tm) of the probes is between 49.7 and 76.7 °C, and 84% of the probes have a Tm between 60 and 70 °C. On the basis of nomenclatural taxonomy presented at RDP II (http://rdp.cme.msu.edu/; Garrity et al., 2007), the probe set targets 19 of 34 bacterial phyla with at least one probe (Table 1). In addition, some phyla, noticeably the Proteobacteria, Firmicutes, Actinobacteria and Bacteroidetes, are covered with probes at lower taxonomical levels, that is class, family, genus and/or species (Supplementary Table 3). Overall, the microarray covers, at the genus and/or species level, (i) most bacterial taxa (that is, about 90 species in 50 genera) known to include potentially plant-beneficial bacteria (based on rrs sequences available in databases) and (ii) a wide range of other bacteria known to be present in soil, including some human pathogens (Berg et al., 2005).
Comparison of rhizobacterial community in black root rot-suppressive and -conducive soils
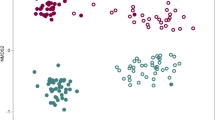
When rhizosphere samples from both T. basicola-inoculated and -non-inoculated tobacco plants were analyzed with the microarray, the number of positive probes was 144–152 per sample in soil MS8 and 137–153 in soil MC112. Hierarchical clustering performed on microarray data distinguished clearly between soils MS8 and MC112 (Figure 1). Inoculation with the phytopathogen T. basicola had no impact, regardless of whether soil MS8 or MC112 was considered. Concordant results were obtained with principal component analysis, where soils were clearly separated along the first principal axis (confirmed by analysis of variance of treatment replicates), whereas no separation was observed between inoculated and non-inoculated samples for any of the two soils (not shown).

Comparison of suppressive soil MS8 and conducive soil MC112 based on clustering of microarray hybridization data for the 199 positive bacterial rRNA gene probes (using Euclidean distance matrix and UPGMA algorithm). Plants inoculated with the pathogen T. basicola are indicated with black squares and non-inoculated plants with open squares. The heatmap shows hybridization levels only for the 76 probes that gave statistically different signals between the treatments (that is, the four soil × inoculation combinations). The taxonomic significance of probes is given in Table 2. UPGMA, Unweighted Pair Group with Mathematical Average.
The differences between the two soils were based mainly on variations in signal intensity rather than on the presence/absence of signals. As many as 69 of all 199 positive probes gave statistically different hybridization levels between the two soils, 34 of them higher for MS8 and the 35 others lower (Table 2). Twelve probes yielded significantly different signals as a consequence of T. basicola inoculation, and these differences were insufficient to make the whole community statistically different. In addition, the interaction between soil and T. basicola inoculation was significant for 13 probes.
Analysis of probes discriminating between black root rot-suppressive and -conducive soils
The Pseudomonas probes contributed substantially to the discrimination between the two soils (Figure 1). One probe for the Pseudomonas genus (that is, Pseu1) and two probes (that is, PseubC2BC3-2 and PseubC2-10) targeting fluorescent pseudomonads from clusters C3, C4 and C5 (Sanguin et al., 2008) gave significantly higher signals with suppressive soil (Table 2). In contrast, the probe Pseu33 targeting Pseudomonas citronellolis/P. nitroreducens displayed a higher signal with conducive soil.
Besides Pseudomonas, probes for various bacteria also contributed significantly to separation of soils, MS8 and MC112, according to principal component analysis (not shown) and analysis of variance (Figure 1 and Table 2). Probes giving higher signals in suppressive soil targeted noticeably (Table 2) (i) the Alphaproteobacteria family Sphingomonadaceae, genera Gluconacetobacter and Azospirillum, and species Azospirillum lipoferum, (ii) the Betaproteobacteria genera Nitrosospira/Nitrosovibrio and Comamonas, and species Burkholderia glathei/B. multivorans/B. cepacia/B. andropogonis, B. glumae and Herbaspirillum seropedicae, (iii) the Gammaproteobacteria family Xanthomonadaceae, and genera Stenotrophomonas/Xanthomonas, Photorhabdus, Methylosarcina and Methylomonas, (iv) the Deltaproteobacteria family Polyangiaceae, (v) the Actinobacteria genera Agromyces and Collinsella, (vi) the Firmicutes species Paenibacillus alginolyticus (and closely related species), (vii) the Cyanobacteria genus Lyngbia and (viii) Acidobacteria. Probes giving higher signals in conducive soil included in particular those targeting (i) the Alphaproteobacteria family Rhodobacteraceae, genera Rhodospirillum, Bradyrhizobium, Xanthobacter, Ehrlichia and Azospirillum irakense/Rhodocista spp., (ii) the Gammaproteobacteria species Pantoea (ex Erwinia) toletana and Pantoea agglomerans, (iii) the Deltaproteobacteria genus Thermodesulforhabdus, (iv) the Mollicutes genera Spiroplasma and Mycoplasma, (v) the Actinobacteria in general and the Actinobacteria genus Mycobacterium and (vi) the Firmicutes species Bacillus vedderi, Bacillus fortis (and closely related species) and Bacillus circulans.
Validation of selected microarray results by cloning/sequencing of PCR products and/or qPCR
The presence of selected taxa, for which the corresponding probes (i) gave positive signals both with soils MS8 and MC112, but (ii) contributed significantly to soil discrimination, was verified by cloning/sequencing (Supplementary Table 4). The genera Azospirillum, Burkholderia, Bradyrhizobium, Nitrosospira/Nitrosovibrio, Mycobacterium and genera within the Sphingomonadaceae family were successfully evidenced in both soils. The presence of Herbaspirillum (rather than the species H. seropedicae targeted by probe Hefri2) was confirmed only in soil MS8, as only clones of its close relative Janthinobacterium were found in soil MC112. Overall, with the exception of Herbaspirillum primers, the majority of clones were indeed affiliated to the expected taxonomic group, indicating that probe specificity was reliable.
qPCR procedures were developed and their specificity validated by cloning–sequencing (Supplementary Table 4) for a few selected taxa. qPCR results (Table 3) confirmed microarray findings that bacteria affiliated to Sphingomonadaceae were more prevalent in the tobacco rhizosphere in suppressive soil and Mycobacterium in conducive soil. For two plants grown in MS8, bacteria affiliated to Bradyrhizobium were less prevalent than in conducive soil, as expected from microarray results, but for the third plant in MS8, the abundance was comparable with that in the rhizosphere of plants grown in MC112. For all the taxa tested, the between-plant variability within a soil was higher with qPCR than with microarray.
Discussion
The majority of studies on disease-suppressive soils have been restricted to one effective microbial population, omitting the rest of the microbial communities (Mazzola, 2002; Nel et al., 2006; Weller, 2007; Frapolli et al., 2008). In the rhizosphere, however, the microbial community is diverse, which means that different types of microbial populations might protect the plant. In addition, complex interactions take place between microorganisms and roots, and the effective plant-protecting populations may be influenced by accompanying microbiota (Duijff et al., 1999; Raaijmakers et al., 2008). This is especially relevant in the case of black root rot-suppressive soils from Morens, in which the antagonistic Pseudomonas populations do not differ extensively from those in conducive soils (Ramette et al., 2003; Frapolli et al., 2008).
The new 16S rRNA gene-based taxonomic microarray developed here combines previous prototypes, which provided an extensive coverage of bacterial diversity at higher taxonomical units (19 of 34 bacterial phyla can be detected) and have been validated for analysis of soil/rhizosphere samples (Sanguin et al., 2006a, 2006b, 2008; Demanèche et al., 2008; Kyselková et al., 2008). To access rhizobacterial populations potentially relevant in suppressive soils, however, the microarray had to be significantly expanded (Table 1). This was achieved by designing several 100 additional probes, to target low-taxonomic-level taxa (that is, genus/species) known to include at least certain strains involved in beneficial interactions with plants and/or biocontrol of plant pathogens (Rodriguez and Fraga, 1999; Raaijmakers et al., 2002, 2008; Kennedy et al., 2004; Bally and Elmerich, 2005). The level of unexpected hybridizations with pure strains (2.3%, often corresponding to weak nonspecific signals) was well in line with that in other microarray studies (Bodrossy et al., 2003; Loy et al., 2005; Sanguin et al., 2006a; Kyselková et al., 2008).
To identify taxa indicative of soil suppressive status, we compared tobacco rhizobacterial communities from a suppressive and a conducive soil. Data showed that they differed rather in terms of probe signal intensity than in the number of positive probes. This is in accordance with the fact that suppressive and neighboring conducive soils near Morens are largely similar in physicochemical properties (except clay mineralogy; Stutz et al., 1989) and subjected to the same type of crop rotation and farming practices (Stutz et al., 1985; Ramette et al., 2003). Indeed, only a few taxa (corresponding to six probes) were detected in one soil only, and when so, probe signals were weak (often close to detection limit). It was the case for P. toletana (described as a secondary invader of olive knots; Rojas et al., 2004) found only in conducive soil, and Agromyces (rather common soil actinomycete), Collinsella (human intestine bacterium; Zoetendal et al., 2004), Selenomonas noxia/S. flueggei/S. infelix (anaerobes associated with periodontal pockets; Moore et al., 1987), Paenibacillus alginolyticus/P. chondroitinus and Cohnella (all three are phytopolymer degraders from the Paenibacillaceae family; Nakamura, 1987; Yoon et al., 2007) detected only in suppressive soil. Similarly, the presence/absence of signals from one plant to the next within a soil concerned only weakly hybridized probes.
As many as 69 of the 199 probes positive with both soils (that is, 35%) gave statistically significant differences in signal intensities between suppressive and conducive soil. Variability in signal intensity of a probe may be due to a different quantity of target molecules, and/or weak nonspecific binding of non-targets (especially in the absence of target molecules), as shown with pure strains. Therefore, for selected probes that discriminated between the two soils, we (i) verified that the targets were actually present in both soils (by cloning/sequencing) and/or (ii) confirmed the higher prevalence of the corresponding taxa in one of the soils (by qPCR). A good agreement between microarray and cloning/sequencing results was already found with partial probe sets (Sanguin et al., 2006a, 2006b, 2008; Kyselková et al., 2008). Therefore, the differences of signal intensities between soils MS8 and MC112 may be generally interpreted as differences in taxa abundance.
For about half of the probes discriminating between Morens suppressive and conducive soils, the mean signal intensity was 2–60 times higher or lower in one of the soils (Figure 1), pointing to important quantitative differences in community composition. Comparable results were obtained in two studies assessing whole rhizobacterial communities (i) of clubroot suppressive soil after amendments with, for example, chitin (Hjort et al., 2007) or (ii) in neighboring soils suppressive or not to ectoparasitic nematodes (where rhizobacterial community structure differed despite a similar soil composition; Rimé et al., 2003). In the two studies, however, the fingerprinting methodology did not enable direct identification of the corresponding taxa.
Though a function cannot be directly attributed to bacteria identified with rrs microarray, it is interesting to note that several taxa associated with suppressive soil MS8 are known to include strains with biocontrol capacity (Raaijmakers et al., 2008). Besides fluorescent pseudomonads, for which Phl+ strains have already been studied in Morens (for example, Stutz et al., 1986; Ramette et al., 2003; Frapolli et al., 2008), it is the case for the Burkholderia spp., for which certain strains produce various antifungal compounds and can antagonize fungal phytopathogens (Hwang et al., 2002; Compant et al., 2008). It applies also to taxa for which biocontrol ability has been less studied, noticeably Azospirillum spp. (Bashan and de-Bashan, 2002; Russo et al., 2008) and Comamonas (Thompson et al., 1998). In addition, probes for several taxa extensively studied for direct stimulation of plant growth (Baldani et al., 1986; Mehnaz and Lazarovits, 2006) also gave higher signals in the suppressive soil. It is the case for the Azospirillum spp. well known for associative nitrogen fixation, auxin production and/or ACC deaminase activity (Dobbelaere et al., 1999; Blaha et al., 2006), as well as certain strains from genera Herbaspirillum (Baldani et al., 1986), Sphingomonas (Adhikari et al., 2001) and Gluconacetobacter (Kennedy et al., 2004) studied for associative nitrogen fixation. This can be relevant for plant health because an enhanced plant vigor is important for resistance to pathogen attacks (Agrios, 1997). These taxa are of interest as bioindicators of disease suppressiveness, and they would merit an assessment of their potential functional role in suppressive soils. Comparably fewer taxa known to include bacteria with plant-beneficial properties were associated with the conducive soil MC112, for example Bradyrhizobium and P. agglomerans, which contain biocontrol strains (Siddiqui and Shaukat, 2002; Stockwell et al., 2002).
Root necrosis causes leakage of organic root constituents, which leads to distinct ecological conditions of rhizosphere colonization (Yang et al., 2001; Chapon et al., 2002). The shifts in various rhizosphere populations that can take place due to higher disease levels in conducive soil (Sarniguet and Lucas, 1992; McSpadden Gardener and Weller, 2001) may represent a bias when comparing suppressive and conducive soils (Ramette et al., 2003). In this work, however, root damage by added T. basicola did not affect rhizobacterial community composition, even in conducive soil. This result strengthens and expands the previous observation derived from the sole analysis of Pseudomonas populations (Frapolli et al., 2008). Perhaps lytic activity of T. basicola was not very strong when infecting tobacco roots (Hood and Shew, 1997), which means that root damage could have taken place without extensive nutrient leakage from diseased roots.
In conclusion, microarray comparison of a suppressive and conducive soil from Morens revealed previously unseen differences in the predominance of a large number of bacterial taxa between the two soils. We suppose that such differences in rhizobacterial community composition may be found for different types of suppressive soils and some of the taxa identified in this study could prove useful as disease suppressiveness indicators. In addition, many of these taxa are known to include strains with biocontrol or plant growth-promoting properties, and they represent key new targets whose potential functional role in disease suppression will need to be assessed.
Accession codes
References
Adhikari TB, Joseph CM, Yang GP, Phillips DA, Nelson LM . (2001). Evaluation of bacteria isolated from rice for plant growth promotion and biological control of seedling disease of rice. Can J Microbiol 47: 916–924.
Agrios GN . (1997). Plant Pathology. Academic Press: San Diego, CA, USA.
Alabouvette C, Höper C, Lemanceau P, Steinberg C . (1996). Soil suppressiveness to diseases induced by soilborne plant pathogens. In: Stotsky G, Bollag JM (eds). Soil Biochemistry. Marcel Dekker Inc.: New York. pp 371–413.
Ashelford KE, Weightman AJ, Fry JC . (2002). PRIMROSE: a computer program for generating and estimating the phylogenetic range of 16S rRNA oligonucleotide probes and primers in conjunction with the RDP-II database. Nucleic Acids Res 30: 3481–3489.
Baldani JI, Baldani VLD, Seldin L, Döbereiner J . (1986). Characterization of Herbaspirillum seropedicae gen. nov, sp. nov, a root-associated nitrogen-fixing bacterium. Int J Syst Bacteriol 36: 86–93.
Bally R, Elmerich C . (2005). Biocontrol of plant diseases by associative and endophytic nitrogen-fixing bacteria. In: Elmerich C, Newton WE (eds). Associative and Endophytic Nitrogen-fixing Bacteria and Cyanobacterial Associations. Kluwer Academic Publishers: Dordrecht. pp 171–190.
Bashan Y, de-Bashan LE . (2002). Reduction of bacterial speck (Pseudomonas syringae pv. tomato) of tomato by combined treatments of plant growth-promoting bacterium, Azospirillum brasilense, streptomycin sulfate, and chemo-thermal seed treatment. Eur J Plant Pathol 108: 821–829.
Berg G, Eberl L, Hartmann A . (2005). The rhizosphere as a reservoir for opportunistic human pathogenic bacteria. Environ Microbiol 7: 1673–1685.
Berling CH, Défago G, Kern H . (1984). Population-dynamics of Thielaviopsis basicola in soils conducive and suppressive to black root-rot disease. Phytopathology 74: 867.
Blaha D, Prigent-Combaret C, Mirza MS, Moënne-Loccoz Y . (2006). Phylogeny of the 1-aminocyclopropane-1-carboxylic acid deaminase-encoding gene acdS in phytobeneficial and pathogenic Proteobacteria and relation with strain biogeography. FEMS Microbiol Ecol 56: 455–470.
Bodrossy L, Stralis-Pavese N, Murrell JC, Radajewski S, Weilharter A, Sessitsch A . (2003). Development and validation of a diagnostic microbial microarray for methanotrophs. Environ Microbiol 5: 566–582.
Borneman J, Becker JO . (2007). Identifying microorganisms involved in specific pathogen suppression in soil. Annu Rev Phytopathol 45: 153–172.
Bruce KD, Hiorns WD, Hobman JL, Osborn AM, Strike P, Ritchie DA . (1992). Amplification of DNA from native populations of soil bacteria by using the polymerase chain reaction. Appl Environ Microbiol 58: 3413–3416.
Chapon A, Guillerm AY, Delalande L, Lebreton L, Sarniguet A . (2002). Dominant colonisation of wheat roots by Pseudomonas fluorescens Pf29A and selection of the indigenous microflora in the presence of the take-all fungus. Eur J Plant Pathol 108: 449–459.
Compant S, Nowak J, Coenye T, Clément C, Ait Barka E . (2008). Diversity and occurrence of Burkholderia spp. in the natural environment. FEMS Microbiol Rev 32: 607–626.
Demanèche S, Sanguin H, Pote J, Navarro E, Bernillon D, Mavingui P et al. (2008). Antibiotic-resistant soil bacteria in transgenic plant fields. Proc Natl Acad Sci USA 105: 3957–3962.
Dobbelaere S, Croonenborghs A, Thys A, Vande Broek A, Vanderleyden J . (1999). Phytostimulatory effect of Azospirillum brasilense wild type and mutant strains altered in IAA production on wheat. Plant Soil 212: 153–162.
Duijff BJ, Recorbet G, Bakker PAHM, Loper JE, Lemanceau P . (1999). Microbial antagonism at the root level is involved in the suppression of Fusarium wilt by the combination of nonpathogenic Fusarium oxysporum Fo47 and Pseudomonas putida WCS358. Phytopathology 89: 1073–1079.
Frapolli M, Moënne-Loccoz Y, Meyer J, Défago G . (2008). A new DGGE protocol targeting 2,4-diacetylphloroglucinol biosynthetic gene phlD from phylogenetically contrasted biocontrol pseudomonads for assessment of disease-suppressive soils. FEMS Microbiol Ecol 64: 468–481.
Garbeva P, van Veen JA, van Elsas JD . (2004). Microbial diversity in soil: selection of microbial populations by plant and soil type and implications for disease suppressiveness. Annu Rev Phytopathol 42: 243–270.
Garrity GM, Lilburn TG, Cole JR, Harrison SH, Euzeby J, Tindall BJ . (2007). The Taxonomic Outline of Bacteria and Archaea. TOBA Release 7.7, March 2007. Michigan State University Board of Trustees. http://www.taxonomicoutline.org/.
Gasser R, Défago G . (1981). Case of certain soils which are suppressive to the black root-rot of tobacco caused by Thielaviopsis basicola. Botanica Helvetica 91: 75–80.
Hjort K, Lembke A, Speksnijder A, Smalla K, Jansson JK . (2007). Community structure of actively growing bacterial populations in plant pathogen suppressive soil. Microb Ecol 53: 399–413.
Hood ME, Shew HD . (1997). Initial cellular interactions between Thielaviopsis basicola and tobacco root hairs. Phytopathology 87: 228–235.
Hwang J, Chilton W, Benson DM . (2002). Pyrrolnitrin production by Burkholderia cepacia and biocontrol of Rhizoctonia stem rot of poinsettia. Biol Control 25: 56–63.
Kennedy IR, Choudhury ATMA, Kecskes ML . (2004). Non-symbiotic bacterial diazotrophs in crop-farming systems: can their potential for plant growth promotion be better exploited? Soil Biol Biochem 36: 1229–1244.
Kyselková M, Kopecký J, Felfoldi T, Čermák L, Omelka M, Grundmann GL et al. (2008). Development of a 16S rRNA gene-based prototype microarray for the detection of selected actinomycetes genera. Antonie Van Leeuwenhoek 94: 439–453.
Lebreton L, Lucas P, Dugas F, Guillerm AY, Schoeny A, Sarniguet A . (2004). Changes in population structure of the soilborne fungus Gaeumannomyces graminis var. tritici during continuous wheat cropping. Environ Microbiol 6: 1174–1185.
Lemanceau P, Alabouvette C . (1991). Biological control of fusarium diseases by fluorescent Pseudomonas and non-pathogenic Fusarium. Crop Prot 10: 279–286.
Loy A, Schulz C, Lucker S, Schopfer-Wendels A, Stoecker K, Baranyi C et al. (2005). 16S rRNA gene-based oligonucleotide microarray for environmental monitoring of the betaproteobacterial order Rhodocyclales. Appl Environ Microbiol 71: 1373–1386.
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res 32: 1363–1371.
Mazzola M . (2002). Mechanisms of natural soil suppressiveness to soilborne diseases. Anton Leeuw 81: 557–564.
McSpadden Gardener BB, Weller DM . (2001). Changes in populations of rhizosphere bacteria associated with take-all disease of wheat. Appl Environ Microbiol 67: 4414–4425.
Mehnaz S, Lazarovits G . (2006). Inoculation effects of Pseudomonas putida, Gluconacetobacter azotocaptans, and Azospirillum lipoferum on corn plant growth under greenhouse conditions. Microb Ecol 51: 326–335.
Moore LVH, Johnson JL, Moore WEC . (1987). Selenomonas noxia sp. nov., Selenomonas flueggei sp. nov., Selenomonas infelix sp. nov., Selenomonas dianae sp. nov., and Selenomonas artemidis sp. nov., from the human gingival crevice. Int J Syst Bacteriol 37: 271–280.
Nakamura LK . (1987). Bacillus alginolyticus sp. nov. and Bacillus chondroitinus sp. nov., two alginate-degrading species. Int J Syst Bacteriol 37: 284–286.
Nel B, Steinberg C, Labuschagne N, Viljoen A . (2006). Isolation and characterization of nonpathogenic Fusarium oxysporum isolates from the rhizosphere of healthy banana plants. Plant Pathol 55: 207–216.
Onofri A . (2006). Enhancing Excel capability to perform statistical analyses in agriculture applied research. In: International Association for statistical Computing (ed). Computational Statistics and Data Analysis & Statistical Software Newsletters, 15/02/2006. http://www.csdassn.org/softlist.cfm.
Peterson LE . (2002). CLUSFAVOR 5.0: hierarchical cluster and principal component analysis of microarray-based transcriptional profiles. Genome Biol 3: software 0002.1–0002.8.
Pfaffl MW . (2001). A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res 29: e45.
Pruesse E, Quast C, Knittel K, Fuchs B, Ludwig W, Peplies J et al. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35: 7188–7196.
Raaijmakers JM, Paulitz TC, Alabouvette C, Steinberg C, Moënne-Loccoz Y . (2008). The rhizosphere: a playground and battlefield for soilborne pathogens and beneficial microorganisms. Plant Soil advance online publication, 23 February 2008; doi:10.1007/s11104-008-9568-6.
Raaijmakers JM, Vlami M, de Souza JT . (2002). Antibiotic production by bacterial biocontrol agents. Anton Leeuw 81: 537–547.
Ramette A, Moënne-Loccoz Y, Défago G . (2003). Prevalence of fluorescent pseudomonads producing antifungal phloroglucinols and/or hydrogen cyanide in soils naturally suppressive or conducive to tobacco black root rot. FEMS Microbiol Ecol 44: 35–43.
Ramette A, Moënne-Loccoz Y, Défago G . (2006). Genetic diversity and biocontrol potential of fluorescent pseudomonads producing phloroglucinols and hydrogen cyanide from Swiss soils naturally suppressive or conducive to Thielaviopsis basicola-mediated black root rot of tobacco. FEMS Microbiol Ecol 55: 369–381.
Rimé D, Nazaret S, Gourbière F, Cadet P, Moënne-Loccoz Y . (2003). Comparison of sandy soils suppressive or conducive to ectoparasitic nematode damage on sugarcane. Phytopathology 93: 1437–1444.
Rodriguez H, Fraga R . (1999). Phosphate solubilizing bacteria and their role in plant growth promotion. Biotechnol Adv 17: 319–339.
Rojas AM, García de los Rios JE, Fischer-Le Saux M, Jimenez P, Reche P, Bonnea S et al. (2004). Erwinia toletana sp. nov., associated with Pseudomonas savastanoi-induced tree knots. Int J Syst Evol Microbiol 54: 2217–2222.
Russo A, Vettori L, Felici C, Fiaschi G, Morini S, Toffanin A . (2008). Enhanced micropropagation response and biocontrol effect of Azospirillum brasilense Sp245 on Prunus cerasifera L. clone Mr.S 2/5 plants. J Biotechnol 134: 312–319.
Sanguin H, Herrera A, Oger-Desfeux C, Dechesne A, Simonet P, Navarro E et al. (2006a). Development and validation of a prototype 16S rRNA-based taxonomic microarray for Alphaproteobacteria. Environ Microbiol 8: 289–307.
Sanguin H, Kroneisen L, Gazengel K, Kyselková M, Remenant B, Prigent-Combaret C et al. (2008). Development of a 16S rRNA microarray approach for the monitoring of rhizosphere Pseudomonas populations associated with the decline of take-all disease of wheat. Soil Biol Biochem 40: 1028–1039.
Sanguin H, Remenant B, Dechesne A, Thioulouse J, Vogel TM, Nesme X et al. (2006b). Potential of a 16S rRNA-based taxonomic microarray for analyzing the rhizosphere effects of maize on Agrobacterium spp. and bacterial communities. Appl Environ Microbiol 72: 4302–4312.
Sarniguet A, Lucas P . (1992). Evaluation of populations of fluorescent pseudomonads related to decline of take-all patch on turfgrass. Plant Soil 145: 11–15.
Siddiqui I, Shaukat S . (2002). Mixtures of plant disease suppressive bacteria enhance biological control of multiple tomato pathogens. Biol Fertil Soils 36: 260–268.
Stockwell VO, Johnson KB, Sugar D, Loper JE . (2002). Antibiosis contributes to biological control of fire blight by Pantoea agglomerans strain Eh252 in orchards. Phytopathology 92: 1202–1209.
Stralis-Pavese N, Sessitsch A, Weilharter A, Reichenauer T, Riesing J, Csontos J et al. (2004). Optimization of diagnostic microarray for application in analysing landfill methanotroph communities under different plant covers. Environ Microbiol 6: 347–363.
Stutz EW, Défago G, Hantke R, Kern H . (1985). Effect of parent materials derived from different geological strata on suppressiveness of soils to black root rot of tobacco. In: Parker CA, Rovira AD, Moore KJ, Wong PTW, Kollmorgen JF (eds). Ecology and Management of Soil-borne Plant Pathogens. Academic Press, Inc.: New York. pp 215–217.
Stutz EW, Défago G, Kern H . (1986). Naturally occurring fluorescent pseudomonads involved in suppression of black root rot of tobacco. Phytopathology 76: 181–185.
Stutz EW, Kahr G, Défago G . (1989). Clays involved in suppression of tobacco black root rot by a strain of Pseudomonas fluorescens. Soil Biol Biochem 21: 361–366.
Thioulouse J, Chessel D, Dolédec S, Olivier JM . (1997). Ade-4: a multivariate analysis and graphical display software. Stat Comput 7: 75–83.
Thompson DC, Kobayashi DY, Clarke BB . (1998). Suppression of summer patch by rhizosphere competent bacteria and their establishment on Kentucky bluegrass. Soil Biol Biochem 30: 257–263.
van Bruggen AHC, Semenov AM . (2000). In search of biological indicators for soil health and disease suppression. Appl Soil Ecol 15: 13–24.
van Elsas JD, Jansson JK, Trevors JT . (2007). Modern Soil Microbiology. CRC Press: Boca Raton, FL, USA.
Weller DM . (2007). Pseudomonas biocontrol agents of soilborne pathogens: looking back over 30 years. Phytopathology 97: 250–256.
Weller DM, Raaijmakers JM, McSpadden Gardener BB, Thomashow LS . (2002). Microbial populations responsible for specific soil suppressiveness to plant pathogens. Annu Rev Phytopathol 40: 309–348.
Yang C, Crowley DE, Menge JA . (2001). 16S rDNA fingerprinting of rhizosphere bacterial communities associated with healthy and Phytophthora infected avocado roots. FEMS Microbiol Ecol 35: 129–136.
Yoon MH, Ten LN, Im WT . (2007). Cohnella panacarvi sp nov., a xylanolytic bacterium isolated from ginseng cultivating soil. J Microbiol Biotechnol 17: 913–918.
Zoetendal EG, Collier CT, Koike S, Mackie RI, Gaskins HR . (2004). Molecular ecological analysis of the gastrointestinal microbiota: a review. J Nutr 134: 465–472.
Acknowledgements
This work was supported in part by CORESTA (Paris, France), the bi-national PHC program Barrande (Czech Republic and France) and the Ministère Français de la Recherche. Some of the probes were designed in the European project MicroMaize (FW6 STREP). We are grateful to M-A Poirier and J Haurat (UMR CNRS 5557 Ecologie Microbienne), H Sanguin (University of Manchester, UK), C Deschamps and C Gauthier (PRABI), J Bernillon (DTAMB) and C Oger (DTAMB/PRABI) for technical help and/or discussion. We thank J Frère (Université de Poitier, France), K Shreiner and M Schloter (German Research Center for Environmental Health, Helmholtz Zentrum München, Germany) for gift of strains. This work made use of the DTAMB technical platform and PRABI/Rhône-Alpes bioinformatics center at IFR 41 in Université Lyon 1.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website (http://www.nature.com/ismej)
Rights and permissions
About this article
Cite this article
Kyselková, M., Kopecký, J., Frapolli, M. et al. Comparison of rhizobacterial community composition in soil suppressive or conducive to tobacco black root rot disease. ISME J 3, 1127–1138 (2009). https://doi.org/10.1038/ismej.2009.61
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2009.61
Keywords
This article is cited by
-
Testing the stress gradient hypothesis in soil bacterial communities associated with vegetation belts in the Andean Atacama Desert
Environmental Microbiome (2023)
-
Quantifying and visualizing Nitrospirillum amazonense strain CBAmC in sugarcane after using different inoculation methods
Plant and Soil (2023)
-
First Study Case of Microbial Biocontrol Agents Isolated from Aquaponics Through the Mining of High-Throughput Sequencing Data to Control Pythium aphanidermatum on Lettuce
Microbial Ecology (2023)
-
Exploring rhizo-microbiome transplants as a tool for protective plant-microbiome manipulation
ISME Communications (2022)
-
Microbial dysbiosis together with nutrient imbalance cause the replant problem of upper six flue-cured tobacco in Central Henan
Journal of Plant Diseases and Protection (2021)
