
Abstract
Dysbiosis resulting in gut-microbiome alterations with reduced butyrate production are thought to disrupt intestinal immune homeostasis and promote complex immune disorders. However, whether and how dysbiosis develops before the onset of overt pathology remains poorly defined. Interleukin-15 (IL-15) is upregulated in distressed tissue and its overexpression is thought to predispose susceptible individuals to and have a role in the pathogenesis of celiac disease and inflammatory bowel disease (IBD). Although the immunological roles of IL-15 have been largely studied, its potential impact on the microbiota remains unexplored. Analysis of 16S ribosomal RNA-based inventories of bacterial communities in mice overexpressing IL-15 in the intestinal epithelium (villin-IL-15 transgenic (v-IL-15tg) mice) shows distinct changes in the composition of the intestinal bacteria. Although some alterations are specific to individual intestinal compartments, others are found across the ileum, cecum and feces. In particular, IL-15 overexpression restructures the composition of the microbiota with a decrease in butyrate-producing bacteria that is associated with a reduction in luminal butyrate levels across all intestinal compartments. Fecal microbiota transplant experiments of wild-type and v-IL-15tg microbiota into germ-free mice further indicate that diminishing butyrate concentration observed in the intestinal lumen of v-IL-15tg mice is the result of intrinsic alterations in the microbiota induced by IL-15. This reconfiguration of the microbiota is associated with increased susceptibility to dextran sodium sulfate-induced colitis. Altogether, this study reveals that IL-15 impacts butyrate-producing bacteria and lowers butyrate levels in the absence of overt pathology, which represent events that precede and promote intestinal inflammatory diseases.
Similar content being viewed by others


Introduction
The mammalian microbiota consists of four dominant phyla: Firmicutes, Bacteroidetes, Actinobacteria and Proteobacteria (Eckburg et al., 2005). Although these broad taxonomic groups are the dominant phyla in the mammalian gastrointestinal tract, fluctuations in their relative abundance can occur not only in response to particular endogenous or exogenous perturbations (Dethlefsen et al., 2007; Maslowski and Mackay, 2011; Honda and Littman, 2012) but also according to specific gut compartment (Mowat and Agace, 2014). In a state of dysbiosis, an increase in pathobionts and/or a decrease in symbionts can disrupt intestinal immune homeostasis (Round and Mazmanian, 2009; Hill and Artis, 2010) and set the stage for autoimmune disorders such as rheumatoid arthritis (Wu et al., 2010), type-1 diabetes (Wen et al., 2008; Mathis and Benoist, 2012), multiple sclerosis (Lee et al., 2011) and intestinal inflammatory disorders, such as inflammatory bowel disease (IBD) and celiac disease (CeD) (Round and Mazmanian, 2009; Maynard et al., 2012; Verdu et al., 2015). Furthermore, an important immune regulatory role played by the microbiota is linked to its ability to produce short chain fatty acids (SCFAs), in particular butyrate (Turnbaugh et al., 2009; Maslowski and Mackay, 2011). Bacterial butyrate production is a key function attributed to a ‘healthy’ gut microbiota (eubiosis) owing to the preferred use of this SCFA as an energy source for intestinal epithelial cells (IECs; Hamer et al., 2008; Sartor, 2010). Low levels of butyrate caused by a reduction of butyrate-producing bacteria in the gut have been implicated in IBD pathogenesis (Galvez et al., 2005; Frank et al., 2007). Furthermore, it has been shown that treatment of IBD patients with butyrate ameliorates signs of inflammation (Harig et al., 1989, Scheppach et al., 1992).
It remains unclear whether gut dysbiosis is a causative factor or a consequence of intestinal inflammatory disorders. More specifically, the precise mechanisms leading to dysbiosis and reduced levels of butyrate concentration are still being defined. Interleukin-15 (IL-15) is a cytokine that belongs to the four alpha-helix bundle family of cytokines that has proinflammatory properties and is upregulated by myeloid and non-myeloid cells under conditions of tissue stress and during infection (Waldmann and Tagaya, 1999; Fehniger and Caligiuri, 2001; Waldmann, 2006; Jabri and Abadie, 2015). It is known that IL-15 is overexpressed in the epithelium of the small bowel and colon in CeD (Abadie and Jabri, 2014), Crohn’s disease (CD) and ulcerative colitis (UC) patients (Liu et al., 2000; Leon et al., 2009). Although the role of IL-15 in immunity has been largely studied (Fehniger and Caligiuri, 2001; Waldmann, 2006; Jabri and Abadie, 2015), the impact this cytokine has on the composition or function of the microbiota has yet to be investigated. Importantly, IL-15 was reported to be upregulated in not only active CeD patients but also in the epithelium of ‘at-risk’ family members in the absence of villous atrophy (Setty et al., 2015), and in both involved and uninvolved areas of the intestine in patients with IBD (Liu et al., 2000; Leon et al., 2009). Together, this suggests dysregulated expression of IL-15 may be an early and predisposing event in the disease process before, and in absence of, intestinal tissue damage.
In this study, we explore the impact of IL-15 on the microbiota in the ileum and the colon using villin-IL-15 transgenic (v-IL-15tg) mice that, similar to patients with intestinal inflammatory disorders, overexpress IL-15 in IECs throughout the small and large intestine (Pinto et al., 1999; DePaolo et al., 2011 and Supplementary Figure S1). Importantly, these mice do not develop intestinal lesions (DePaolo et al., 2011 and Supplementary Figure S2), allowing for assessment of how IL-15 alters the microbiota in the absence of ongoing tissue damage. Overall, our study provides new insights into mechanisms that may cause dysbiosis and alter production of butyrate in the small bowel and the colon. As IL-15 is closely linked to autoimmune and inflammatory disorders, we propose that changes in IL-15 levels alter the functional properties of the gut microbiota, which in turn affect its metabolic capacity to produce host protective molecules such as butyrate. These events may predispose at-risk individuals to the development of various intestinal inflammatory disorders.
Materials and methods
Human tissue samples
Intestinal tissue samples from patients with ileitic CD or UC were identified in the University of Chicago Medicine’s Epic patient database and obtained from the University of Chicago’s Human Tissue Resource Center. The study was approved by the institutional review board of the National Cancer Institute (NCI), Bethesda, MD, USA. One representative slide from each group out of six patients per group is shown (Supplementary Figure S1).
Mice
Specific pathogen-free (SPF) v-IL-15tg mice have been previously described (DePaolo et al., 2011). To establish littermate controls, mice were bred to C57BL/6 mice in our colony and heterozygous mice were bred to homozygous transgenic mice. Mice were co-housed until weaning and then single housed before collection of samples for sequencing or analysis of butyrate in luminal contents by gas-chromatography mass spectrometry (GC-MS). For all experiments, mice analyzed were between 6 and 12 weeks of age. All mice were kept under standard conditions of temperature and light, and were fed with standard laboratory chow and water ad libitum. All mice were maintained in a SPF environment at the University of Chicago and experimental guidelines were approved by the Institutional Animal Care and Use Committee (IACUC). Germ-free (GF) WT C57BL/6 and v-IL-15tg mice were housed at the University of Chicago gnotobiotic facility under strict GF conditions. GF status was assayed weekly by aerobic and anaerobic plating and by 16S ribosomal RNA (rRNA)-based quantitative PCR (qPCR).
Immunohistochemical staining
Immunohistochemical staining was performed as previously described (Tang et al., 2015). Briefly, biopsy specimens were fixed in 10% buffered neutral formalin, paraffin embedded and sections (4 μm) were stained with anti-IL-15 antibody (Abcam, Cambridge, MA, USA). Slides were stained using Dako EnVision+ System-HRP kit (Dako, Carpinteria, CA, USA). Antibody revelation was performed with 3,3′-diaminobenzidine chromogen (Dako), and counterstain was completed with hematoxylin (Thermo Fisher Scientific, Waltham, MA, USA). Slides were evaluated on a DM 2500 microscope (Leica, Buffalo Grove, IL, USA) using an HC Plan Apochromat 20 × /0.70 lens (Leica). Images were acquired by a Retiga Exi FAST 1394 camera (QImaging, Surrey, BC, Canada), processed by Image-Pro Plus 7.0 software (Media Cybernetics, Bethesda, MD, USA) and exported in TIFF format. Slides demonstrate x10 and x20 magnification.
DSS colitis model
DSS colitis was induced in mice as described (Devkota et al., 2012). In brief, mice received dextran sodium sulfate (DSS; MW: 36.000–50.000; MP Biomedicals, Santa Ana, CA, USA) 2% (w/v) in drinking water for 7 days and were then switched to regular drinking water. Weight was recorded daily until mice were killed on day 8. Length of the colon was measured and inspected visually. Colitis severity and inflammation-associated damage was blindly scored on hematoxylin and eosin-stained colonic sections by a gastrointestinal pathologist. Areas of erosion were quantified as percentage of erosion relative to the total mucosal surface area per sample.
Sample collection, DNA extraction and qPCR
Gut contents and feces were collected and snap frozen at −80 °C. Bacterial DNA was then extracted as previously described (Wang et al., 2010). qPCR of 16S rRNA-encoding genes was performed on a Roche Lightcycler 480 (Roche Scientific, Indianapolis, IN, USA) using the primers 1132F (5′-atggYtgtcgtcagctcgtg-3′) and 1108R (5′-gggttgcgctcgttgc-3′) (Leigh et al., 2007). All reactions were carried out in LightCycler 480 96 well plates sealed with LightCycler 480 sealing foil (Roche). In brief, each 20 μl reaction contained 10 μl 2x SYBR Green PCR Master Mix (Roche Scientific), 1 μl of each primer (10 μM concentration, 0.5 μM final), 1 μl template DNA and 7 μl of Hypure Molecular Biology grade water (Thermo Fisher Scientific). Reaction conditions were as follows: 95 °C for 2 min (initial denaturation), 40 cycles at 95 °C for 5 s, 60 °C for 30 s (qPCR amplification conditions). Specific amplification of targets was quantified using dilution curves of a purified pCR4-TOPO vector (Invitrogen, Waltham, MA, USA) containing a cloned 16S rRNA-encoding gene from Escherichia coli as a standard. Standards ranging in concentration from 108 to 100 plasmid copies μl–1 were run in parallel with our samples during each qPCR run. Using those results, a standard curve was generated to quantify the number of targets within the samples.
16S rRNA amplicon library preparation and sequencing
The V4 region of the 16S rRNA-encoding gene (515F-806R) was amplified using standard protocols to survey the total bacterial community in the extracted samples (Caporaso et al., 2011, 2012). Extracted bacterial DNA template was PCR amplified in triplicate using the Earth Microbiome Project barcoded primer set adapted for Illumina Miseq (http://www.earthmicrobiome.org/emp-standard-protocols/) and sequencing was performed at the Next-Generation Sequencing Core at Argonne National Laboratory, Argonne, IL USA. Amplicons were then sequenced on a 151 bp × 12 bp × 151 bp MiSeq run using customized sequencing primers and procedures (Caporaso et al., 2012). High-quality sequence reads were then analyzed using validated analysis tools, namely QIIME and phyloseq (Caporaso et al., 2010b; McMurdie and Holmes 2013).
Sequence data analysis
Paired-end sequences were joined together and classified using QIIME (Caporaso et al., 2010b). Sequences were clustered into operational taxonomic units (OTUs) using UCLUST (Edgar, 2010) at 97% similarity (de novo OTU picking). Representative sequences were aligned via PyNAST (Caporaso et al., 2010a), a consensus taxonomy from the Greengenes reference database (McDonald et al., 2012) was assigned to each sequence using the UCLUST taxonomy assigner, and a phylogenetic tree built using FastTree (Price et al., 2010). For beta diversity calculations, any OTUs containing only a single sequence (singletons) were discarded and samples were rarefied to even sequencing depth. Beta diversity comparisons between samples were made using the weighted UniFrac algorithm (Hamady et al., 2010; Price et al., 2010, Lozupone et al., 2011). Additional analyses and statistical tests were conducted in QIIME (Caporaso et al., 2010b) and packages in R v.3.2.0 (R Core Team, 2015) including phyloseq (McMurdie and Holmes, 2013) as indicated in the text and figure legends. The data are publicly available via a project specific page in MG-RAST based at Argonne, including instant availability of the sequence data, bioinformatic analyses and tools (http://metagenomics.anl.gov/linkin.cgi?project=18895).
Measurement of SCFAs in luminal contents or feces by GC-MS
Measurement of SCFA in luminal contents and feces of mice was performed as previously described (Renom et al., 2001). In brief, fecal/luminal contents were collected, weighed, homogenized in 600 μl water and centrifuged at 13 000 × g to separate particulates from supernatant containing SCFA. In all, 500 μl of supernatant was acidified with 100 μl 50% H2SO4 and an internal standard (isobutyric acid; Sigma, St Louis, MO, USA) was added to each sample. Extraction of SCFA was performed three times; each with 500 μl of diethyl ether. In total, 1 ml of pooled diethyl ether extract was derivatized for 2–24 h with 250 μl of N-tert-butyldimethylsilyl-N-methyltrifuoroacetamide with 1% tert-butyldimethylchlorosaline (Sigma) and 1 μl was injected with a 25 μl Hamilton syringe and run on a Saturn 2000 GC-MS (Varian, Walnut Creek, CA, USA). The SCFAs were identified on chromatograms by their specific retention times. Individual calibration curves were obtained for each compound using a standard 1 m SCFA mixture (acetate, propionate and butyrate). SCFA concentrations were normalized to the weight of intestinal contents and displayed as μMol g–1 contents.
Fecal microbiota transplantation (FMT)
Fecal pellets of SPF v-IL-15tg or wild-type (WT) littermate controls were weighed, homogenized in phosphate-buffered saline by mashing the pellet with a pipet tip and vortexed vigorously for 5 min. Tubes were centrifuged at 400 × g for 5 min to pellet large debris and the supernatant was filtered through a sterile 70 μm cell strainer (Thermo Fisher Scientific) and transferred to a new tube. Tubes were again centrifuged at 400 × g for 5 min and supernatant was used for FMT. Six- to 8-week-old GF v-IL-15tg mice or GF WT mice were orally inoculated with 200 μl of either WT littermate or v-IL-15tg fecal supernatant, were housed in separate gnotobiotic isolators for the duration of the experiment and analyzed 7 and 21 days after colonization.
Histology for hematoxylin and eosin and periodic acid–Schiff staining from mouse tissue
Small and/or large intestine was flushed with cold phosphate-buffered saline followed by immediate flushing with 10% formalin. A section of ~3 cm from ileum/colon was cut with a razor blade, cut open longitudinally and pinned out with needles in wax boxes. Tissue was fixed for 24 h at room temperature and pre-embedded in 2% agar (Sigma). Hematoxylin and eosin and periodic acid–Schiff staining were performed on 5 μm paraffin-embedded intestinal sections. Slides were analyzed under a Leica DM 2500 microscope with a HC PLAN APO 320/0.7 NA and a HCX PL APO 3100/1.40–0.70 objectives.
Analysis of IL-15tg gene expression of fluorescence-associated cell sorting cells
To determine the mRNA expression of IL-15 in the epithelium of SPF and GF mice, epithelial cells of the small intestine were isolated as described in (DePaolo et al., 2011). The cell suspension was stained for 20 min with fluorochrome-labeled cell-surface antibodies for CD45 (eBioscience, San Diego, CA, USA) and EpCAM (eBioscience). In all, 5x105–5x106 CD45−EpCAM+ (epithelial cells) cells were sorted using the FacsAria (BD Biosciences, San Jose, CA, USA). IL-15 mRNA expression of CD45−EpCAM+ cells was determined by RT-PCR and normalized relative to the expression of GAPDH as described (Wachowicz et al., 2014). Primer sequence: mouse IL-15 primer pair: mIL-15R: 5′-TCTCCTCCAGCTCCTCACAT-3′; mIL-15F: 5′-TGCAATGAACTGCTTTCTCC-3′.
Statistical analysis
Unless otherwise stated, unpaired Student’s t-test was performed with Prism (Graph Pad, La Jolla, CA, USA); *P<0.05, **P<0.01, ***P<0.001.
Results
Overexpression of IL-15 in the intestinal epithelium increases susceptibility to colitis
We generated v-IL-15tg mice (DePaolo et al., 2011) that mimic IL-15 overexpression in IECs found in patients with UC and ileitic CD (Liu et al., 2000; Leon et al., 2009) (Supplementary Figure 1), to determine whether dysregulated expression of IL-15 could set the stage for IBD by promoting the development of intestinal dysbiosis. We first assessed that the overexpression of IL-15 in IECs was indeed associated with increased susceptibility to colitis using a classical model of chemically induced colitis (Chassaing et al., 2014). It was previously reported that IL-15 knockout mice were protected against DSS-induced colitis (Yoshihara et al., 2006). Strikingly, DSS-treated v-IL-15tg mice exhibited a significant increase in weight loss (Figure 1a) and a more marked reduction in colonic length (Figure 1b) when compared with DSS-treated WT littermate control mice at day 8 of the experimental period. In stark contrast to DSS-treated WT littermate controls, v-IL-15tg mice showed a more severe histopathology in the colon, accompanying massive inflammation (Figure 1c). Histological scores also revealed a significantly higher percentage of areas of erosion in colons of DSS-treated v-IL-15tg mice (Figure 1d), which was associated with a significantly lower concentration of butyrate in luminal contents of the cecum (Figure 1e).

Overexpression of IL-15 in the intestinal epithelium promotes colitis. WT littermate control and v-IL-15tg mice were either control treated or received 2% DSS into drinking water for 7 days and were then switched to water until mice were killed at day 8. (a) Time course of percent body weight changes. (b) Analysis of colon length at day 8 (left panel) and representative picture of colon (right panel) from DSS-treated WT littermate control and v-IL-15tg mice. (c) Histology (hematoxylin and eosin-stained (H&E), upper panel; periodic acid–Schiff (PAS) staining lower panel) of colon from control and DSS-treated mice. (d) Areas of erosion were quantified by a gastrointestinal pathologist and displayed as percentage of erosion relative to the total mucosal surface area. Data are from two independent experiments (WT (n=5), v-IL-15tg (n=7)). (e) Levels of butyrate were measured by GC-MS in cecal contents of DSS-treated v-IL-15tg mice or WT littermate controls and are displayed as μM per g luminal contents. (a–c) (WT water (n=4), v-IL-15tg water (n=2), WT DSS (n=8), v-IL-15tg DSS (n=10). (a–d) Data are from two independent experiments. Error bars, mean±s.e.m.; *P<0.05; **P<0.01; ***P<0.001.
Taken together, these data demonstrate that similar to a suggested role for IL-15 in the development of IBD in humans, v-IL-15tg mice are significantly more prone to DSS-induced colitis.
Epithelial overexpression of IL-15 alters the composition of major gut bacterial phyla in all intestinal compartments
Steady-state v-IL-15tg mice exhibit normal intestinal histology (Supplementary Figure S2), allowing us to dissect the impact of IL-15 on the microbiota in the absence of confounding factors such as tissue damage and inflammation. An emphasis has been placed on profiling the fecal microbiome as a proxy for colonic bacterial communities; however, there is an increasing recognition that the microbiota in the small bowel also has an important role in health and disease (Verdu et al., 2015). Furthermore, IBDs of the colon and the small bowel have common, but also distinct, pathological features (Cuthbert et al., 2002; Abraham and Cho 2009; Khor et al., 2011). In addition, there may be compartment-specific changes in bacterial populations in response to IL-15, as differences between the bacterial communities of the small and large bowel have been shown to parallel not only marked differences in IECs (Campbell et al., 1994) but various immune cell populations as well (Abadie et al., 2012; Maynard et al., 2012).
Bacterial communities were collected from the feces, cecum and ileal lumen of v-IL-15tg and WT littermate controls, and analyzed using non-cultivation-based approaches across and between gut compartments. In order to describe the compositional and structural differences in these location-specific microbial communities, we generated amplicon libraries targeting the 16S rRNA-encoding gene that were then sequenced using the Illumina MiSeq platform (Caporaso et al., 2012). Inspection of the taxonomic composition of the bacterial communities showed, as expected, dominance of the Bacteroidetes and Firmicutes phyla (Figure 2a), regardless of genotype or intestinal compartment (Ley et al., 2008). In addition, the proportion of Firmicutes successively increased in both sets of mice going from the feces to the cecum to the ileum (WT littermate—feces: 20.35%, cecum: 40.89%, ileum: 94.24%; v-IL-15tg—feces: 21.05%, cecum: 40.03%, ileum: 73.14%). When principal coordinates analysis based on weighted UniFrac was performed on the samples according to anatomical site (feces, cecum or ileum), we found that there were statistical differences using permutational multivariate analyses of variance (PERMANOVA; significance based on P⩽0.05) between the bacterial communities of WT and littermate v-IL-15tg mice, with the most pronounced significance (P=0.001) in the ileum (feces: pseudo-F=4.8213, P=0.005; cecum: pseudo-F=2.3784, P=0.050; ileum: pseudo-F=6.5427, P=0.001; Figure 2b). Statistically supported differences were found for relative proportions of major gut bacterial phyla between the two groups of mice in specific sites; Bacteroidetes (false discovery rate (FDR)<0.05), Firmicutes (FDR<0.01) and Proteobacteria (FDR<0.05) in the cecum, and Actinobacteria (FDR<0.01), Bacteroidetes (FDR<0.05) and Tenericutes (FDR<0.05) in the ileum (see Supplementary Table S1 (phylum composition across compartments)). Significance was based on tagwise tests using the exact negative binomial test (Robinson and Smyth, 2008) with a FDR (Benjamini–Hochberg) significance threshold of 0.05 (Benjamini and Hochberg 1995; Bergstrom et al., 2012). An overall decrease of the Bacteroidetes was observed in the feces and cecum of v-IL-15tg mice (from 77.0% to 68.1% and 54.4% to 52.0% (FDR<0.05), respectively), characterized by a decrease of the S24-7 family (from 62.1% to 46.2% and 40.2% to 36.6%, respectively). Despite the decrease of the S24-7 family in v-IL-15tg mice, the Bacteroidaceae family expanded in both of these compartments (from 1.36% to 3.99% (FDR<0.01) and 2.00% to 6.85% (FDR<0.05), respectively) but not in the ileum of v-IL-15tg mice. Conversely, in the luminal contents derived from the ileum of v-IL-15tg mice, a significant increase of Bacteroidetes was observed, when compared with WT littermate controls (16.2% compared with 2.64% (FDR<0.05)); Figure 2a; Supplementary Table S1). In particular, the S24-7 family significantly expanded in the ileum (from 2.48% to 13.6% (FDR<0.05); Supplementary Table S2) of v-IL-15tg mice, mimicking the prominence of this family in the lower gastrointestinal tract. Furthermore, members of the Firmicutes dominated the ileal communities in both the WT littermate and v-IL-15tg mice as expected, however, with large contrasting shifts in the proportion of members of the Lactobacillaceae (orange, cross-hatch) and Clostridiaceae (orange, solid) between genotypes. In the WT ileum, the Clostridiaceae comprised 65.3% of the community compared with 23.0% in v-IL-15tg mice, whereas the Lactobacillaceae were dominant in v-IL-15tg ileum (43.4% compared with 20.8%) in WT littermates. Differences in the Firmicutes within both the cecum and feces were much less pronounced (see Supplementary Table S2).

Dysregulated expression of IL-15 in the epithelium induces dysbiosis across intestinal compartments. (a) Bacterial community composition is organized by site (feces, cecum and ileum) and mouse genotype (side-by-side columns). The phyla identified in the samples are according to the GreenGenes taxonomy. The number of samples (n) per intestinal compartment-genotype is noted at the top. Percent composition is the average according to n (see main text and Supplementary Table S1 for statistical support). (b) Principal coordinates analysis (PCoA) based on the weighted UniFrac distance between the bacterial communities from samples according to host genotype (WT (gray squares) and v-IL-15tg mice (open circles)) by site (feces, cecum or ileum). Distinctions between genotype were considered significant by statistical analyses of the underlying UniFrac distances using PERMANOVA at P⩽0.05 based on 999 permutations. (a, b) Data are from two independent experiments; WT: feces (n=9); cecum (n=10), ileum (n=7); v-IL-15tg: feces (n=7), cecum (n=6), ileum (n=6).
To further assess how IL-15 expression impacts the composition of the microbiota among specific intestinal compartments, we performed principal coordinates analysis based on the weighted UniFrac distance between the bacterial communities from the feces and different intestinal compartments (cecum and ileum) within each host genotype. The microbiome profile of WT littermate mice exhibited clear demarcations between the three sites, and the ileum could be clearly separated from the feces and cecum along the first principal coordinates axis (PC1) (Figure 3a). These distinctions were significant as demonstrated by statistical analyses using PERMANOVA of the weighted UniFrac distances used to drive the principal coordinates analyses (WT littermate: pseudo-F=45.8072, P=0.001; based on 999 permutations). Interestingly, in v-IL-15tg mice, although the three sites were also clearly delineated (v-IL-15tg: pseudo-F=9.7823, P=0.001; based on 999 permutations), the samples of the ileum did not cluster as tightly as in WT littermate control mice. Furthermore, two-sample t-tests of the weighted UniFrac distance of the WT littermate ileal, fecal and cecal samples revealed that there was a significant difference between ileal and fecal/cecal samples in WT (P<0.001, Bonferroni-corrected in both cases), but not v-IL-15tg mice, suggesting that the ileal microbiota of v-IL-15tg mice acquired colonic-like features (Figure 3a). This latter point is further suggested by the expansion of the Bacteroidetes (specifically the S24-7 family) in the ileum of v-IL-15tg mice noted earlier that dominate in both, the cecum and feces of WT littermate mice (Figure 2a).

Overexpression of IL-15 in the epithelium mediates compartment-specific changes in bacterial community diversity. (a) Principal coordinates analysis (PCoA) based on the weighted UniFrac distance between the bacterial communities from each site (feces (blue circles), cecum (red squares) and ileum (orange triangles)) within each host genotype (WT littermate and v-IL-15tg mice). (b) Boxplots of measure of alpha diversity (Shannon) of bacterial community diversity within each site (feces, cecum, and ileum) according to mouse genotype (WT littermate or v-IL-15tg). (a, b) Data are from two independent experiments; WT: feces (n=9); cecum (n=10), ileum (n=7); v-IL-15tg: feces (n=7), cecum (n=6), ileum (n=6). Significant differences between genotype are noted based on Mann–Whitney U-test at *P<0.05; *P<0.01. The color reproduction of this figure is available at the The ISME Journal online.
Intestinal inflammatory disorders are classically associated with a decrease in alpha diversity (Kostic et al., 2014). To investigate the impact of IL-15 on overall microbial community structure within compartments, we compared the alpha diversity derived from the underlying distribution and abundances of OTUs based on the Shannon index between host genotypes in the feces and intestinal compartments (Figure 3b). As expected, differences were observed between intestinal compartments in each mouse genotype marked by an overall decrease in OTU number and diversity in the ileum relative to the cecum and feces. Interestingly, although we observed a significant decrease in alpha diversity for the v-IL-15tg fecal community (P<0.01 (0.0036); based on Mann–Whitney U-test), a significant increase in diversity was observed in the v-IL-15tg ileal community compared with WT littermate mice (P<0.05 (0.0101); based on Mann–Whitney U-test). The increased microbial diversity observed in the ileum of v-IL-15tg mice is in accordance with the principal coordinates analysis-based observation of a decreased distinction between the ileum and feces. These differences were independent of changes in microbial biomass between genotypes, as no significant differences were detected by qPCR of 16S rRNA-encoding genes (Supplementary Figure S3).
Together, these data indicate that overexpression of IL-15 in IECs has an impact on the composition and structure of the microbial community. Although some of these effects were observed across all compartments, others differed depending on the intestinal compartment in which IL-15 was overexpressed. Interestingly, overexpression of IL-15 in the ileum led to changes in bacterial community composition that result in features similar to that of the cecum in WT animals.
IL-15 promotes changes within the Bacteroidetes and abundance of butyrate-producing bacteria
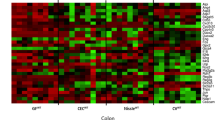
To further assess the impact of IL-15 on the microbiota, we investigated changes in specific bacterial populations (OTUs) between WT littermate and v-IL-15tg mice (Figure 4). More specifically, we characterized changes in specific OTUs between host genotype in the feces and gut compartments using phyloseq and DESeq2 (McMurdie and Holmes, 2013; Love et al., 2014), and plotted changes in significant OTUs as a series of heatmap (log2-fold changes based on a FDR significance threshold of 0.05; Benjamini and Hochberg, 1995). Representative sequences for each of these OTUs were then used to search the Ribosomal Database Project’s set of quality-controlled, aligned and annotated bacterial 16S rRNA sequences using SeqMatch (Cole et al., 2014) in order to obtain nearest neighbor matches with cultured representatives at the genus-species level. Using this approach, we found that a large fraction of OTUs that changed significantly between WT littermates and v-IL-15tg mice across all compartments were members of the Bacteroidetes.

Analysis of OTUs reveals that IL-15 overexpression influences the presence and abundance of butyrate-producing bacteria. The figure depicts significant log2-fold changes (based on a FDR (Benjamini–Hochberg) significance threshold of 0.05; Benjamini and Hochberg, 1995) in the relative abundance of specific OTUs between host genotype (WT and v-IL-15tg) in feces, cecum and ileum as heatmaps. On the left are the phylum and family classifications of the OTUs and on the right are the putative genus/species designation based on best similarity matches from the RDP using SeqMatch (numbers in parentheses indicate SeqMatch score (Sab)). OTUs and SeqMatch genus/species assignments in bold italics indicate that the OTU had an increased log2-fold change in the v-IL-15tg mice. The panel to the right of the heatmaps indicate whether butyrate kinase (buk) or butyryl-CoA:acetate CoA-transferase (but) encoding genes have been detected in the genomes of the SeqMatch genus/species assignments based on the buk and but entries in the RDP FunGene Functional Gene Pipeline and Repository. A closed black circle indicates presence of a buk or but gene in that genome, whereas a closed gray circle indicates an alternate annotation (defined at the bottom of the panel). An open circle indicates that no match was found in the RDP FunGene. Data are from two independent experiments; WT: feces (n=9); cecum (n=10), ileum (n=7); v-IL-15tg: feces (n=7), cecum (n=6), ileum (n=6).
In the ileum of v-IL-15tg mice, OTUs classified as members of the S24-7 family solely accounted for the underlying increase in the Bacteroidetes in our data set (Figure 4, bottom panel). Although putatively assigned based on SeqMatch, the majority of these OTUs were most closely associated with the Barnesiella genus (B. intestinihominis and B. viscericola). Furthermore, we observed an increase in members of the Firmicutes in the ileum of v-IL-15tg mice, mainly the Lactobacillaceae. Only a single OTU, classified as a member of the Clostridiaceae (‘Candidatus Arthromitus’, also referring to segmented filamentous bacteria), was enriched in WT littermate controls.
In the cecum, the distribution of significantly changing OTUs was relatively uniform between Bacteroidetes and Firmicutes (Figure 4, middle panel), reflecting the overall gross phylum-level proportions described in Figure 2a and Supplementary Table S1. The majority of the OTUs classified as Bacteroidetes that significantly decreased were members of the S24-7 family, including Barnesiella instestinihominis, Barnesiella viscericola and Bacteroides instestinalis. Members of the Firmicutes classified as Clostridia (either unclassified at the family level or members of the Lachnospiraceae) were also decreased in the ceca of v-IL-15tg mice, although the overall proportion of Firmicutes was not significantly different between mouse genotypes (Figure 4, middle panel; right-hand side).
Focusing on the feces, we again observed that the majority of the significantly changing OTUs were members of the Bacteroidetes (Figure 4, top panel). We additionally observed that the change in OTU abundance was marked by a loss of OTUs in v-IL-15tg mice belonging to members of the S24-7 family and to a lesser extent a loss of members of the Rikenellaceae family (both families belong to the phylum Bacteroidetes). As observed in the ceca of v-IL-15tg mice, the nearest match for many of the S24-7-derived OTUs that decreased in the feces from v-IL-15tg mice was B. intestinihominis, whereas for the Rikenellaceae-derived OTUs they were Alistipes finegoldii (Figure 4, top panel). This matches the trend observed at the overall phylum and family level noted earlier where the Bacteroidetes, and more specifically the S24-7 family, were shown to decrease in both the feces and cecum of v-IL-15tg mice. Of note, members of the Rikenellaceae and S24-7 families (Bacteroidetes) are known to convert pyruvate to butyrate using the acetyl-CoA pathway (Buckel and Barker, 1974; Barker et al., 1982; Gerhardt et al., 2000).
As known butyrate-producing bacteria were found to be decreased in the feces and cecum of v-IL-15tg mice, and butyrate is known to have a key role in intestinal immune homeostasis and health (Sartor, 2010), results from SeqMatch were used to mine the RDP FunGene Functional Gene Pipeline and Repository databases for butyrate kinase (buk) and butyryl-CoA:acetate CoA-transferase (but), two key genes involved in butyrate production by bacteria, based on genus/species matches. This cross-referencing between the SeqMatch results and FunGene is depicted next to the heatmap as columns of definitive (black circles), putative (gray circles), or absent (open circles) annotation of buk and/or but in representative genomes. Surprisingly, roughly two-thirds (67.2%) of the significantly changing OTUs that differed between genotypes appear to be associated with differences in the presence of the buk and/or but genes (Figure 4). In contrast, there were no significant overall changes within the Firmicutes, in particular the well-characterized ‘classical’ butyrate-producing bacteria typically found within Clostridium clusters IV and XIVa (such as Clostridium scindens (XIVa), Clostridium hathewayi (XIVa), Clostridium aldenense (XIVa) and Clostridium leptum (IV)) (Pryde et al., 2002; Rainey et al., 2009). qPCR targeting buk and but genes developed primarily from these groups (Vital et al., 2013) also did not show significant differences in gene abundances between WT and v-IL-15tg mice (data not shown).
Taken together, these findings indicate that IL-15 induces changes principally in specific bacterial populations (members of the Bacteroidetes) that contain genes involved in butyrate production. In contrast, there were no significant changes in the well-characterized ‘classical’ butyrate-producing groups of the Firmicutes.
IL-15-induced dysbiosis results in reduced levels of gut butyrate concentration
Given the IL-15-dependent changes in specific bacterial groups implicated in the production of butyrate (Figure 4), we investigated whether microbiome differences translated to alterations in available butyrate concentrations at each anatomical site as measured by GC-MS. In accordance with data suggesting a decrease in butyrate-producing bacteria in the feces and cecum (Figures 2a and 4), we found that butyrate concentrations, both in the luminal contents of the cecum and the feces, were significantly decreased in v-IL-15tg mice as compared with WT littermate control mice in the same compartment (Figure 5a). Unexpectedly, a similar reduction was observed in the ileum of v-IL-15tg mice (Figure 5a). In contrast, IL-15 did not consistently impact measured luminal levels of other SCFAs including acetate (Supplementary Figure 4a) or propionate (Supplementary Figure 4b). Only propionate was detected at a significantly higher level in the cecum of v-IL-15tg mice.

IL-15-induced microbial alterations are associated with a significant reduction in luminal (ileum and cecum) and fecal butyrate concentration across intestinal compartments and a reduction of butyrate-producing bacteria. (a) Levels of butyrate were measured by GC-MS in feces or luminal contents (cecum and ileum) of v-IL-15tg mice or WT littermate controls and are displayed as μM g–1 luminal content. Data are from three independent experiments (n=7–14). (b) GF v-IL-15tg mice received fecal microbiota of either SPF WT littermate or v-IL-15tg mice. Panel depicts experimental design for FMT experiment. (c) Graph shows measured concentrations of butyrate in feces or luminal contents (cecum or ileum) from recipient mice by GC-MS at day 7 post-FMT (GF v-IL-15tg mice transplanted with WT littermate feces, n=5; GF v-IL-15tg mice transplanted with v-IL-15tg feces, n=8). (d) qPCR targeting the 16S rRNA-encoding gene was used to measure bacterial load in the feces of mice receiving FMT. No statistical difference was found between both groups. (e) The heatmap depicts significant log2-fold changes in the relative abundance of specific OTUs between recipients based on donor host genotype (WT or v-IL-15tg) in feces (for detailed description of statistics see Figure 4). As in Figure 4, phylum and family classifications of the OTUs are on the left and on the right are the putative genus/species designation based on best similarity matches from the RDP using SeqMatch. OTUs and SeqMatch genus/species assignments in bold italics indicate that the OTU had an increased log2-fold change in the recipient mice transplanted with feces from v-IL-15tg mice; GF v-IL-15tg mice transplanted with WT littermate feces, n=3; GF v-IL-15tg mice transplanted with v-IL-15tg feces, n=5. One representative analysis out of two independent experiments is shown. (a, c) Error bars, mean±s.e.m.; *P<0.05; **P<0.01.
To explore the physiological effects of this observation, we performed microbial transplants into GF v-IL-15tg mice using feces harvested from either WT littermate control or v-IL-15tg mice (Figure 5b). Of note, SPF and GF v-IL-15tg mice that were between 6 and 12 weeks of age showed a 24- and 34-fold increase in IL-15 mRNA expression in IECs as compared with their WT counterparts, respectively (Supplementary Figure 5). Analysis of luminal butyrate concentrations was performed at day 7 (Figure 5c) and day 21 (Supplementary Figure 6a) post-transplantation. Using qPCR targeting 16S rRNA-encoding genes, we verified the restoration of the bacterial load within both sets of recipients at similar levels at day 7 post-FMT (Figure 5d) and day 21 post-FMT (Supplementary Figure 6b). In general, monitoring the establishment of the FMT community of recipient mice over time (days 7 and 21 post-FMT), we observed the community composition to be increasingly similar to that of the respective donor (Supplementary Figure 6c) and recapitulated many features observed in the feces and ceca from SPF v-IL-15tg mice (Figure 2a). This included changes in the composition and abundance of members of the Bacteroidetes, particularly contraction of the S24-7 family (from 58.6% to 41.0%) and expansion of the Bacteroidaceae (from 4.67% to 9.30%) in the v-IL-15tg microbiota by day 21 post-FMT. However, we did observe proportional differences within the Firmicutes primarily characterized by an increase in the fraction of unassigned Clostridial families (from 6.06% to 18.0%), which in the SPF v-IL-15tg mice only occurred within the ileum.
Inspecting the shifts in populations at an OTU level between FMT groups and cross-referencing between SeqMatch results and FunGene, we further confirmed alterations in the proportions of ‘non-traditional’ butyrate-producing bacteria belonging to members of the S24-7 family that contain buk and/or but genes in GF v-IL-15tg mice transplanted with v-IL-15tg (but not WT) microbiota at day 7 (Figure 5e) and day 21 (Supplementary Figure 6d). Many of the nearest matches to specific organisms were similar to what was observed in SPF v-IL-15tg mice (Figure 4). At day 21 post-FMT, we also observed an increase in the abundance of specific OTUs belonging to the Proteobacteria including members of the Desulfovibrionaceae (Supplementary Figure 6d; see OTUs 2353 and 16 875) and alterations in the composition of Firmicutes, specifically expansions of the Lachnospiraceae (<0.1% to 2.56%) and, as noted earlier, unassigned Clostridial families in mice receiving v-IL-15tg microbiota. Expansions of these same groups were found in upstream compartments (cecum (Lachnospiraceae) and ileum (unassigned Clostridial families)) within the SPF v-IL-15tg model (Figure 2a).
Within the Firmicutes there were fewer instances of but and/or buk genes associated with OTUs compared with the Bacteroidetes, but when they occurred they were almost exclusively associated with those OTUs found to be more abundant within the WT FMT group (see OTUs 315, 322, 8835 in Supplementary Figure 6d). Importantly, the reduction in the proportions of ‘non-traditional’ butyrate-producing bacteria belonging to members of the S24-7 family in GF v-IL-15tg mice transplanted with v-IL-15tg microbiota was associated 7 days (Figure 5e) and 21 days (Supplementary Figure 6d) post-FMT with significantly lower levels of luminal butyrate concentration in the ileum, cecum and feces when compared with those of GF v-IL-15tg mice receiving WT microbiota. The wider spread of butyrate concentrations observed in the FMT experiments may be an indicator of the ongoing dynamic occurring in the WT donor microbiota as it establishes itself in the v-IL-15tg host (Figure 5c). Finally, the converse transplantation experiment consisting in the transplantation of WT GF mice with WT littermate control or v-IL-15tg mice feces (Supplementary Figure 7a) also showed that the butyrate concentrations found in transplanted GF WT mice were those of the respective fecal donors (Supplementary Figure 7b).
All together, these experiments suggest that IL-15 re-configures the structure of the microbiome in a way that impacts not only its function (that is, decreased butyrate concentration) but promotes a stable change that persists following fecal transplantation into GF animals. Furthermore, these results, in combination with our compartment-specific analyses, suggest that different mechanisms may be responsible for the reduction in butyrate levels in the ileum and cecum.
Discussion
Our data reveal that IL-15, an inflammatory cytokine closely linked with autoimmune and intestinal inflammatory disorders, induces dysbiosis characterized by a decrease in abundance of ‘non-classical’ butyrate-producing bacteria (especially members of the S24-7 family) and a concomitant decrease in luminal (ileal and cecal) and fecal butyrate concentrations in the absence of intestinal tissue damage. Furthermore, our data suggest that the intestinal location impacts how IL-15 alters the composition of the microbiota. Altogether, our data provide a mechanism by which dysbiosis can occur in the context of sterile inflammation and in the absence of overt intestinal pathology, and reveal an unexpected property of IL-15 that unravels a new pathway through which IL-15 could contribute to intestinal inflammatory disorders and autoimmunity.
Two alterations taking place across all intestinal compartments under conditions of IL-15 overexpression are particularly relevant. One alteration is the shift in abundance and composition of the Bacteroidetes and Firmicutes phyla. These include changes in the composition and abundance of members of the Bacteroidetes (particularly contraction of the S24-7 family in the v-IL-15tg microbiota regardless of gut compartment) and Firmicutes (decrease in the Clostridiaceae that becomes increasingly pronounced in the ileum). The second alteration is the change in abundance of butyrate-producing bacteria associated with a decrease in the concentration of butyrate, a SCFA that not only has an important role in intestinal epithelial health (Hague et al., 1997; Sartor 2010), but also in immune tolerance because of its major histone deacetylase inhibitory properties (Waldecker et al., 2008). The decrease in butyrate concentration in the cecum and feces parallels the decrease in the groups of organisms other than those (Clostridium clusters IV and XIVa) typically associated with this activity (Vital et al., 2013, 2015). However, this is not the case in the ileum, suggesting that alternative mechanisms need to be invoked, one of which is an increase in butyrate consuming bacteria. Supporting that intrinsic alterations in the microbiota have a role in the reduction in butyrate concentration is the key finding that intestinal transplantation of WT microbiota into v-IL-15tg mice leads to levels of butyrate concentration comparable to those found in WT littermate mice under SPF conditions and significantly higher than after transplantation of v-IL-15tg microbiota into v-IL-15tg mice.
The potential compartment-specific effects of IL-15 are also illustrated by the observation that the S24-7 family is significantly expanded in the ileum, but decreased in the cecum, whereas Bacteroidaceae are expanded in the cecum and feces but not the ileum of v-IL-15tg mice. Furthermore, IL-15 overexpression leads to decreased and increased alpha diversity in the cecum and ileum, respectively. The increase in diversity and expansion of the Bacteroidetes (S24-7 family) observed in the ileum is in line with the observation that in v-IL-15tg mice the microbiota of the ileum is more similar to that of the large bowel than in WT mice. These observations highlight both how dysregulated expression of a cytokine can distinctively affect the microbiota in the small intestine and cecum, as well as the importance of studying the microbiota in a location-specific manner. More specifically, our data suggest that analysis of the fecal microbiota incompletely reflects changes observed in the ileum under conditions of dysregulated IL-15 expression.
Dysbiosis is thought to have a critical role in autoimmunity and intestinal inflammatory disorders (Mazmanian et al., 2008; Maynard et al., 2012). Unresolved questions include deciphering whether dysbiosis found in patients with IBD and CeD precedes or is secondary to tissue damage, as well as uncovering what initiates dysbiosis in patients with autoimmune disorders. IL-15 is a cytokine, which is found to be upregulated in numerous autoimmune and intestinal inflammatory disorders, and is thought to have a critical role in their pathogenesis (Abadie and Jabri 2014). Our unexpected finding that IL-15 induces dysbiosis in absence of overt intestinal damage while increasing susceptibility to colitis is highly relevant given that IL-15 was shown to be upregulated in uninvolved areas of the intestine of patients with IBD (Leon et al., 2009) and in individuals with a family history of CeD but normal intestinal histology (Setty et al., 2015). As IL-15 was shown to be increased in the blood-serum of patients with type-1 diabetes (Chen et al., 2013) and dysbiosis is thought to have a role in the pathogenesis of type-1 diabetes (Pozzilli et al., 1993; Wen et al., 2008), one can pose the question as to whether dysregulated IL-15 expression in the absence of overt intestinal pathology may promote development of type-1 diabetes. Together, we hypothesize that IL-15 overexpression in the intestine could precede and precipitate both, intestinal and extraintestinal complex immune disorders, through its impact on the microbiota and in particular through its impact on butyrate-producing bacteria and luminal butyrate concentration.
In summary, our results shed an unexpected light on the contribution of IL-15 to disease by showing that in addition to its impact on immune cells (Abadie and Jabri, 2014), IL-15 promotes dysbiosis. Furthermore, our study puts forward the concept that a cytokine linked to tissue distress can lead to the reduction in the abundance of ‘non-classical’ butyrate-producing bacteria (S24-7 family) and a decrease in intestinal butyrate concentration, which is relevant given their association with IBD and potentially autoimmunity (Arpaia et al., 2013; Furusawa et al., 2013; Vaarala, 2013; Davis-Richardson and Triplett, 2015). Antibodies neutralizing IL-15 and/or blocking IL-15 signaling are being considered for the treatment of CeD and autoimmune inflammatory disorders (Baslund et al., 2005; DePaolo et al., 2011; Abadie and Jabri, 2014; Jabri and Abadie, 2015). Future studies will determine whether blocking IL-15 alters the microbiota and levels of butyrate concentration and how these alterations correlate with clinical improvement. The drivers of IL-15 upregulation in complex immune and inflammatory disorders, and the mechanisms by which IL-15 induces dysbiosis remain to be determined.
References
Abadie V, Discepolo V, Jabri B . (2012). Intraepithelial lymphocytes in celiac disease immunopathology. Semin Immunopathol 34: 551–566.
Abadie V, Jabri B . (2014). IL-15: a central regulator of celiac disease immunopathology. Immunol Rev 260: 221–234.
Abraham C, Cho JH . (2009). Inflammatory bowel disease. N Engl J Med 361: 2066–2078.
Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P et al. (2013). Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504: 451–455.
Barker H, Kahn J, Hedrick L . (1982). Pathway of lysine degradation in Fusobacterium nucleatum. J Bacteriol, 201–207.
Baslund B, Tvede N, Danneskiold-Samsoe B, Larsson P, Panayi G, Petersen J et al. (2005). Targeting interleukin-15 in patients with rheumatoid arthritis: a proof-of-concept study. Arthritis Rheum 52: 2686–2692.
Benjamini Y, Hochberg Y . (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B (Methodological), 289–300.
Bergstrom A, Licht TR, Wilcks A, Andersen JB, Schmidt LR, Gronlund HA et al. (2012). Introducing GUt low-density array (GULDA): a validated approach for qPCR-based intestinal microbial community analysis. FEMS Microbiol Lett 337: 38–47.
Buckel W, Barker HA . (1974). Two pathways of glutamate fermentation by anaerobic bacteria. J Bacteriol 117: 1248–1260.
Campbell AP, Merrett MN, Kettlewell M, Mortensen NJ, Jewell DP . (1994). Expression of colonic antigens by goblet and columnar epithelial cells in ileal pouch mucosa: their association with inflammatory change and faecal stasis. J Clin Pathol 47: 834–838.
Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R . (2010a). PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26: 266–267.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al. (2010b). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336.
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci USA 108 (Suppl 1): 4516–4522.
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6: 1621–1624.
Chassaing B, Aitken JD, Malleshappa M, Vijay-Kumar M . (2014). Dextran sulfate sodium (DSS)-induced colitis in mice. Curr Protoc Immunol 104: Unit 15: 25.
Chen J, Feigenbaum L, Awasthi P, Butcher DO, Anver MR, Golubeva YG et al. (2013). Insulin-dependent diabetes induced by pancreatic beta cell expression of IL-15 and IL-15Ralpha. Proc Natl Acad Sci USA 110: 13534–13539.
Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y et al. (2014). Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res 42: D633–D642.
Cuthbert AP, Fisher SA, Mirza MM, King K, Hampe J, Croucher PJ et al. (2002). The contribution of NOD2 gene mutations to the risk and site of disease in inflammatory bowel disease. Gastroenterology 122: 867–874.
Davis-Richardson AG, Triplett EW . (2015). A model for the role of gut bacteria in the development of autoimmunity for type 1 diabetes. Diabetologia 58: 1386–1393.
DePaolo RW, Abadie V, Tang F, Fehlner-Peach H, Hall JA, Wang W et al. (2011). Co-adjuvant effects of retinoic acid and IL-15 induce inflammatory immunity to dietary antigens. Nature 471: 220–224.
Dethlefsen L, McFall-Ngai M, Relman DA . (2007). An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature 449: 811–818.
Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A et al. (2012). Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature 487: 104–108.
Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M et al. (2005). Diversity of the human intestinal microbial flora. Science 308: 1635–1638.
Edgar RC . (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460–2461.
Fehniger TA, Caligiuri MA . (2001). Interleukin 15: biology and relevance to human disease. Blood 97: 14–32.
Frank DN St, Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR . (2007). Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA 104: 13780–13785.
Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D et al. (2013). Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504: 446–450.
Galvez J, Rodriguez-Cabezas ME, Zarzuelo A . (2005). Effects of dietary fiber on inflammatory bowel disease. Mol Nutr Food Res 49: 601–608.
Gerhardt A, Cinkaya I, Linder D, Huisman G, Buckel W . (2000). Fermentation of 4-aminobutyrate by Clostridium aminobutyricum: cloning of two genes involved in the formation and dehydration of 4-hydroxybutyryl-CoA. Arch Microbiol 174: 189–199.
Hague A, Singh B, Paraskeva C . (1997). Butyrate acts as a survival factor for colonic epithelial cells: further fuel for the in vivo versus in vitro debate. Gastroenterology 112: 1036–1040.
Hamady M, Lozupone C, Knight R . (2010). Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J 4: 17–27.
Hamer HM, Jonkers D, Venema K, Vanhoutvin S, Troost FJ, Brummer RJ . (2008). Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther 27: 104–119.
Harig JM, Soergel KH, Komorowski RA, Wood CM . (1989). Treatment of diversion colitis with short-chain-fatty acid irrigation. N Engl J Med 320: 23–28.
Hill DA, Artis D . (2010). Intestinal bacteria and the regulation of immune cell homeostasis. Annu Rev Immunol 28: 623–667.
Honda K, Littman DR . (2012). The microbiome in infectious disease and inflammation. Annu Rev Immunol 30: 759–795.
Jabri B, Abadie V . (2015). IL-15 functions as a danger signal to regulate tissue-resident T cells and tissue destruction. Nat Rev Immunol 15: 771–783.
Khor B, Gardet A, Xavier RJ . (2011). Genetics and pathogenesis of inflammatory bowel disease. Nature 474: 307–317.
Kostic AD, Xavier RJ, Gevers D . (2014). The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology 146: 1489–1499.
Lee YK, Menezes JS, Umesaki Y, Mazmanian SK . (2011). Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA 108 (Suppl 1): 4615–4622.
Leigh MB, Pellizari VH, Uhlik O, Sutka R, Rodrigues J, Ostrom NE et al. (2007). Biphenyl-utilizing bacteria and their functional genes in a pine root zone contaminated with polychlorinated biphenyls (PCBs). ISME J 1: 134–148.
Leon AJ, Gomez E, Garrote JA, Bernardo D, Barrera A, Marcos JL et al. (2009). High levels of proinflammatory cytokines, but not markers of tissue injury, in unaffected intestinal areas from patients with IBD. Mediators Inflamm 2009: 580450.
Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS et al. (2008). Evolution of mammals and their gut microbes. Science 320: 1647–1651.
Liu Z, Geboes K, Colpaert S, D'Haens GR, Rutgeerts P, Ceuppens JL . (2000). IL-15 is highly expressed in inflammatory bowel disease and regulates local T cell-dependent cytokine production. J Immunol 164: 3608–3615.
Love MI, Huber W, Anders S . (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550.
Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R . (2011). UniFrac: an effective distance metric for microbial community comparison. ISME J 5: 169–172.
Maslowski KM, Mackay CR . (2011). Diet, gut microbiota and immune responses. Nat Immunol 12: 5–9.
Mathis D, Benoist C . (2012). The influence of the microbiota on type-1 diabetes: on the threshold of a leap forward in our understanding. Immunol Rev 245: 239–249.
Maynard CL, Elson CO, Hatton RD, Weaver CT . (2012). Reciprocal interactions of the intestinal microbiota and immune system. Nature 489: 231–241.
Mazmanian SK, Round JL, Kasper DL . (2008). A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 453: 620–625.
McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A et al. (2012). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6: 610–618.
McMurdie PJ, Holmes S . (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PloS One 8: e61217.
Mowat AM, Agace WW . (2014). Regional specialization within the intestinal immune system. Nat Rev Immunol 14: 667–685.
Pinto D, Robine S, Jaisser F, El Marjou FE, Louvard D . (1999). Regulatory sequences of the mouse villin gene that efficiently drive transgenic expression in immature and differentiated epithelial cells of small and large intestines. J Biol Chem 274: 6476–6482.
Pozzilli P, Signore A, Williams AJ, Beales PE . (1993). NOD mouse colonies around the world—recent facts and figures. Immunol Today 14: 193–196.
Price MN, Dehal PS, Arkin AP . (2010). FastTree 2—approximately maximum-likelihood trees for large alignments. PloS One 5: e9490.
Pryde SE, Duncan SH, Hold GL, Stewart CS, Flint HJ . (2002). The microbiology of butyrate formation in the human colon. FEMS Microbiol Lett 217: 133–139.
R Core Team. (2015) R: A language and environment for statistical computing. R Foundation for Statistical Computing: Vienna, Austria, Available at: https://www.R-project.org/.
Rainey FA, Hollen BJ, Small A, Genus I . Clostridium. In: De Vos P, Garrity GM, Jones D, Krieg NR, Ludwig W, Rainey FA, Schleifer K-H, Whitman WB (eds) (2009) Bergey’s manual of systematic bacteriology. 2. The Firmicutes. Springer: New York, NY, USA, pp 738–830.
Renom G, Bulois P, Hafraoui S, Colombel JF, Degand PM . (2001). Simple gas chromatography analysis of faecal butyrate: application to patients at risk of pouchitis. Clin Chem Lab Med 39: 15–19.
Robinson MD, Smyth GK . (2008). Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics 9: 321–332.
Round JL, Mazmanian SK . (2009). The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol 9: 313–323.
Sartor RB . (2010). Genetics and environmental interactions shape the intestinal microbiome to promote inflammatory bowel disease versus mucosal homeostasis. Gastroenterology 139: 1816–1819.
Scheppach W, Sommer H, Kirchner T, Paganelli GM, Bartram P, Christl S et al. (1992). Effect of butyrate enemas on the colonic mucosa in distal ulcerative colitis. Gastroenterology 103: 51–56.
Setty M, Discepolo V, Abadie V, Kamhawi S, Mayassi T, Kent A et al. (2015). Distinct and synergistic contributions of epithelial stress and adaptive immunity to functions of intraepithelial killer cells and active celiac disease. Gastroenterology 149: 681–691 e610.
Tang F, Sally B, Lesko K, Discepolo V, Abadie V, Ciszewski C et al. (2015). Cysteinyl leukotrienes mediate lymphokine killer activity induced by NKG2D and IL-15 in cytotoxic T cells during celiac disease. J Exp Med 212: 1487–1495.
Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI . (2009). The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med 1: 6ra14.
Vaarala O . (2013). Human intestinal microbiota and type 1 diabetes. Curr Diab Rep 13: 601–607.
Verdu EF, Galipeau HJ, Jabri B . (2015). Novel players in coeliac disease pathogenesis: role of the gut microbiota. Nat Rev Gastroenterol Hepatol 12: 497–506.
Vital M, Penton CR, Wang Q, Young VB, Antonopoulos DA, Sogin ML et al. (2013). A gene-targeted approach to investigate the intestinal butyrate-producing bacterial community. Microbiome 1: 8.
Vital M, Gao J, Rizzo M, Harrison T, Tiedje JM . (2015). Diet is a major factor governing the fecal butyrate-producing community structure across Mammalia, Aves and Reptilia. ISME J 9: 832–843.
Wachowicz K, Hermann-Kleiter N, Meisel M, Siegmund K, Thuille N, Baier G . (2014). Protein kinase C theta regulates the phenotype of murine CD4+ Th17 cells. PloS One 9: e96401.
Waldecker M, Kautenburger T, Daumann H, Busch C, Schrenk D . (2008). Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J Nutr Biochem 19: 587–593.
Waldmann TA, Tagaya Y . (1999). The multifaceted regulation of interleukin-15 expression and the role of this cytokine in NK cell differentiation and host response to intracellular pathogens. Annu Rev Immunol 17: 19–49.
Waldmann TA . (2006). The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat Rev Immunol 6: 595–601.
Wang Y, Devkota S, Musch MW, Jabri B, Nagler C, Antonopoulos DA et al. (2010). Regional mucosa-associated microbiota determine physiological expression of TLR2 and TLR4 in murine colon. PloS One 5: e13607.
Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC et al. (2008). Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature 455: 1109–1113.
Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y et al. (2010). Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 32: 815–827.
Yoshihara K, Yajima T, Kubo C, Yoshikai Y . (2006). Role of interleukin 15 in colitis induced by dextran sulphate sodium in mice. Gut 55: 334–341.
Acknowledgements
This work was supported by grants from the Digestive Diseases Research Core Center (DK42086) at the University of Chicago to DAA and BJ, the US National Institutes of Health (R01DK078938) to SKM, (RO1DK67180) to BJ, and FWF Austrian Science Fund (project no.: J 3418-B19) to MM. The submitted manuscript has been created by UChicago Argonne, LLC, Operator of Argonne National Laboratory (‘Argonne’). Argonne, a US Department of Energy Office of Science laboratory, is operated under contract no. DE-AC02-06CH11357. The US Government retains for itself, and others acting on its behalf, a paid-up nonexclusive, irrevocable worldwide license in said article to reproduce, prepare derivative works, distribute copies to the public, and perform publicly and display publicly, by or on behalf of the Government.
Author contributions
MM, TM, HF-P, JCK, SLO, RH, KL, SK, RB, and LC performed the studies. CRW performed the histological analysis and scoring. MM, TM, BJ and DAA analyzed the data. SKM helped with the generation of germ free mice, and the design of germ-free experiments. MM, DAA and BJ wrote the manuscript. DAA, and BJ coordinated and supervised the project. DAA designed and performed analysis of the microbiota. BJ conceived and designed the overall project.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Meisel, M., Mayassi, T., Fehlner-Peach, H. et al. Interleukin-15 promotes intestinal dysbiosis with butyrate deficiency associated with increased susceptibility to colitis. ISME J 11, 15–30 (2017). https://doi.org/10.1038/ismej.2016.114
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2016.114
This article is cited by
-
Probiotics ameliorate endocrine disorders via modulating inflammatory pathways: a systematic review
Genes & Nutrition (2024)
-
Evidence of innate immune dysfunction in first-episode psychosis patients with accompanying mood disorder
Journal of Neuroinflammation (2022)
-
Cytomegalovirus infection disrupts the influence of short-chain fatty acid producers on Treg/Th17 balance
Microbiome (2022)
-
The double-edged sword of gut bacteria in celiac disease and implications for therapeutic potential
Mucosal Immunology (2022)
-
Early-life gut dysbiosis linked to juvenile mortality in ostriches
Microbiome (2020)

{kind=link}