
Abstract
Microbiota transplantation to germ-free animals is a powerful method to study involvement of gut microbes in the aetiology of metabolic syndrome. Owing to large interpersonal variability in gut microbiota, studies with broad coverage of donors are needed to elucidate the establishment of human-derived microbiotas in mice, factors affecting this process and resulting impact on metabolic health. We thus transplanted faecal microbiotas from humans (16 obese and 16 controls) separately into 64 germ-free Swiss Webster mice caged in pairs within four isolators, with two isolators assigned to each phenotype, thereby allowing us to explore the extent of microbial spread between cages in a well-controlled environment. Despite high group-wise similarity between obese and control human microbiotas, transplanted mice in the four isolators developed distinct gut bacterial composition and activity, body mass gain, and insulin resistance. Spread of microbes between cages within isolators interacted with establishment of the transplanted microbiotas in mice, and contributed to the transmission of metabolic phenotypes. Our findings highlight the impact of donor variability and reveal that inter-individual spread of microbes contributes to the development of metabolic traits. This is of major importance for design of animal studies, and indicates that environmental transfer of microbes between individuals may affect host metabolic traits.
Similar content being viewed by others


Introduction
The global burden of obesity represents a worldwide public health challenge. Since the first report on the gut microbiota as an environmental factor that regulates fat storage (Bäckhed et al., 2004), the understanding of links between gut microbiota and obesity has been elaborated. Gut microbiota transplantations from both humans and mice with genetic or diet-induced obesity into germ-free (GF) mice, and the resulting development of recipient phenotypes with higher weight gain, have revealed that the gut microbiota plays a causal role in obesity (Turnbaugh et al., 2006, 2008; Vijay-Kumar et al., 2010; Ridaura et al., 2013; Goodrich et al., 2014). Studies in transplanted GF mice allow elimination of confounding factors present in human studies, and have led to identification of specific microbial configurations and activities causally related to obesity (Turnbaugh et al., 2008; Ridaura et al., 2013; Goodrich et al., 2014).
From an ecological point of view, transplantation into a GF mouse gut implies that the human microbial community must establish and colonise in a new habitat. Only a subset of human bacterial phylotypes will establish in mice, and some phylotypes change abundance in the new host (Licht et al., 2007; Wos-Oxley et al., 2012). The fraction of bacterial taxa from human faecal microbiota that establishes in transplanted mice is 50–90% at the genus level (Turnbaugh et al., 2009; Ridaura et al., 2013). Moreover, the well-described huge interpersonal variability between human gut microbiotas suggests that a broad coverage of donor microbiotas is required in transplantation studies to give representative and statistically meaningful results.
Mice are coprophagous, thus transfer of gut microbes through the faecal–oral route happens between mice housed in the same cage, resulting in transfer of an obese phenotype (Ridaura et al., 2013). Self-grooming additionally facilitates the ingestion of microbes from the surrounding environment. The recently established catalogue of the mouse gut metagenome shows that the environment, determined by mouse provider and housing laboratory, has a more pronounced effect on the conventional gut microbiota than seen for mouse strain, feed or gender (Xiao et al., 2015). Environmental reprogramming of microbiota by breeding mice in a new site (Ussar et al., 2015) or altering housing conditions (Müller et al., 2015) is shown to change the development of the metabolic syndrome. However, the extent of microbial spread within a specific environment and the effects of this spread on the development of metabolic phenotypes have currently not been studied.
To elucidate the selective establishment of human gut microbes in the GF mouse gut, as well as the resulting transfer of metabolic phenotypes from humans to mice, we inoculated 64 GF Swiss Webster mice, caged in pairs receiving the same donor microbes, with the individual faecal microbiotas from 16 obese children/adolescents and 16 matched controls. By housing the transplanted mice of each donor group in two independent isolators, we aimed to explore the extent of microbial spread between cages in a well-controlled environment, and the derived development of metabolic phenotypes in the mice. Additionally, we determined the longitudinal changes in the microbiota during establishment in the GF mice. Our findings are highly relevant for design of animal studies, and reveal that metabolic traits may be affected by spread of microbes between separately caged animals.
Materials and methods
Physiological measurements of human donors and faecal collection
Sixteen obese and 16 normal-weight children and adolescents (controls), age range 6.8–17.8 years, were recruited from The Danish Childhood Obesity Biobank. Determination of body mass index z-score as well as plasma glucose, serum insulin, serum triglycerides, serum total cholesterol, high-density lipoprotein cholesterol and low-density lipoprotein cholesterol concentrations were performed as previously reported (Fonvig et al., 2015). Total body/fat/bone mass was measured by whole-body dual-energy X-ray absorptiometry. Total body fat percentage was calculated as fat mass/(total body mass−total bone mass). The homoeostatic model assessment of insulin resistance (HOMA-IR) was calculated as insulin (pmol l−1) × glucose (mmol l−1)/(22.5 × 6).
The procedure for obtaining the human donor consent was approved by the Regional Health Research Ethics Committee for Region Zealand in trial SJ-104. Faecal samples were homogenised either 1:1 in pre-reduced 50% glycerol for use in transplantation studies, or 1:1 in water for DNA extraction, and stored at −80 °C.
Preparation of faecal bacterial suspension
Human faecal samples were thawed at 4 °C and transferred to an anaerobic chamber. One millilitre of each sample was mixed with 4 ml of cold pre-reduced 0.9% saline in a 15 ml tube, vortexed for 3 min and centrifuged at 200 g for 3 min at 4 °C. The supernatant was transferred to a new vial and kept at 4 °C for maximally 3 h until inoculation into the mice. Bacterial concentrations in the inocula were determined by anaerobic cultivation for 2 days at 37 °C on Wilkins Chalgren anaerobic agar with 5% horse blood (OXOID, Roskilde, Denmark).
Animals and experimental setup
Sixty-four male Swiss Webster mice bred at the GF facility of the Technical University of Denmark were separated from their mothers at age 5–6 weeks, stratified based on body weight and litter, and randomly distributed into four GF isolators (Scanbur, Karlslunde, Denmark). Two isolators were dedicated to the mice inoculated with human control samples and designated Control 1 and 2, while the other two isolators contained mice inoculated with samples from obese humans and were designated Obese 1 and 2. Human samples within each group (obese/control) were randomly distributed between the two isolators. Two mice, inoculated by oral gavage with 50 μl faecal bacterial suspension from the same donor, were housed in each cage. All mice were fed ad libitum with a standard chow diet (metabolisable energy 11.9 MJ kg−1; Altromin 1324; Lage, Germany) sterilised by irradiation. Individual body weight and food intake per cage were recorded at least weekly.
Animal experiments were approved by the Danish Animal Experiments Inspectorate. To maintain a controlled environment, the outer surface of all material transferred into the isolator was sterilised by exposure to chlorine dioxide for 20 min in the attached transfer airlock. To limit bacterial cross-contamination within isolators, gloves were disinfected with ethanol between handling of each cage. Six mice died due to the inoculation, and one mouse died 49 days post colonisation.
Sampling from mice
Faeces were collected from one mouse in each cage at 1, 2, 4, 7, 28, 42 and 49 days post colonisation (dpc). At the end point (51/52 dpc), the mice were fasted for 5–6 h prior to anaesthesia. Blood was then sampled from the eye vein, left to settle at room temperature for half an hour and subsequently centrifuged twice at 2000 g, 4 °C for 10 min to obtain serum. Following blood sampling, mice were killed by cervical dislocation and dissected. Epididymal fat pads were weighed, and luminal contents from ileum, caecum and colon from one mouse in each cage were snap-frozen. All samples were stored at −80 °C until further analyses. The reason for sampling microbiota (faeces and intestinal contents) from only one mouse per cage was that mice in the same cage were inoculated with the same donor microbiota, and additionally, mice practice coprophagy and thus ‘mix’ their microbiotas when co-caged.
Analysis of blood parameters in mice
Glycated haemoglobin was measured in tail vein blood using a DCA Vantage Analyser (Siemens, Erlangen, Germany), while blood glucose was measured with a glucose analyser Accu-Chek (Roche Diagnostics, Mannheim, Germany). Serum insulin and leptin were analysed using a Mouse Metabolic Kit (Meso Scale Diagnostics, Rockville, MD, USA). Serum cholesterol, triglyceride and non-esterified fatty acids (NEFA) were analysed using commercial kits LabAssay Cholesterol, LabAssay Triglyceride and NEFA-HR(2), respectively (Wako Chemicals, Neuss, Germany).
16S rRNA gene sequencing and data analysis
DNA extraction and 16S rRNA gene sequencing were performed essentially as described before (Tulstrup et al., 2015). Sequences were de-multiplexed and trimmed using the CLC Genomic Workbench v7.0.3 (Qiagen, Arhus, Denmark). Each sequence was classified to the lowest possible taxonomic rank using the Ribosomal Database Project Classifier v2.10.1 (Wang et al., 2007), and collapsed to different taxonomic levels, resulting in a phylotype data table. Operational taxonomic units (OTUs) were generated using UPARSE v8.0.1623 (Edgar, 2013).
Microbial α-diversity (Shannon index and OTU richness) and β-diversity were analysed based on the OTU table using Qiime 1.8.0 (Caporaso et al., 2010), and ADONIS tests were performed to assess the differential clustering of microbial communities using the vegan R package v2.3–0 (Oksanen et al., 2015). Analyses at genus/family/phylum levels were based on the phylotype data table. Principle component analysis was performed using the LatentiX 2.12 software (Latent5, http://www.latentix.com). The co-occurrence network of bacterial genera was built using SparCC (Friedman and Alm, 2012) and Cytoscape v3.2.1 (Shannon et al., 2003). Identification of bacterial features differentially abundant between groups was performed by permutation tests or Kruskal–Wallis tests followed by Dunn's multiple comparisons test.
Biochemical analyses
Faecal gross energy was assessed using a bomb calorimeter C6000 (IKA, Staufen, Germany). Short-chain fatty acids (SCFAs) were analysed by gas chromatography flame ionisation detector as previously described (Nejrup et al., 2015). Ultra-performance liquid chromatography mass spectrometry was applied for profiling of serum bile acids (Supplementary Tables S1 and S2).
Statistical analyses
Student’s t-test, Mann–Whitney test, one-way ANOVA followed by Tukey's multiple comparisons test, Kruskal–Wallis test followed by Dunn's multiple comparisons test, Spearman’s rank correlation or other statistics were used as noted in the figure legends. P-values from multiple testing were adjusted (q-value) using the Benjamini–Hochberg false discovery rate (Benjamini and Hochberg, 1995). P- or q-values lower than 0.05 were considered significant unless otherwise specified.
A more detailed method description is provided in the Supplementary Material.
Results
Human donors were different with respect to obesity and blood lipids, but not to faecal microbiota
Obese and control donors were similar in age and gender (Obese, age range 6.8–17.2 years, median 11.6 years, 7 girls and 9 boys; controls, age range 7.3–17.8 years, median 10.1 years, 7 girls and 9 boys). The obese donors had distinctly higher body mass index z-score (P<0.001; Figure 1a) and fat percentage (P<0.001; Figure 1b) than the controls. One particular control (donor 13) had remarkably high HOMA-IR and fasting insulin, which caused an absence of statistical significance of the differences between HOMA-IR and fasting insulin values obtained from obese and controls (Figures 1c and d, donor 13 is encirculated). No difference in fasting glucose was detected between the two groups (Figure 1e). Obese donors had higher levels of blood lipids than the controls, including triglycerides (P<0.05; Figure 1f), total cholesterol (P<0.01; Figure 1g), low-density lipoprotein cholesterol (P<0.01; Figure 1h) and higher ratio of total cholesterol to high-density lipoprotein cholesterol (P<0.05; Figure 1i), while no difference in high-density lipoprotein cholesterol was detected (Figure 1j).

Metabolic syndrome parameters and faecal microbiotas of obese human donors and controls. (a) Body mass index z-score, (b) fat percentage, (c) HOMA-IR, (d) fasting insulin in plasma, (e) fasting glucose in plasma. (f) Fasting triglycerides, (g) total cholesterol, (h) low-density lipoprotein (LDL) cholesterol, (i) ratio of total cholesterol to high-density lipoprotein (HDL) cholesterol and (j) HDL cholesterol in serum. PCoA based on unweighted (k) and weighted (i) UniFrac distances between faecal microbiotas of 16 obese human donors and 16 controls. In (a–j), the mean of each donor group is shown by a horizontal line, and asterisks represent significant differences between the two groups (*P<0.05, **P<0.01, ***P<0.001, unpaired t-test or Mann–Whitney test). In (k) and (l), P-values are listed for differential clustering assessed by ADONIS test and R2 values represent the percentage of variation explained by donor phenotype.
Despite metabolic differences between obese and controls, no differences were found in their faecal microbial communities. Microbial α-diversity was similar in control and obese donors, as assessed both by the Shannon index (5.99±0.46 vs 6.13±0.25, P=0.44) and by richness estimation (340.90±38.88 vs 329.70±38.48 observed OTUs per 10000 reads, P=0.42). Similarly, no clustering of obese and control donors was found by principle coordinate analysis (PCoA) (Figures 1k–l). No bacterial phyla, families, genera or OTUs, which discriminated the bacterial composition of obese donors from that of the controls, were identified.
Faecal microbiotas from human donors affected the physiology of transplanted mice differently
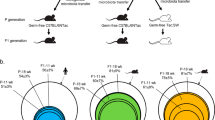
Transplanted mice were housed in four isolators with two groups representing each of the human phenotypes (Control 1, Control 2, Obese 1 and Obese 2; Figure 2a). Despite the absence of detectable differences between microbiotas of obese and control donors, these microbiotas had different effects on the metabolism of transplanted mice. Mice colonised with microbiotas from the obese donors had gained more weight by dpc 7 than mice colonised with microbiotas from the controls, and the groups remained significantly different until termination at dpc 51/52 (Figure 2b). Interestingly, weight gain of colonised mice was positively correlated with fat percentages of the corresponding human donors (Spearman r=0.42, P=0.001; Figure 2c), suggesting a contribution of gut microbiota features to body mass development of both human donors and transplanted mice.

Phenotypes of mice colonised with faecal microbiota from obese human donors or controls. (a) Schematic overview of the experimental design indicating study groups and time points of sampling. (b) Post-colonisation weight gain development of mice in the control and obese groups (n=28–29 per group). (c) Spearman correlation between fat percentage of human donors and weight gain of mice at the end point (n=57 mice and 32 human donors). (d) Weight gain over time and (e) weight gain at the end point shown by isolator groups (n=13–15 per group). (f) Total energy intake per mouse, (g) average faecal gross energy content at dpc 42 and 49 and (h) feed conversion ratio (n=8 per group). Spearman correlations between faecal gross energy and either (i) weight gain or (j) feed conversion ratio (n=8 per group). (k) HOMA-IR, (l) serum fasting insulin, (m) NEFA and (n) triglycerides (n=13–15 per group). The mouse transplanted with microbiota from human donor no. 13 is encirculated with a dashed line (the other mouse in the same cage died after inoculation). In (b) and (d), the mean of each group is shown, and the error bar represents s.e.m.; in (e–h) and (k–n), the mean of each group is shown by a horizontal line. In (b), asterisks represent significant differences between the two groups (*P<0.05, **P<0.01, unpaired t-test or Mann–Whitney test); in (e–h) and (k–n), asterisks represent significant differences between two groups (*P<0.05, **P<0.01, ***P<0.001, Tukey's multiple comparisons test or Dunn's multiple comparisons test). In (c, i and j), correlation coefficients, r, and significance levels, P, from Spearman correlations are listed and linear regression lines are shown.
Although the phenotypes of human donors did not differ between the two obese groups or between the two control groups applied for inoculation of the mice (Figure 2a and Supplementary Figure S1), transplanted mice in the two control groups developed distinct phenotypes. Mice in Control 1 gained less weight compared with the two obese groups (Figures 2d and e), which contributed to the overall differences in weight gain observed between obese and control transplanted mice (Figure 2b). Mice in all groups had similar amounts of energy intake from feed (Figure 2f), while mice of Control 1 tended to excrete a higher amount of energy in faeces than Obese 1 (Figure 2g), and had a higher feed conversion ratio than both obese groups (Figure 2h). Across all groups, faecal gross energy content was negatively correlated with weight gain (Figure 2i) and positively correlated with the feed conversion ratio (Figure 2j).
Although mice in Control groups 1 and 2 did not differ significantly in weight gain (Figure 2e), the latter group had lower insulin resistance, as shown by significantly lower levels of HOMA-IR, fasting insulin, as well as of two circulating lipids closely related to insulin resistance, NEFA and triglycerides (Julius, 2003; Kahn et al., 2006; Karpe et al., 2011) (Figures 2k–n). Additionally, mice in Obese 2 had significantly higher levels of fasting insulin (Figure 2l), circulating NEFA (Figure 2m) than that of mice in Control 2. No differences were detected between the four groups with respect to total circulating cholesterol (Supplementary Figure S2a), fasting glucose (Supplementary Figure S2b) or glycated haemoglobin (Supplementary Figure S2c). Moreover, no differences were found in the weight of epididymal fat pad (Supplementary Figure S2d) or the level of circulating leptin (Supplementary Figure S2e), indicating a similar obesity level among groups.
Gut microbiotas of the four groups of mice differed
Microbiotas of the transplanted mice differed markedly among the four groups, although the faecal microbiotas of human donors were very similar among groups in terms of α-diversity, β-diversity and bacterial taxa (Figures 1k–l and Figure 3). α- Diversity as determined by Shannon index and OTU richness differed between groups in the ileal and caecal mouse microbiotas, and was lowest in the Obese 1 group (Figures 3a and b). No differences in α-diversity were observed in the colonic microbiota. PCoA of mouse ileal, caecal and colonic microbiotas revealed clustering into each of the four isolator groups (Figure 3c). Already 7 days after separation into different isolators, Controls 1 and 2 tended to form two clusters in the PCoA plot (Supplementary Figure S3).

Gut microbiotas of human donors and transplanted mice shown by isolator groups. Alpha diversity as calculated by (a) Shannon index and (b) OTU richness of human donor samples and mouse ileal, caecal and colonic samples. Asterisks represent significant differences between groups (*P<0.05, **P<0.01, ***P<0.001, Tukey's multiple comparisons test or Dunn's multiple comparisons test), while (c) represents PCoAs based on unweighted UniFrac distance matrices of mice ileal, caecal and colonic microbiotas (three separate models). Pie charts (d) show relative abundances of bacterial phyla in human faecal microbiotas, and mouse ileal, caecal and colonic microbiotas. Each group has a significance indication including four characters representing the four groups Control 1, Control 2, Obese 1 and Obese 2. A dash (-) in a given position indicates that the phylum abundance is not different from the rest of the groups, while an asterisk (*) indicates significant difference from the group at that position (P<0.05, Dunn's multiple comparisons test). Heatmaps (e) show the relative abundances of ileal, caecal and colonic genera differing among groups, as well as the relative abundances of these genera in human samples. Asterisks represent FDR-adjusted P-values as calculated by Kruskal–Wallis testing (*q<0.05, **q<0.01, ***q<0.001). n=8 per group. Information about which of the specific groups that differs from the other groups is given in Supplementary Figure S4.
At phylum level, mice in the two control groups were rather similar, while mice in the Obese 2 group had lower relative abundance of Verrucomicrobia in ileal samples than found in Obese 1 (Figure 3d). In general, mice in the obese groups had lower relative abundance of Proteobacteria in the ileum, less Firmicutes in the caecum and colon, but more Bacteroidetes in the caecum and colon than seen in the control groups (Figure 3d). At genus level, each of the four groups harboured a distinct microbial profile (Figure 3e and Supplementary Figure S4). Several differences were observed, of which the most significant ones occurred within the Firmicutes, particularly the Clostridiales order. In specific gut compartments, Control 1 had >100-fold higher levels of Pseudoflavonifractor and Parasporobacterium, Control 2 had >100-fold higher levels of Sarcina and Clostridium XI, Obese 1 had >100-fold lower levels of Lachnospiracea IS and Obese 2 had >100-fold higher levels of Streptococcus and >100-fold lower levels of Anaerostipes than other groups (Figure 3e and Supplementary Figure S4).
Taken together, although the two control groups and the two obese groups, respectively, had similar microbial patterns at the phylum level with respect to Firmicutes and Bacteroidetes, the spread of microbes between cages in the same isolators rapidly caused different patterns within each isolator group in terms of composition at the genus level and microbial diversity.
Phenotype and gut microbiota of donor 13 and the corresponding colonised mouse
Donor 13 had high insulin resistance (Figures 1c and d), accompanied by the highest levels of circulating triglycerides, total cholesterol, ratio of total cholesterol to high-density lipoprotein cholesterol, and low-density lipoprotein cholesterol measured in donors from Control 2 (Supplementary Figures S1f–i). Interestingly, the mouse transplanted with the microbiota from donor 13 additionally showed the highest epididymal fat weight and circulating leptin of all mice in the control groups (Supplementary Figures S2d–e), while HOMA-IR, fasting insulin, circulating NEFA and circulating triglycerides were the highest among the mice in the Control 2 group, but average compared with Control 1 (Figure 2). We identified 21 faecal bacterial genera that differed (q<0.05) between donor 13 and the other control donors (Supplementary Figure S5a). Similarly, we identified 18 ileal/caecal/colonic genera that differed between the mouse transplanted with microbiota from donor 13, and the other mice in the Control 2 group (Supplementary Figures S5b–d). Among these, some were found to be shared between donor and mouse, including lower relative abundance of Bacteroides, and higher abundance of Clostridium XI, Pseudobutyrivibrio and Coprococcus in donor 13 as well as in the transplanted mouse (Supplementary Figure S5).
Gut microbial activities were different between groups
Given the significant differences in microbiotas between the four isolated groups, we investigated whether the gut microbial activity also differed between these groups (Supplementary Figure S6). Principle component analysis of 13 murine bile acids in serum, quantified by ultra-performance liquid chromatography mass spectrometry (Supplementary Figure S7 and Figure 4), revealed that the ratio between conjugated and unconjugated bile acids was significantly lower in Obese 2 than in all other groups (Figure 4b), indicating elevated microbial de-conjugation of bile acids in the Obese 2 group. Generally, mice in Control 1 had significantly higher serum levels of several bile acids, including the secondary bile acids tauro-ω-muricholic acid, taurohyodeoxycholic acid and ω-muricholic acid (ωMCA) than the other groups (Figures 4c–o).

Serum bile acids in the four separately isolated mouse groups. (a) Score and loading plots of principle component analysis (PCA) of bile acids in serum based on auto-scaled data; (b) ratios of conjugated bile acids to unconjugated bile acids; (c) TCA, taurocholic acid; (d) TβMCA, tauro-β-muricholic acid; (e) TωMCA, tauro-ω-muricholic acid; (f) TDCA, taurodeoxycholic acid; (g) TCDCA, taurochenodeoxycholic acid; (h) THDCA, taurohyodeoxycholic acid; (i) CA, cholic acid; (j) DCA, deoxycholic acid; (k) UDCA, ursodeoxycholic acid; (l) CDCA, chenodeoxycholic acid; (m) βMCA, β-muricholic acid; (n) ωMCA, ω-muricholic acid; and (o) αMCA, α-muricholic acid (n=12–15 per group). In (b–o), the mean of each group is shown by a horizontal line, and asterisks indicate significant differences between groups (*P<0.05, **P<0.01, ***P<0.001, Tukey's multiple comparisons test or Dunn's multiple comparisons test). The average levels of the 13 measured bile salts in all mice are shown in Supplementary Figure S7.
Quantification of seven SCFAs in caecal contents by the gas chromatography flame ionisation detector showed that valeric acid was markedly depleted in Control 2 animals (Figure 5a). Individual differences between the four groups were not significant for butyric and iso-butyric acid (Figure 5b and c); however, when comparing the combined obese groups (Obese 1 and 2) to the combined controls (Control 1 and 2), butyric acid and iso-butyric acid were higher in the controls than that in the obese (2.18±0.90 vs 1.50±0.44, 0.35±0.12 vs 0.25±0.07, respectively, P-values <0.05). No differences were found in acetic acid, propionic acid, iso-valeric acid, caprioic acid or the sum of the seven SCFAs (Supplementary Figures S8a–e).

SCFAs in caecal content. (a) Valeric acid, (b) butyric acid and (c) iso-butyric acid. The mean of each group is shown by a horizontal line, and asterisks represent significant differences between groups (*P<0.05, **P<0.01, ***P<0.001, Tukey's multiple comparisons test or Dunn's multiple comparisons test). Spearman correlations between faecal gross energy and (d) the sum of caecal SCFAs, (e) acetic acid, (f) propionic acid and (g) butyric acid (n=8 per group). Correlation coefficients, r, and significance levels, P, from Spearman correlations are listed and linear regression lines are shown (d–g).
As SCFAs have been suggested as markers of energy harvesting by the microbiota (Murphy et al., 2010), we looked for correlations between faecal gross energy content and caecal SCFAs. Gross energy content was positively correlated with the sum of SCFAs as well as with acetic acid, and tended towards correlation with propionic and butyric acid (Figures 5d–g). The latter three were the most abundant SCFAs, accounting for >90% of total SCFAs. Associations with the remaining SCFAs were not significant (Supplementary Figures S8f–i).
Associations between microbiota, metabolites and host phenotypes
Spearman’s rank correlation analyses were performed and followed by correction for multiple testing. In the following, correlations with q-values lower than 0.1 are reported while those with absolute Spearman r values larger than 0.4 are mentioned as tendencies for correlation.
Mouse body weight was negatively associated with caecal Anaerostipes, and tended to be negatively associated with ileal Anaerostipes, ileal Pseudoramibacter and ileal Klebsiella (Figure 6a). These three genera formed a tight co-occurring cluster with Clostridium XVIII in the ileum (Figure 6b). Body weight gain was negatively associated with Ileal Actinobacteria and tended to be negatively associated with caecal Dorea, and correspondingly, these two bacterial groups were positively associated with the feed conversion ratio (Figure 6a). HOMA-IR and fasting insulin were strongly negatively associated with ileal Anaerofustis and positively associated with caecal OTU richness. Additionally, circulating NEFA was negatively associated with ileal Asaccharobacter (Figure 6a).

Heatmaps and network analysis Spearman correlations between (a) metabolic features of mice and characteristics of their ileal and caecal microbiota, (c) caecal SCFAs and caecal bacterial taxa, (d) serum bile acids and ileal bacterial taxa. FDR correction was performed phenotype-wise (a) or metabolite-wise (c, d) on correlation P-values, generating q-values. Microbiota characteristics with at least one correlation coefficient <−0.4 or >0.4 are shown. Hierarchical clustering is presented for bacterial taxa based on the Euclidean distances between their Spearman coefficients. ‘°’ indicates coefficient <−0.4 or >0.4, and ‘+’ indicates q-values below 0.1. Asterisks in labels of rows and columns indicate significant differences between isolator groups as determined by one-way ANOVA or Kruskal–Wallis test. P-values are indicated as *P<0.05, **P<0.01, ***P<0.001, while for bacterial genera and families, FDR-adjusted P-values are shown as *q<0.05, **q<0.01, ***q<0.001 (n=29–32). A co-occurrence network (b) was built from SparCC correlation coefficients between abundances of ileal genera, and correlations with P<0.002 are shown. The nods represent genera and the edges represent the correlation coefficients between genera. Nods are coloured according to the significance of the Spearman correlations between genus and body weight. Edges are coloured red for a positive correlation and grey for a negative correlation, and weights of the edges reflect strength of the correlation. Nods are positioned using an organic layout in Cytoscape (n=29–32).
Butyric acid tended to be positively associated (Figure 6c) with the well-described butyrate-producing Clostridia clade within Lachnospiraceae, Clostridium XIVa (Pryde et al., 2002; Louis and Flint, 2009). However, a much stronger positive correlation was found between butyric acid and the genus of Ruminococcaceae, Oscillibacter, species of which have been reported to produce either butyric acid or valeric acid (Iino et al., 2007; Lee et al., 2012, 2013). Both acetic acid and butyric acid were positively correlated with Ruminococcaceae, which are known to degrade several types of polysaccharides and represent the first step of microbiome-driven carbohydrate metabolism (Morgan et al., 2012). Additionally, a very strong negative correlation was found between valeric acid and the family Clostridiaceae 1, within which more than 90% of the reads were represented by the genus Sarcina. As mentioned above, Sarcina was >100-fold more abundant in Control 2 than in other groups, and may be responsible for the depletion of valeric acid in Control 2. Iso-butyric acid, which mainly originates from fermentation of branched amino acids (Smith and Macfarlane, 1997; Macfarlane and Macfarlane, 2003; Davila et al., 2013), tended towards a positive association with Alistipes.
As most bile acids (~95%) are known to be absorbed in the ileum (Ridlon et al., 2006), we looked for associations between abundances of ileal bacterial phylotypes and serum bile acids (Figure 6d). Pseudoflavonifractor was positively correlated to ωMCA, suggesting that this bacterial group could be contributing to the C6-epimerization of βMCA into ωMCA. Streptococcus was negatively associated with TDCA, while Barnesiella and Flavonifractor were negatively associated with the conjugated/unconjugated ratio. No significant associations were detected between the SCFAs/bile acids and metabolic measures (data not shown).
Establishment of human gut microbes in GF mice
Anaerobic cultivation showed that a total of 3 × 106-2 × 108 CFUs were inoculated to each GF mouse. The structure of the bacterial communities in the new hosts evolved extensively during the first week after colonisation. PCoA based on unweighted UniFrac distances between faecal microbiotas (Figure 7a) showed that with time, the microbiotas of the mice gradually became more similar to the original human inoculum. Principle component analysis based on genera revealed specific changes in the microbial communities occurring during the establishment (Figure 7b). Facultative anaerobic bacteria including Escherichia/Shigella and Enterococcus were present in high relative abundance in the faeces at dpc 1 and dpc 2, which is consistent with the fact that the GF gut is relatively rich in oxygen (Bornside et al., 1976). The numbers of observed faecal genera with relative abundances >0.01% were lowest at dpc 1, and thereafter increased (Figure 7c), reflecting a low microbial diversity at the early stage of colonisation as previously reported (El Aidy et al., 2012, 2013). The mucin-degrading bacterium Akkermansia (Everard et al., 2013) increased in abundance after the early colonisation phase. This is in agreement with the finding that the mucus layer in the gut of GF mice in not mature, and that during colonisation, the ileal mucus becomes more detached, increasing the relative amount of available mucin in the colon (Johansson et al., 2015). Interestingly, human donors were distinguished from transplanted mice by harbouring a cluster of genera, many of which are potential butyrate producers, including Roseburia, Eubacterium (within the family Eubacteriaceae), Coprococcus, Faecalibacterium, Pseudobutyrivibrio and Lachnospira (Rode et al., 1981; Pryde et al., 2002; Paillard et al., 2007; Louis and Flint, 2009). Instead, another group of potential butyrate producers, for example, Anaerostipes, Butyricicoccus, Anaerotruncus and Anaerofustis (Pryde et al., 2002; Eeckhaut et al., 2008; Louis and Flint, 2009), established in transplanted mice. Additionally, human Bifidobacterium was inefficiently established in the mice.

Colonisation and establishment of human microbiota in GF mice. (a) PCoA based on unweighted UniFrac distances between faecal microbiotas from human donors and transplanted mice, (b) score and loading plots of a principle component analysis (PCA) based on faecal genera of human donors and transplanted mice. Ruminococcus belonged to the family Ruminococcaceae, Ruminococcus2 belonged to the family Lachnospiraceae and Eubacterium belonged to the family Eubacteriaceae. Relative abundances were log-transformed and auto-scaled. Additional analyses included (c) numbers of detected genera (relative abundance >0.01%) in humans and mice, (d) overlap between detected OTUs/genera with relative abundances >0.01% in human faecal microbiotas and mouse colonic microbiotas, (e) Shannon index and (f) OTU richness of the microbiotas. In (e) and (f), asterisks represent significant differences between humans and mice (***P<0.001, Wilcoxon test or paired t-test). n=32 humans or mice.
We investigated the fractions of bacterial phylotypes identified both in a given human faecal inoculum and in the colonic sample of the corresponding transplanted mice. On average, 19.5±7.8% of all observed OTUs and 44.5±4.9% genera were shared between the human donor and the mouse recipient (Figure 7d). Some bacteria (26.9±6.0% of OTUs and 17.8±4.5% of the genera) reached detectable limits in the mouse, although they were below the detection limit in the human inoculum. The presence/absence of each individual genus in a human donor and the corresponding mouse was recorded (Supplementary Figure S9). Shannon index and OTU richness of mice colonic microbiotas were significantly lower than that of human faecal microbiotas (Figures 7e and f).
Discussion
Gut microbiota transplantation to GF or antibiotic-treated animals is a powerful approach to verify causality of gut microbiota in a broad range of diseases (Bercik et al., 2011; Goodrich et al., 2014; Natividad et al., 2015). However, due to the need for strict containment of each human donor microbiota in separate isolators, it is a challenge for many studies to reach a representative amount of donors. Here, by using sufficient numbers of human donors to place them in two groups (obese and control) based on statistically significant differences in metabolic features (Figures 1a–j), we aimed to elucidate the transmissibility of such features through colonisation of individual mice with the individual human microbiotas. Additionally, we aimed to investigate the necessity of costly separate isolation of individual microbiotas through quantification of environmental microbial transmission between mice caged separately within the well-controlled environment of an isolator. The study design thus included four isolators, two of which each contained 8 cages of mice inoculated with two groups of 8 different microbiotas originating from 16 human obese subjects, while the two others each contained 8 cages of mice inoculated with 16 microbiotas obtained from normal-weight controls.
We found that spread of microbes between cages did indeed occur within each isolator, probably via animal handling, although isolator gloves were treated with ethanol between handling of different cages, suggesting that spread between cages is difficult to avoid. This is obviously relevant for transplantation studies, but may also affect results from studies of conventional mice, even though the well-described colonisation resistance (van der Waaij et al., 1971) probably renders a conventional microbiota less susceptible to establishment of new microbes than an incomplete, host-unspecific transplanted microbiota.
Our findings emphasise the need for explicit descriptions of animal housing conditions in publications involving microbiota studies, but an important additional question is whether the environmental spread is in itself sufficient to affect the phenotype of a host animal, or whether a substantial oral exposure (by transplantation or coprophagy) is needed to be of host phenotypical relevance. Confirming the relevance of environmental spread between cages, we found that the distinct patterns of microbial composition and activities observed in the four different isolators (Figure 3) were indeed accompanied by distinct metabolic phenotypes (Figure 2). The lowest weight gain was observed in one of the groups of mice colonised with microbiotas from control donors with normal-weight and without any indication of metabolic syndrome (Control 1; Figure 2). However, this group was remarkably and significantly different from the other group of mice colonised with control donor faeces (Control 2) with respect to HOMA-IR, fasting insulin and circulating NEFA, triglycerides and bile acids, which highlights the relevance of using a representative donor group size as well as of considering microbial spread. It is important to note that in spite of the differences in weight gain, none of the animals in the study were obese. We therefore suggest that the observed difference in insulin resistance between Control 1 and Control 2 was not driven by obesity, but rather by other factors related to the microbiota, putatively including alterations in inflammatory phenotype, microbial metabolite profiles or the abundance of specific microbial populations such as, for example, Anaerofustis.
Although we did not find any specific associations between bile acids and metabolic measures, we cannot rule out that microbiota-driven differences in metabolic homoeostasis (Figures 2k–n) could be mediated by differences in serum levels of bile acids, since major differences in bile acid profiles were found between the four groups (Figure 4), and bile acids are known to regulate glucose and lipid metabolism (Lefebvre et al., 2009; Tremaroli and Bäckhed, 2012).
Although no statistically significant differences were detected between the faecal microbiotas from obese and control human donor groups (Figures 1k–l), the ileal and caecal (but not colonic) microbiotas differed between the transplanted obese and control mice (Figures 3a and b). This suggests that differences may have existed between the upper gut microbiotas of the obese and control groups of human donors, which were not reflected in faeces. Considering that most differences in genera found in mice colon already occurred in ileum, we find it likely that the ileal microbiota plays an important role in development of the metabolic syndrome (El Aidy et al., 2015), which is in line with the fact that ileum is the main place for bile acid absorption (Dawson et al., 2009) and intestinal immunity (Jung et al., 2010). A strong positive correlation between caecal OTU richness and insulin resistance was observed (Figure 6a), whereas previous reports have linked diversity and gene richness of the faecal microbiome to a healthy metabolic status in humans (Lozupone et al., 2012; Le Chatelier et al., 2013).
Caecal SCFA levels correlated positively with faecal gross energy, suggesting that high amounts of faecal energy were not caused by inefficiency of microbial polysaccharide conversion, but that other factors were contributing to the efficiency of energy extraction from feed. One such factor is likely to be the SCFA-regulated excretion of incretin hormone glucagon-like peptide-1. Under conditions of low SCFA production in caecum, a low energy availability in colon stimulates the secretion of glucagon-like peptide-1, which slows the small intestinal transit and allows greater nutrient absorption, resulting a low faecal energy content (Wichmann et al., 2013).
Many microbes are host-specific, and only a subset of human gut microorganisms is capable of colonising the GF mouse gut (Licht et al., 2007; Goodman et al., 2011). We found that at the genus level, approximately 60% of the human microbiota established in the mice (Figure 7d), and that the species richness was typically lower in the individual colonised mouse than observed in its corresponding human donor (Figure 7f). We additionally noted that the potential butyrate-producing genera that were poorly transferred from humans to mice, including Roseburia, Eubacterium, Coprococcus and Faecalibacterium, are in fact among the top 20 most abundant commensals in conventional mouse (Xiao et al., 2015), indicating a strong host-specific selection on the species/strain level. Importantly, we observed that the transplanted communities kept evolving with time, and that the highest similarity to the human microbiotas was not reached until 28 days after inoculation (Figure 7a). We speculate that particularly during the first week after inoculation, this development was partly driven by an increasing depletion of oxygen in the originally GF mouse gut, gradually allowing the anaerobic species to proliferate and establish.
In one particular case, we found that colonisation with the microbiota of a normal weight, but outlier insulin resistant donor (donor 13, Figure 1) correlated to the most detrimental metabolic syndrome parameters observed in the given isolator to the specific transplanted mouse (Figures 2k–n and Supplementary Figures S2d–e). If the microbiota from donor 13 was indeed causal for the high insulin resistance in the transplanted mouse, it is noteworthy that this microbiota did not drive the insulin resistance level of the whole isolator to a higher level than other isolators. Nevertheless, the microbial spread within that isolator was reflected in a unique microbial composition (Figures 3c and e) and activity (Figure 5a). We therefore speculate that specific microbe(s) may have been related to insulin resistance in donor 13, but were not transferred to other mice in the isolator.
Considerable care should be taken when extrapolating data from animal experiments to the human physiology. However, if we assume that specific microbes are capable of elevating insulin resistance upon human intestinal colonisation, either alone or in collaboration with the community that they encounter, and if some of these microbes are spread through common inter-individual interaction, this has major consequences for our understanding of insulin resistance as a non-transmissible feature.
References
Benjamini Y, Hochberg Y . (1995). Controlling the false discovery rate - a practical and powerful approach to multiple testing. J R Stat Soc Ser B 57: 289–300.
Bercik P, Denou E, Collins J, Jackson W, Lu J, Jury J et al. (2011). The intestinal microbiota affect central levels of brain-derived neurotropic factor and behavior in mice. Gastroenterology 141: 599–609; 609.e1–3.
Bornside GH, Donovan WE, Myers MB . (1976). Intracolonic tensions of oxygen and carbon dioxide in germfree, conventional, and gnotobiotic rats. Proc Soc Exp Biol Med 151: 437–441.
Bäckhed F, Ding H, Wang T, Hooper L V, Koh GY, Nagy A et al. (2004). The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA 101: 15718–15723.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336.
Davila A-M, Blachier F, Gotteland M, Andriamihaja M, Benetti P-H, Sanz Y et al. (2013). Intestinal luminal nitrogen metabolism: role of the gut microbiota and consequences for the host. Pharmacol Res 68: 95–107.
Dawson PA, Lan T, Rao A . (2009). Bile acid transporters. J Lipid Res 50: 2340–2357.
Edgar RC . (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10: 996–998.
Eeckhaut V, Van Immerseel F, Teirlynck E, Pasmans F, Fievez V, Snauwaert C et al. (2008). Butyricicoccus pullicaecorum gen. nov., sp. nov., an anaerobic, butyrate-producing bacterium isolated from the caecal content of a broiler chicken. Int J Syst Evol Microbiol 58: 2799–2802.
El Aidy S, Derrien M, Merrifield CA, Levenez F, Doré J, Boekschoten M V et al. (2013). Gut bacteria-host metabolic interplay during conventionalisation of the mouse germfree colon. ISME J 7: 743–755.
El Aidy S, van Baarlen P, Derrien M, Lindenbergh-Kortleve DJ, Hooiveld G, Levenez F et al. (2012). Temporal and spatial interplay of microbiota and intestinal mucosa drive establishment of immune homeostasis in conventionalized mice. Mucosal Immunol 5: 567–579.
El Aidy S, van den Bogert B, Kleerebezem M . (2015). The small intestine microbiota, nutritional modulation and relevance for health. Curr Opin Biotechnol 32: 14–20.
Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB et al. (2013). Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci USA 110: 9066–9071.
Fonvig CE, Chabanova E, Andersson EA, Ohrt JD, Pedersen O, Hansen T et al. (2015). 1H-MRS measured ectopic fat in liver and muscle in Danish lean and obese children and adolescents. PLoS One 10: e0135018.
Friedman J, Alm EJ . (2012). Inferring correlation networks from genomic survey data. PLoS Comput Biol 8: e1002687.
Goodman AL, Kallstrom G, Faith JJ, Reyes A, Moore A, Dantas G et al. (2011). Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc Natl Acad Sci USA 108: 6252–6257.
Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R et al. (2014). Human genetics shape the gut microbiome. Cell 159: 789–799.
Iino T, Mori K, Tanaka K, Suzuki K, Harayama S . (2007). Oscillibacter valericigenes gen. nov., sp. nov., a valerate-producing anaerobic bacterium isolated from the alimentary canal of a Japanese corbicula clam. Int J Syst Evol Microbiol 57: 1840–1845.
Johansson ME V, Jakobsson HE, Holmén-Larsson J, Schütte A, Ermund A, Rodríguez-Piñeiro AM et al. (2015). Normalization of host intestinal mucus layers requires long-term microbial colonization. Cell Host Microbe 18: 582–592.
Julius U . (2003). Influence of plasma free fatty acids on lipoprotein synthesis and diabetic dyslipidemia. Exp Clin Endocrinol Diabetes 111: 246–250.
Jung C, Hugot J-P, Barreau F . (2010). Peyer’s patches: the immune sensors of the intestine. Int J Inflam 2010: 823710.
Kahn SE, Hull RL, Utzschneider KM . (2006). Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444: 840–846.
Karpe F, Dickmann JR, Frayn KN . (2011). Fatty acids, obesity, and insulin resistance: time for a reevaluation. Diabetes 60: 2441–2449.
Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G et al. (2013). Richness of human gut microbiome correlates with metabolic markers. Nature 500: 541–546.
Lee G-H, Kumar S, Lee J-H, Chang D-H, Kim D-S, Choi S-H et al. (2012). Genome sequence of Oscillibacter ruminantium strain GH1, isolated from rumen of Korean native cattle. J Bacteriol 194: 6362.
Lee G-H, Rhee M-S, Chang D-H, Lee J, Kim S, Yoon MH et al. (2013). Oscillibacter ruminantium sp. nov., isolated from the rumen of Korean native cattle. Int J Syst Evol Microbiol 63: 1942–1946.
Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B . (2009). Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev 89: 147–191.
Licht TR, Madsen B, Wilcks A . (2007). Selection of bacteria originating from a human intestinal microbiota in the gut of previously germ-free rats. FEMS Microbiol Lett 277: 205–209.
Louis P, Flint HJ . (2009). Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol Lett 294: 1–8.
Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R . (2012). Diversity, stability and resilience of the human gut microbiota. Nature 489: 220–230.
Macfarlane S, Macfarlane GT . (2003). Regulation of short-chain fatty acid production. Proc Nutr Soc 62: 67–72.
Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward D V et al. (2012). Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol 13: R79.
Murphy EF, Cotter PD, Healy S, Marques TM, O’Sullivan O, Fouhy F et al. (2010). Composition and energy harvesting capacity of the gut microbiota: relationship to diet, obesity and time in mouse models. Gut 59: 1635–1642.
Müller VM, Zietek T, Rohm F, Fiamoncini J, Lagkouvardos I, Haller D et al. (2015). Gut barrier impairment by high-fat diet in mice depends on housing conditions. Mol Nutr Food Res 60: 897–908.
Natividad JM, Pinto-Sanchez MI, Galipeau HJ, Jury J, Jordana M, Reinisch W et al. (2015). Ecobiotherapy rich in firmicutes decreases susceptibility to colitis in a humanized gnotobiotic mouse model. Inflamm Bowel Dis 21: 1883–1893.
Nejrup RG, Bahl MI, Vigsnæs LK, Heerup C, Licht TR, Hellgren LI . (2015). Lipid hydrolysis products affect the composition of infant gut microbial communities in vitro. Br J Nutr 114: 63–74.
Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB et al. (2015). vegan: Community Ecology Package.
Paillard D, McKain N, Chaudhary LC, Walker ND, Pizette F, Koppova I et al. (2007). Relation between phylogenetic position, lipid metabolism and butyrate production by different Butyrivibrio-like bacteria from the rumen. Antonie Van Leeuwenhoek 91: 417–422.
Pryde SE, Duncan SH, Hold GL, Stewart CS, Flint HJ . (2002). The microbiology of butyrate formation in the human colon. FEMS Microbiol Lett 217: 133–139.
Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL et al. (2013). Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341: 1241214.
Ridlon JM, Kang D-J, Hylemon PB . (2006). Bile salt biotransformations by human intestinal bacteria. J Lipid Res 47: 241–259.
Rode LM, Genthner BR, Bryant MP . (1981). Syntrophic association by cocultures of the methanol- and CO(2)-H(2)-utilizing Species Eubacterium limosum and pectin-fermenting Lachnospira multiparus during growth in a pectin medium. Appl Environ Microbiol 42: 20–22.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504.
Smith EA, Macfarlane GT . (1997). Dissimilatory amino acid metabolism in human colonic bacteria. Anaerobe 3: 327–337.
Tremaroli V, Bäckhed F . (2012). Functional interactions between the gut microbiota and host metabolism. Nature 489: 242–249.
Tulstrup MV-L, Christensen EG, Carvalho V, Linninge C, Ahrné S, Højberg O et al. (2015). Antibiotic treatment affects intestinal permeability and gut microbial composition in Wistar rats dependent on antibiotic class. PLoS One 10: e0144854.
Turnbaugh PJ, Bäckhed F, Fulton L, Gordon JI . (2008). Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 3: 213–223.
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI . (2006). An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444: 1027–1031.
Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI . (2009). The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med 1: 6ra14.
Ussar S, Griffin NW, Bezy O, Fujisaka S, Vienberg S, Softic S et al. (2015). Interactions between gut microbiota, host genetics and diet modulate the predisposition to obesity and metabolic syndrome. Cell Metab 22: 516–530.
van der Waaij D, Berghuis-de Vries JM, Lekkerkerk-van der Wees JEC . (1971). Colonization resistance of the digestive tract in conventional and antibiotic-treated mice. J Hyg (Lond) 69: 405–411.
Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S et al. (2010). Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 328: 228–231.
Wang Q, Garrity GM, Tiedje JM, Cole JR . (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73: 5261–5267.
Wichmann A, Allahyar A, Greiner TU, Plovier H, Lundén GÖ, Larsson T et al. (2013). Microbial modulation of energy availability in the colon regulates intestinal transit. Cell Host Microbe 14: 582–590.
Wos-Oxley M, Bleich A, Oxley APA, Kahl S, Janus LM, Smoczek A et al. (2012). Comparative evaluation of establishing a human gut microbial community within rodent models. Gut Microbes 3: 234–249.
Xiao L, Feng Q, Liang S, Sonne SB, Xia Z, Qiu X et al. (2015). A catalog of the mouse gut metagenome. Nat Biotechnol 33: 1103–1108.
Acknowledgements
This work was funded by the Danish Council for Independent Research (Mobility Grant No. 09-067572) and by the Innovation Fund Denmark (Grants Nos 11-116163, 0603-00457B and 0603-00484B). The Novo Nordisk Foundation Center for Basic Metabolic Research is an independent Research Center at the University of Copenhagen partially funded by an unrestricted donation from the Novo Nordisk Foundation. We thank Anne-Marie Ørngreen, Maja Danielsen, Kenneth Worm, Bodil Madsen, Kate Vibefeldt, Jannie Agersten, Lisbeth Rosholm and Thor Láadal for excellent technical assistance, as well as Monica Tulstrup and Louise Vigsnæs for the kind help with mouse dissection. Additionally, Linfei Zhou is acknowledged for help with data analysis.
Author contributions
TRL, MIB, TH, J-CH and LZ conceived and designed the study. LZ, HMR, CEF and HLF performed the experiments. LZ, HMR and TRL wrote the paper. TH, J-CH, OP and LIH contributed with sample material and techniques. All authors participated in analysis and interpretation of the data, and commented on the manuscript.
Data deposition
The microbial DNA sequences encoding bacterial 16 S rRNA V3 region reported in this paper have been deposited in the Sequence Read Archive (SRA) under the accession number SRP071872.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Zhang, L., Bahl, M., Roager, H. et al. Environmental spread of microbes impacts the development of metabolic phenotypes in mice transplanted with microbial communities from humans. ISME J 11, 676–690 (2017). https://doi.org/10.1038/ismej.2016.151
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2016.151
This article is cited by
-
Current Trends and Challenges of Microbiome Research in Bladder Cancer
Current Oncology Reports (2024)
-
Candida expansion in the gut of lung cancer patients associates with an ecological signature that supports growth under dysbiotic conditions
Nature Communications (2023)
-
Stool energy density is positively correlated to intestinal transit time and related to microbial enterotypes
Microbiome (2022)
-
From germ-free to wild: modulating microbiome complexity to understand mucosal immunology
Mucosal Immunology (2022)
-
Antibiotics create a shift from mutualism to competition in human gut communities with a longer-lasting impact on fungi than bacteria
Microbiome (2020)
