
Abstract
Ticks are of medical importance owing to their ability to transmit pathogens to humans and animals. The Rocky Mountain wood tick, Dermacentor andersoni, is a vector of a number of pathogens, including Anaplasma marginale, which is the most widespread tick-borne pathogen of livestock. Although ticks host pathogenic bacteria, they also harbor bacterial endosymbionts that have a role in tick physiology, survival, as well as pathogen acquisition and transmission. The goal of this study was to characterize the bacterial microbiome and examine the impact of microbiome disruption on pathogen susceptibility. The bacterial microbiome of two populations of D. andersoni with historically different susceptibilities to A. marginale was characterized. In this study, the microbiome was disrupted and then ticks were exposed to A. marginale or Francisella novicida to determine whether the microbiome correlated with pathogen susceptibility. Our study showed that an increase in proportion and quantity of Rickettsia bellii in the microbiome was negatively correlated to A. marginale levels in ticks. Furthermore, a decrease in Francisella endosymbionts was associated with lower F. novicida infection levels, demonstrating a positive pathogen–endosymbiont relationship. We demonstrate that endosymbionts and pathogens have varying interactions, and suggest that microbiome manipulation may provide a possible method for biocontrol by decreasing pathogen susceptibility of ticks.
Similar content being viewed by others

Introduction
Ticks are obligate hematophagous arthropods that feed on vertebrates and are of medical importance owing to their ability to inflict harm to humans and animals (Jongejan and Uilenberg, 2004). In particular, ticks transmit pathogens throughout all continents and are labeled as principle vectors of zoonotic pathogens (Clay and Fuqua, 2010; Ahantarig et al., 2013). In fact, >15 new tick-borne bacterial pathogens have been discovered since Lyme disease was characterized in 1982 (Jongejan and Uilenberg, 2004).
Dermacentor andersoni, the Rocky Mountain wood tick, has been labeled a ‘veritable Pandora’s box of disease-producing agents’ (Cooley, 1938) and transmits several pathogens, including Rickettsia rickettsii and Francisella tularensis. It is also a vector of Anaplasma marginale, the agent of anaplasmosis, and the most widespread tick-borne pathogen of livestock worldwide (Kocan et al., 2003). Economic losses due to anaplasmosis in cattle are estimated to be >$300 million per year in the United States (Rochon et al., 2012).
Many microorganisms colonize ticks, with the majority being non-pathogenic endosymbionts. The endosymbiont community has a role in tick physiology, survival, and importantly, pathogen acquisition and transmission (Clay and Fuqua, 2010). Amblyomma americanum exhibits decreased fitness in all life stages when the microbiome is manipulated by antibiotic exposure (Zhong et al., 2007). In insects, bacterial species that are required for survival and are maintained within the population transovarially are classically considered to be primary endosymbionts, whereas secondary endosymbionts are beneficial or non-threatening and colonize insects through environmental exposure (Ahantarig et al., 2013). Currently, the extent to which environmental conditions shape the microbiome is not known. There have been varied reports in the literature of environmental impact on the tick microbiome, which suggests the idea that there is not a conserved microbiome. For example, geographic location has been shown to affect the microbial composition of Ixodes ricinus ticks (Halos et al., 2010), however, there was not a habitat-related difference in the microbiome of Dermacentor variabilis and Ixodes scapularis ticks (Hawlena et al., 2012).
The bacterial microbiome, in addition to other functions, is thought to aid tick defense against pathogens. Evidence of endosymbiont-pathogen-inhibiting abilities include the inhibition of R. rickettsii transovarial transmission by Rickettsia peacockii (Telford, 2009). Interestingly, geographically distinct populations of D. andersoni have been shown to experience intraspecies variation in pathogen acquisition: D. andersoni populations from Lake Como, MT, USA have lower Anaplasma marginale acquisition rates (12–30%) than populations from Burns, Oregon (54–62%) (Scoles et al., 2005). In addition, A. marginale acquisition rates vary over time, within a location. We hypothesize that the tick microbiome affects pathogen susceptibility, resulting in differences in pathogen acquisition between tick populations. We tested this hypothesis by examining the microbiome of two geographically distinct populations of D. andersoni, altering the microbiome, and testing for pathogen acquisition with two pathogens, A. marginale and Francisella novicida.
Materials and methods
Tick collection and colonization
Questing adult D. andersoni were collected by flag and drag techniques at sites in Burns, Oregon and Lake Como, Montana (Scoles et al., 2005). Field-collected ticks were used to establish laboratory colonies, which were reared and maintained at the Holm Research Center at the University of Idaho in Moscow, ID, USA (Washington State University IACUC #04440-004 and University of Idaho IACUC #2013-66) (Scoles et al., 2005). Unless there is an ongoing pathogenic infection, the host blood meal does not affect the arthropod microbiome (Hawlena et al., 2012). Furthermore, we screened any bovine blood used in our experiments for the presence of bacteria before colony feeds by running PCR with universal 16S primers.
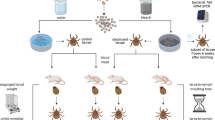
For tick microbiome analysis (Figure 1), a cohort of adult F1 or F2 male ticks from each colony was fed on a Holstein calf and dissected to collect midguts (MG) and salivary glands (SG) for genomic DNA isolation. Adult males were analyzed given that they are the primary source of A. marginale transmission, as the males move from infected to naive hosts in search of mates (Scoles et al., 2005). In addition, MG and SG are the tissues colonized by A. marginale during acquisition and transmission, respectively (Ueti et al., 2007).

The experimental design for microbiome analysis and pathogen acquisition. P1 ticks were collected from the field and reared one generation to produce F1 adults. F1 adult male ticks were exposed to a broad-spectrum antibiotic (oxytetracycline; Tet treated) by feeding on an antibiotic-treated calf to disrupt the bacterial community and to produce altered F2 adults (F2 Alt; red-colored ticks). A cohort of F1 adults were reared identically to F2 Alt ticks, but were not exposed to the antibiotic producing the F2 WT adults. F2 (WT and Alt) adult male ticks (green-colored ticks) were fed on an A. marginale-infected calf (green-colored calf) and cohorts were collected at 2 and 6 days, dissected to collect MG and SG, which were used to assess pathogen infection. Larvae were fed on laboratory rats and nymphs and adults were fed on cattle. The 12-month scale represents the time it takes to rear one generation of ticks under laboratory conditions.
DNA preparation and sequencing
F1 and F2 generation adult male ticks were fed for 7 days and then dissected within 24 h. Before dissection, the ticks were surface sterilized and all dissection tools were sterilized between each dissection (Scoles et al., 2005). Tick MG and SG were dissected and pooled in groups of 30 with three biological replicates. Tissues were stored in Cell Lysis Solution (Qiagen, Valencia, CA, USA) and Proteinase K (1.25 mg ml−1). Genomic DNA was isolated using the PureGene Extraction kit (Qiagen) according to the manufacturer’s specifications (Scoles et al., 2005). In total, 16S recombinant DNA was amplified using Platinum Pfx DNA Polymerase (Invitrogen, Carlsbad, CA, USA) and the primers described below at an annealing temperature of 54 °C. Each sample was amplified in at least three technical replicates with the same sample barcode. To pool our samples, we used barcoded sample-specific primers following the Amplicon Fusion Primer format (Andreotti et al., 2011). To target as many 16S variable regions as technically possible, we used modified universal primers 27F (AGAGTTTGATCMTGGCTCAGAACG) and 1435R (CGATTACTAGCGATTCCRRCTTCA) (Turner et al., 1999; Lane, 1991). The DNA concentration of each sample was measured with a bioanalyzer (Agilent, Santa Clara, CA, USA) and samples were pooled in equimolar amounts. Samples were submitted to Washington State University’s Sequencing Core for Pacific Bioscience (PacBio, Menlo Park, CA, USA) Circular Consensus Sequencing.
Sequence analysis
Sequence reads were analyzed using the Ribosomal Database Program (Cole et al., 2009) and CLC Genomics Workbench. Raw sequence data were filtered by PacBio software according to the expected sequence size range and 99% accuracy. After filtering, reads were blasted against National Center for Biotechnology Information BLASTn database. Blast results were filtered at a minimum of 1275 bp in length and 98% identity. Reads that were below 98% identity were reported at genus level, whereas 98% and above were classified at the species level (Jones et al., 2009; Bonnet et al., 2014; Budachetri et al., 2014). Operational taxonomic units that accounted for 1% or less of the total proportion of the microbiome were grouped together in a ‘rare’ category. The average number of reads/sample after filtering was 3444 sequences, which satisfied a rarefaction curve allowing us to be confident that all operational taxonomic units were captured. Raw microbiome sequence data were deposited in the sequence read archive at National Center for Biotechnology Information (SRR2049110—2049116 and SRR2049119—2049159).
Pathogens used in this study
The Saint Maries strain of A. marginale is the prototypical efficiently tick transmitted strain and the type strain for A. marginale (Dumler et al., 2001). The genome of the St Maries strain has been completely sequenced, and this strain has been used in numerous studies of vector efficiency (Futse et al., 2003; Brayton et al., 2005; Scoles et al., 2005). F. tularensis subspecies novicida strain U112 is a rare human pathogen, causing mild disease. Our interest in this pathogen is that it behaves similarly to F. tularensis and provides a model system to study tick transmission (Reif et al., 2011).
Microbiome disruption
The microbiome was disrupted to study the consequences of an altered microbiome on pathogen acquisition. To alter the microbiome, a cohort of F1 ticks from both Burns and Lake Como colonies were fed on either antibiotic-treated calves (which produced ‘Alt’ microbiomes) or untreated calves (which produced wild-type ‘WT’ microbiomes). The antibiotic-treated calves received a therapeutic dose (11 mg kg−1 of body weight) of Liquamycin LA-200 oxytetracycline injections on −4, −1, +3 and +5 days post application of ticks, whereas untreated calves did not receive any injections. This broad-spectrum antibiotic is commonly used to treat a broad range of bacterial infections in cattle, as we intended to broadly target the microbiome. Ticks were grouped into an ‘experimental cohort’, 100 male ticks that were dissected and processed for sequencing following 7 days of feeding, or a ‘colony cohort’ that included 100 males and 100 females that were allowed to feed to repletion. In both colony cohorts, females were allowed to oviposit to continue a second generation of the WT and Alt microbiomes (F2 generation, Figure 1). Note that a single discreet antibiotic treatment was used, and the F2 Alt generation arose from F1 adults that were exposed to antibiotics. The F2 ticks were not subjected to antibiotics.
Acquisition of A. marginale
F2 WT and Alt adult male ticks from Burns and Lake Como cohorts were fed on a Holstein calf that was persistently infected with A. marginale St. Maries strain (6.55 × 105 organisms ml−1 of whole blood). Ninety ticks from each of the four treatment groups were fed in a group-specific stockinette patch. After 12 h all unattached ticks were removed. Cohorts of each group were removed from the calf at 2, 4 and 6 days post application. After removal, ticks were held for 7 days at 26 °C and 98% relative humidity to allow for digestion of the blood meal and then the MG and SG were dissected. Day 2 represents the minimal amount of time that it takes for the tick to become infected with A. marginale, whereas day 6 represents ticks that are ready for transmission feeding (Eriks et al., 1993).
Genomic DNA from acquisition fed ticks was extracted using the PureGene Extraction kit (Qiagen). Pathogen infection rates and levels were analyzed by quantitation of the A. marginale single-copy gene, msp5, using a Bio-Rad Digital Drop PCR (ddPCR; Hercules, CA, USA) machine. The primer set included msp5_for2 (CCGAAGTTGTAAGTGAGGG), msp5_rev2 (CTTATCGGCATGGTCGCC) and msp5_probe (CCTCCGCGTCTTTCGACAATTTGGTT). Thermocycling conditions were as follows: 10 min denaturation at 95 °C; 39 cycles of 94 °C for 30 s, 60 °C for 60 s; and a final cycle of 98 °C for 10 min. Sample preparation and ddPCR protocols followed the manufacturer’s recommendations. Although there is a physical size difference between Burns and Lake Como ticks, A. marginale infection rates and levels are independent of size (Scoles et al., 2005).
Acquisition of F. novicida
To determine whether the tick microbiome affected the ability of ticks to acquire Francisella pathogens, WT and Alt nymphs from F2 Burns and Lake Como were fed on F. novicida U112-infected mice as previously described (Reif et al., 2011). To minimize host-to-host variation, WT and Alt ticks from the same geographic population fed on the same mice, but in separate capsules. Replete nymphs were collected after feeding, incubated at 25 °C and allowed to molt to adults. Midguts were individually harvested from adult ticks, homogenized and the resulting lysate plated (Reif et al., 2014). Enumeration of colony forming units (CFU) was used to calculate the F. novicida infection level in tick MG. Mouse bacteremia was determined by plating blood (Reif et al., 2011).
Quantitative analysis of Rickettsia bellii
To quantify R. bellii, rickA copy numbers were measured using SYBR Green quantitative PCR (Invitrogen) of Lake Como WT and Alt in F1 and F2 ticks. The quantity of R. bellii was determined using qRbRickAF2 (5′-TACGCCACTCCCTGTGTCA-3′) and qRbRickAR (5′-GATGTAACGGTATTACACCAACAG-3′) primers (Oliver et al., 2014). The bacterial quantity was measured in F1 and F2 MG and SG of the pooled samples that were used for PacBio sequencing. The quantities of R. bellii were used to verify proportional changes of R. bellii within the microbiome when comparing WT and Alt compositions, as well as the differences in the microbiome between F1 and F2 generations.
Statistical analysis
Methods for analyzing microbiome data vary greatly and many publications have used paired t-tests for their simplicity and applicability to a wide range of data (Zhong et al., 2007; Clay et al., 2008; Carpi et al., 2011; Budachetri et al., 2014; Bonnet et al., 2014). We used a generalized linear mixed (GLM) model, as well as traditional pairwise comparisons for the A. marginale, R. bellii and F. novicida data. By using both types of statistical analyses we can compare our data with previous studies as well as the two types of statistical methods.
The following data were analyzed with GLM models in the Glimmix Procedure of SAS 9.4 (SAS Institute Inc., Cary, NC, USA): microbiome, A. marginale acquisition, F. novicida and R. bellii. Within the various models, fixed effects included population (L), treatment of calves (T), day of tick feeding (D), organ (O) and generation of tick (G) with a random effect of replicate. Replicate did not account for any variation and was removed from all models. In addition, all models were reduced in a stepwise manner starting with highest order interactions that were not significant. The Tukey–Kramer procedure for unbalanced data were used for all pairwise comparisons.
Microbiome data were transformed to proportion of endosymbiont per pooled sample of ticks to reduce the heterogeneity of variances. In addition, a single count was added to all R. bellii counts prior to being transformed to allow for comparison of the populations, as Burns had only one tick pool with a non-zero count. Endosymbiont data were described using:

A. marginale acquisition data were described using:

F. novicida data were described using:

R. bellii data were described using:

Spearman rank-order correlations (SAS9.4) were calculated between A. marginale and R. bellii within F2 Lake Como ticks. One-way analysis of variance within SPSS (IBM Version 20) was used to calculate pairwise comparisons to analyze statistical differences between and within groups’ pathogen infection rates and levels of A. marginale acquisition, as well as comparisons of tick-fitness measurements. Generalized linear mixed-model output has been included in Supplementary File 1 (Supplementary Tables 1–7).
Results
Bacterial microbiome characterization
We compared tick microbiomes across populations and tissue types. The bacterial composition of both tick populations varied by tissue type. In the population from Burns, the MG had two endosymbiont groups, a mixture of Francisella spp. (20%) and Francisella-like endosymbiont (FLE; 61%), that made up 81% of the microbiome, whereas the SG microbiome was composed mainly of Arsenophonus spp. (95%; Figure 2a). The MG microbiome of the Lake Como population was composed primarily of FLE (60%), Francisella spp. (20%), and a small proportion of a R. bellii (16%), whereas the SG microbiome was composed primarily of R. bellii (82%) and Arsenophonus spp. (11%).

Microbiome of D. andersoni ticks in wild-type and altered populations from Montana and Oregon. The bacterial composition of the midgut (MG) and salivary glands (SG) of adult male Burns (B) and Lake Como (LC) ticks was characterized in (a) F1 ticks and (b) F2 progeny ticks. Species/genera detected included Francisella spp. (blue), FLE (red), Arsenophonus spp. (green), Rickettsia spp. (purple), R. bellii (black), R. peacockii (orange), rare (teal) and unclassified (dark red).
The GLM model found that all endosymbionts varied in the overall proportion of total endosymbionts across both tick populations, except for Francisella spp. (P>0.21) and rare (P>0.56) endosymbionts (Figure 2a; Supplementary File 1 Supplementary Table 4). Burns ticks had a greater proportion of Arsenophonus spp. (45.7%; P<0.01) and R. peacockii (5.1%; P<0.05) compared with Lake Como ticks. A greater proportion of FLE (6.7%; P<0.05), R. bellii (54.2%; P<0.01) and Rickettsia (1.4%; P<0.02) were found in Lake Como ticks compared with Burns ticks. In addition, the proportions of bacteria varied based on tissue type, except for Rickettsia (P>0.06) and rare (P>0.06) endosymbionts (Figure 2a). The proportion of FLE (54.9%; P<0.01), Francisella spp. (31.6%; P<0.01), R. bellii (33.8%; P<0.01) and R. peacockii (4.9%; P<0.05) was greater in MG compared with SG. SG had a greater proportion of Arsenophonus spp. (48.4%; P<0.01) compared with MG.
Microbiome alteration
The complexities of the microbiomes were unchanged by antibiotic exposure; specifically no bacterial species were introduced or lost, although endosymbiont proportions shifted owing to antibiotic exposure (Figure 2a). Antibiotics altered the microbiome in the Burns population, with an increase in the proportion of Arsenophonus spp. (19.3%) and a decrease in the proportion of Francisella spp. (5.0%) and FLE (9.4%) in the SG. In contrast, in the Alt MG, the proportion of Francisella spp. and FLE decreased by 10.5% and 5.2%, respectively, and Arsenophonus spp. increased 2.5%. The response to antibiotic exposure in the Lake Como tick population was different than the response of the Burns tick population. The Lake Como population experienced an increase in the proportion of R. bellii by 12.3%, and a decrease in Arsenophonus spp. by 8.4%, in the SG owing to antibiotic treatment. The MG showed a decrease in the proportion of FLE (8.4%), whereas the proportion of R. bellii increased by 8.9% (Figure 2a), with antibiotics.
Ticks were reared one generation post disruption (F2 ticks) to evaluate whether the alteration to the microbiome was maintained. Burns Alt F2 ticks (Figure 2b) had an increased proportion in Arsenophonus spp. and decreased proportions in both Francisella spp. as well as FLE in the MG without any change in the SG as compared with WT Burns ticks. When comparing F2 Alt and WT Lake Como ticks the bacterial composition of the MG showed an increase in the proportion of R. bellii (15.2%) and Arsenophonus spp. (7.5%) and decreases in Francisella spp. (8.4%) and FLE (15.0%), whereas in the SG there were decreases in the proportion of Francisella spp. (8.5%) and FLE (12.0%) and increased proportion of R. bellii (24.7%).
When the GLM model was applied to the data, FLE (P<0.02) and Francisella spp. (P<0.02) were in a greater proportions in F2 WT ticks compared with Alt ticks, regardless of the tick population or organ type, confirming observations in the unadjusted data. In addition, there were significant (P<0.02) tick population by organ-type interactions for Arsenophonus spp., R. bellii, R. peacockii and rare endosymbionts. With Arsenophonus spp., Burns ticks had a much greater (P<0.01) proportion in the SG (84.9% and 97.5%, F1 and F2, respectively) compared with Lake Como ticks (6.3% and 26.7%, F1 and F2, respectively). In addition, SG of Burns ticks had a greater (P<0.01) proportion of Arsenophonus spp. than MG. In F1 ticks, R. bellii was proportionally different (P<0.01) between Lake Como MG (20.6%) and SG (88.1%), MG between Burns (0.1%) and Lake Como (20.6%), and SG between Burns (0.2%) and Lake Como (88.1%). In F2 ticks, R. bellii was proportionally different (P<0.01) between Lake Como MG (11.1%) and SG (60.9%) and SG between Burns (1.1%) and Lake Como (60.9%). In F2 ticks, R. peacockii had different (P<0.02) proportions between MG of Burns (1.9%) and Lake Como (0.7%) and Burns MG (1.9%) and SG (0.1%). In F2 rare endosymbionts, MG had a different (P<0.03) proportion in Burns (0.4%) compared with Lake Como (0.06%) and Burns MG (0.4%) compared with SG (0.02%).
A. marginale acquisition
For the WT and Alt adult male F2 ticks from Burns and Lake Como that were fed on a calf persistently infected with A. marginale the Alt ticks had significantly less A. marginale (4.42 × 103 A. marginale/tick) than WT ticks (7.22 × 103 A. marginale/tick; P<0.01; Figure 4). Also, A. marginale increased over time, with the highest levels of A. marginale detected at day 6 (P<0.01; Figure 4). All groups had high rates of A. marginale infection by day 2, without a significant difference between days or treatment, with the exception that Lake Como Alt ticks all had a significantly lower infection rate than Burns Alt ticks at day 6 (Supplementary File 1 Supplementary Table 8).

Global analysis of A. marginale infection load. A. marginale load in Lake Como (LC) and Burns (B) ticks is shown for day 2, day 4 and day 6 of acquisition feeding. The pathogen was quantified in the midgut (MG) and salivary glands (SG) using the single-copy msp5 gene in wild-type (WT) and altered (Alt) colonies. A mixed-model approach was used to look at the A. marginale acquisition in its entirety. Error bars indicate the standard error and the double asterisk indicates statistically significant differences between indicated groups at P<0.01.
Tick populations had significantly different levels of acquisition of A. marginale (P<0.006), with Burns ticks having greater A. marginale loads (7.32 × 103 A. marginale/tick) than Lake Como ticks (4.35 × 103 A. marginale/tick), irrespective of tissue or treatment (Supplementary File 1 Supplementary Tables 1). The Burns population did not have any significant differences between treatment groups on day 2 or 6 (Figure 3). Analysis of pathogen load in Lake Como ticks was significantly different between the WT and Alt groups on days 2 and 6 (Figure 3). In the Lake Como ticks, fewer A. marginale occurred in the Alt population as compared with the WT population in the MG on day 2 (P<0.046) and day 6 (P<0.012) as well as in the SG on day 6 (P<0.007; Figure 3).

A. marginale infection load. A. marginale load in Lake Como (LC) and Burns (b) ticks is shown for day 2 (a) and day 6 (b) of acquisition feeding. The pathogen was quantified in the midgut (MG) and salivary glands (SG) using the single-copy msp5 gene in wild-type (WT) and altered (Alt) colonies. A pairwise comparison was used to analyze between and within groups. Error bars indicate the s.e. and the asterisks indicate statistically significant differences between indicated groups.
According to the GLM model (Figure 4), the altered microbiome affected (P<0.01) A. marginale acquisition where Alt ticks had a lower mean (4.45 × 103 A. marginale) than WT ticks (7.21 × 103 A. marginale). This was regardless of the population and tissue type, confirming treatment differences detected in the pairwise analysis. In addition, increased amounts of A. marginale within ticks occurred over time with significant differences detected between day 6 and day 2 (P<0.01) and day 4 (P<0.01; Figure 4). Tick MGs had higher (P<0.01) mean A. marginale loads than SGs. However, one two-way interaction, day by organ, and one three-way interaction, population by treatment by day, were significant (P<0.01). Specifically for the two-way interaction, day 2 MG was different from SG (P<0.05), day 2 was different compared with day 4 (P<0.02) and day 6 (P<0.01) for MG, day 2 was different (P<0.01) than day 6 for SG, MG was different (P<0.01) from SG on day 4 and SG were different (P<0.01) on day 4 compared with day 6. Therefore, A. marginale acquisition increased by day (Figure 4).
Quantitative analysis of R. bellii
The Lake Como population of ticks was colonized by an endosymbiont, R. bellii, which was not detected in the Burns population. The Alt treatment had a significant (P<0.020) effect on R. bellii numbers (Figure 5a), resulting in higher levels of R. bellii in Alt ticks (8.40 × 107 R. bellii per tick) as compared with WT ticks (4.80 × 107 R. bellii per tick; Figure 5). These treatment differences mirror the proportional changes seen in the sequence data. Using the GLM model, tick generation (P>0.51) did not result in detectable differences in R. bellii. However, tissue type was approaching significance (P>0.07) with MG having lower levels of R. bellii compared with SG, similar to what was detected through sequence analysis. In addition, two two-way interactions were significant (P<0.01), treatment by organ type (Figure 5b) and treatment by generation (Figure 5c). The SG from WT ticks had lower levels (P<0.01) of R. bellii than Alt ticks. Furthermore, SG had greater numbers (P<0.01) of R. bellii than the MG from Alt ticks. Antibiotic exposure of F1 ticks impacted R. bellii levels in the F2 generation with Alt ticks having greater (P<0.01) numbers of R. bellii than WT ticks. For Alt ticks, F2 had increased (P<0.04) R. bellii compared with F1. When coupled with the proportion data, it is clear that antibiotic exposure resulted in F2 ticks with higher levels and proportions of R. bellii in the Alt Lake Como microbiome as compared with the WT counterparts (P<0.01; Figure 5c).

Comprehensive quantitation of Rickettsia bellii in Lake Como ticks. The amount of R. bellii in the midgut (MG) and salivary glands (SG) from F1 and F2 ticks is shown. (a) one-way interactions analyzed the treatment, tissue and generation effect on the quantity of R. bellii; (b) two-way interaction of tissue and treatment on the quantity of R. bellii; (c) two-way interaction of treatment and generation on the quantity of R. bellii. Error bars indicate the s.e. and the single asterisk indicates statistically significant differences between indicated groups at P<0.05, the double asterisk indicates P<0.01, whereas the plus sign indicates statistical significance at P<0.07.
Because Lake Como ticks acquired significantly less A. marginale than Burns ticks, and had an endosymbiont, which was not present in Burns ticks (R. bellii), we assessed whether there was a correlation between the numbers of R. bellii in F2 ticks and the A. marginale infection load. A negative correlation was observed with higher levels of R. bellii correlating with lower levels of A. marginale in Alt MG and SG (r2=−0.4472, P<0.55 and −0.8944, P<0.10, respectively). Significance was diminished owing to small sample size (n=3 pools/correlation).
F. novicida acquisition
Next, we determined whether changes to the composition of Francisella spp. within the microbiome altered the ability of F. novicida to colonize the tick. For F2 nymphal ticks that fed on F. novicida-infected mice, the overall nymph-feeding success was 30% for Lake Como ticks and 58% for Burns ticks, averaged across treatment groups (Supplementary File 1 Supplementary Table 9). Although the sample size varied based on population and treatment, nymphs from the WT Burns population had the greatest feeding (χ2(3)=96.08, P<0.001) and molting success rate (χ2(3)=32.71, P<0.001), whereas nymphs from the Alt Lake Como population had the lowest feeding and molting success rate (Supplementary File 1 Supplementary Table 9). The prevalence of F. novicida infection in tick groups was 100%, except for the Lake Como WT group where infection prevalence was 86% (Supplementary File 1 Supplementary Table 9). The lower prevalence in the latter group was due to two ticks that were likely negative owing to feeding on a mouse that developed a very low-level bacteremia (4.0 × 103 CFU ml−1 blood). Figure 6 shows the F. novicida results of tick population and treatment. F. novicida CFU differed (P<0.02) owing to treatment: WT ticks had more F. novicida (8.8 × 106 CFU) compared with Alt ticks (3.74 × 106 CFU), regardless of population. In addition, there was no difference in F. novicida load comparing cohorts from Lake Como and Burns (P>0.13). There was not a significant treatment by population interaction (P>0.87), which means that in both populations, WT ticks had increased F. novicida compared with Alt ticks. Although Lake Como ticks did not have a statistically significant difference in infection level between Alt and WT ticks, this is likely to be a result of high mortality of Lake Como Alt nymphs leading to a small adult sample size—there was an 82% decrease in pathogen load in Lake Como Alt ticks as compared with WT ticks. Similarly, there was a 47% decrease in pathogen load when comparing the Burns Alt cohort with WT.

F. novicida infection load. F. novicida infection level in midgut (MG) of altered (Alt) versus wild-type (WT) Lake Como and Burns tick groups. Error bars indicate the s.e. and the asterisks indicate statistically significant differences between groups and the double asterisk indicates P<0.01.
Discussion
We examined the microbiomes of two tick populations from geographically distinct areas based on a previous observation of differing pathogen susceptibility to A. marginale (Scoles et al., 2005). The tick population from Burns was primarily colonized with Francisella spp. and FLE in the MG and Arsenophonus spp. in the SG, whereas the population from Lake Como was colonized with Francisella spp., FLE and R. bellii in the MG and R. bellii in the SG. In order to associate the microbiome with pathogen acquisition, we altered the microbiome by antibiotic exposure. The microbiome response to alteration varied based on location and tissue type. In the F1 Burns population, the proportion of Arsenophonus spp. increased and the proportion of Francisella spp. and FLE decreased in the MG and SG. However, in the F1 Lake Como population, R. bellii increased in proportion in the MG and SG, whereas Arsenophonus spp. decreased in only the MG, and FLE decreased in only the SG. One generation later, F2, the microbiome was characterized again to understand if the ALT populations would restore WT phenotype. However, that was not the case, with Burns ALT population having an increased proportion in Arsenophonus spp. and decreased proportion of Francisella spp. and FLE in the MG and no compositional difference between WT and ALT in the SG. Furthermore, in F2 Lake Como, there was an increased proportion of R. bellii in the MG and SG, an increased proportion of Arsenophonus spp. in the MG, and a decreased proportion of Francisella spp. and FLE in both MG and SG.
One generation after microbiome alteration, the acquisition susceptibility to A. marginale was analyzed. Our results show that although Burns and Lake Como ticks become infected with different levels of A. marginale, there is not a significant difference in the infection rate, as seen previously (Scoles et al., 2005). Our previous study used a less-sensitive technology (nested PCR) to determine infection rate, whereas we used ddPCR, and therefore the threshold of detection may account for the difference between studies. However, a more likely reason for the difference in results is that there was reorganization of the microbiome of Burns and Lake Como ticks over the 10-year period between sampling. Because the bacterial community was not assessed in the previous study, any changes to the microbiome of these populations between 2004 and 2012 are unknown.
In this study, we provide evidence of a negative correlation between R. bellii, an endosymbiont of D. andersoni found in the Lake Como tick population and A. marginale. As a population, ticks with higher levels of R. bellii had decreased acquisition levels of A. marginale as compared with ticks with lower levels of R. bellii (Figures 4 and 5). Although this result is preliminary and requires additional experimentation for definitive proof, previous studies have shown that Rickettsia endosymbionts have a negative effect on pathogenic rickettsial species in several species of ticks (Niebylski et al., 1997; Macaluso et al., 2002; Telford, 2009; Oliver et al., 2014). These earlier studies have focused on the rickettsial interference in ovarian tissue colonization or transovarial transmission, without investigation of whether rickettsial interference occurs in additional pathogen acquisition- or transmission-associated tissues. These studies identified bacterial interference between species within the genus Rickettsia, and we found that such interference could occur between rickettsial species in different families. Importantly, other factors need to be considered for a complete understanding of the acquisition dynamics, such as tick genetics and complex interactions, which could affect the acquisition of A. marginale.
The theoretical application of rickettsial interference as a method to combat pathogen acquisition and transmission is not a novel idea (Telford, 2009); however, it has remained within the realm of studies on human rickettsial pathogens. Our study demonstrates that this phenomenon could occur in veterinary-relevant pathogens, as seen with a decrease in A. marginale load (Figure 3) in ticks with higher proportion and quantity of R. bellii. Two possible mechanisms of rickettsial interference include a direct and indirect inhibition pathway (Figure 7). The direct mechanism entails R. bellii releasing a molecule that limits replication of A. marginale, whereas the indirect mechanism involves R. bellii preferentially utilizing host resources necessary for pathogen replication (Weiss and Aksoy, 2011). Regardless of pathway, bacterial inhibition appears to affect the susceptibility of ticks to A. marginale.

Mechanisms of interaction between endosymbionts and pathogens. Panels a and b show how the endosymbiont can directly affect the growth of a pathogen, whereas panels c and d show how the endosymbiont can indirectly affect the growth of a pathogen. In panel a, the endosymbiont secretes a compound (black dot) that has a negative effect on pathogen growth. Panel b shows the endosymbiont secretes a compound (black dot) that has a positive effect on pathogen growth. Panel c shows the endosymbiont preferentially taking up nutrients such that the pathogen cannot acquire it, thus leading to poor growth of the pathogen. Panel d depicts the endosymbiont affecting the immune response (symbolized by I) of the tick such that these immune factors cannot act on the pathogen leading to increased growth of the pathogen. The endosymbiont is shown in yellow in all panels. Pathogen is shown in red when it does not grow well in the presence of the endosymbiont and in green when it does grow well in the presence of the endosymbiont.
We tested the acquisition of a second pathogen, F. novicida, in our two populations of ticks to determine whether the microbiome affected acquisition in the same manner as for A. marginale. When microbiome compositions are overlaid with F. novicida acquisition results, our results suggest a positive relationship between the proportion of the microbiome occupied by Francisella endosymbionts and the infection level of F. novicida. This could indicate that Francisella endosymbionts promote a more favorable environment for the related pathogen to replication within the MG. One possible explanation is that the Francisella endosymbionts are causing immunosuppression of the host, which then allows F. novicida to avoid host defenses (Figure 7). This type of symbiotic exploitation is seen in several species of mosquitoes between Wolbachia and Plasmodium (Ze'le et al., 2014), as well as between Wolbachia and Baculoviruses (Graham et al., 2012). In addition, activation of the Toll and Imd immune pathways in Drosophila melanogaster have been shown to increase the amount of Spiroplasma endosymbionts, which results in an increase host susceptibility to other pathogens (Herren and Lemaitre, 2011). Furthermore, Francisella endosymbionts may secrete a molecule, such as a nutrient, that facilitates F. novicida replication (Figure 7). Endosymbionts have been shown to possess the ability to produce many beneficial molecules, such as Blochmannia endosymbionts supplying essential amino acids or Wigglesworthia releasing B vitamins (Kuriwada et al., 2010). Further investigation is needed to tease apart the mechanism of interaction.
In conclusion, a disruption in the microbiome resulted in an increase in the proportion of R. bellii and a decrease of Francisella endosymbionts. These alterations demonstrated two meaningful results: a negative correlation between higher levels of R. bellii and A. marginale acquisition levels and a positive relationship between Francisella endosymbionts and F. novicida acquisition levels. These endosymbiont-pathogen interactions pose an interesting opportunity to explore our ability to decrease vector competence in D. andersoni by microbiome manipulation. Our results warrant further investigation into the bacterial inhibition mechanisms between R. bellii and A. marginale and between Francisella endosymbionts and pathogens, as well as testing these relationships for other pathogens transmitted by D. andersoni. We propose that different mechanisms of interaction can be at play for different pathogen–endosymbiont systems (Figure 7), which will require detailed analysis to elucidate.
References
Ahantarig A, Trinachartvanit W, Baimai V, Grubhoffer L . (2013). Hard ticks and their bacterial endosymbionts (or would be pathogens). Folia Microbiol (Praha) 58: 419–428.
Andreotti R, de León AAP, Dowd SE, Guerrero FD, Bendele KG, Scoles GA . (2011). Assessment of bacterial diversity in the cattle tick Rhipicephalus Boophilus microplus through tag-encoded pyrosequencing. BMC Microbiol 11: 6–6.
Bonnet S, Michelet L, Moutailler S, Cheval J, Hébert C, Vayssier-Taussat M et al. (2014). Identification of parasitic communities within european ticks using next-generation sequencing. PLoS Negl Trop Dis 8: e2753.
Brayton KA, Kappmeyer LS, Herndon DR, Dark MJ, Tibbals DL, Palmer GH et al. (2005). Complete genome sequencing of Anaplasma marginale reveals that the surface is skewed to two superfamilies of outer membrane proteins. PNAS 102: 844–849.
Budachetri K, Browning RE, Adamson SW, Dowd SE, Chao C-C, Ching W-M et al. (2014). An insight into the microbiome of the Amblyomma maculatum (Acari: Ixodidae). J Med Entomol 51: 119–129.
Carpi G, Cagnacci F, Wittekindt NE, Zhao F, Qi J, Tomsho LP et al. (2011). Metagenomic profile of the bacterial communities associated with Ixodes ricinus ticks. PLoS One 6: e25604.
Clay K, Fuqua C . (2010) The Tick Microbiome: Diversity, Distribution and Influence of the Internal Microbial Community for a Blood-Feeding Disease Vector. Critical Needs and Gaps in Understand Prevention, Amelioration, and Resolution of Lyme and Other Tick-Borne Diseases: the Short-Term and Long-Term Outcomes. Institute of Medicine Committee on Lyme Disease and Other Tick-Borne Diseases: The State of the Science: Washington DC, USA. Available from: http://www.iom.edu/~/media/Files/Activity%20Files/Disease/TickBorne/08-The-Tick-Microbiome.pdf.
Clay K, Klyachko O, Grindle N, Civitello D, Oleske D, Fuqua C . (2008). Microbial communities and interactions in the lone star tick Amblyomma americanum. Mol Ecol 17: 4371–4381.
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ et al. (2009). The ribosomal database project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37: D141–D145.
Cooley RA . (1938) The genera Dermacentor and Otocentor (Ixodidae) in the United States, with studies in variation. US Government Printing Office: Washington DC, USA.
Dumler JS, Barbet AF, Bekker CPJ, Dasch GA, Palmer GH, Ray SC et al. (2001). Reorganization of genera in the families Rickettsiaceae and Anaplasmataceae in the order Rickettsiales: unification of some species of Ehrlichia with Anaplasma Cowdria with Ehrlichia and Ehrlichia with Neorickettsia, descriptions of six new species combinations and designation of Ehrlichia equi and 'HGE agent' as subjective synonyms of Ehrlichia phagocytophila. Int J Syst Evol Microbiol 51: 2145–2165.
Eriks IS, Stiller D, Palmer GH . (1993). Impact of persistent Anaplasma marginale rickettsemia on tick infection and transmission. J Clin Microbiol 31: 2091–2096.
Futse JE, Ueti MW, Knowles DP Jr, Palmer GH . (2003). Transmission of Anaplasma marginale by Boophilus microplus: retention of vector competence in the absence of vector-pathogen interaction. J Clin Microbiol 41 (8): 3829–3834.
Graham RI, Grzywacz D, Mushobozi WL, Wilson K . (2012). Wolbachia in a major African crop pest increases susceptibility to viral disease rather than protests. Ecol Letter 15: 993–1000.
Halos L, Bord S, Cotte V, Gasqui P, Abrial D, Barnouin J et al. (2010). Ecological factors characterizing the prevalence of bacterial tick-borne pathogens in Ixodes ricinus ticks in pastures and woodlands. Appl Environ Microbiol 76: 4413–4420.
Hawlena H, Rynkiewicz E, Toh E, Alfred A, Durden LA, Hastriter MW et al. (2012). The arthropod, but not the vertebrate host or its environment, dictates bacterial community composition of fleas and ticks. ISME J 7: 221–223.
Herren JK, Lemaitre B . (2011). Spiroplasma and host immunity: activation of humoral immune responses increases endosymbiont load and susceptibility to certain Gram-negative bacterial pathogens in Drosophila melanogaster. Cell Microbiol 13: 1385–1396.
Jones RT, Knight R, Martin AP . (2009). Bacterial communities of disease vectors sampled across time, space, and species. ISME J 4: 223–231.
Jongejan F, Uilenberg G . (2004). The global importance of ticks. Int J Parasitol 129: S3.
Kocan KM, la Fuente De J, Guglielmone AA, Meléndez RD . (2003). Antigens and alternatives for control of Anaplasma marginale infection in cattle. Clin Microbiol Rev 16: 698–712.
Kuriwada T, Hosokawa T, Kumano N, Shiromoto K, Haraguchi D et al. (2010). Biological role of Nardonella Endosymbiont in its Weevil Host. PLoS One 5: e13101.
Lane DJ . (1991) 16S/23S rRNA sequencing. In: Stackebrandt E, Goodfellow M (eds), Nucleic acid techniques in bacterial systematics. John Wiley and Sons: New York, NY, USA, pp 115–175.
Macaluso KR, Sonenshine DE, Ceraul SM, Azad AF . (2002). Rickettsial infection in Dermacentor variabilis (Acari: Ixodidae) inhibits transovarial transmission of a second rickettsia. J Med Entomol 39: 809–813.
Niebylski ML, Peacock MG, Fischer ER, Porcella SF, Schwan TG . (1997). Characterization of an endosymbiont infecting wood ticks, Dermacentor andersoni, as a member of the genus Francisella. Appl Environ Microbiol 63: 3933–3940.
Oliver JD, Burkhardt NY, Felsheim RF, Kurtti TJ, Munderloh UG . (2014). Motility characteristics are altered for Rickettsia bellii transformed to overexpress a heterologous rickA gene. Appl Environ Microbiol 80: 1170–1176.
Reif KE, Palmer GH, Crowder DW, Ueti MW, Noh SM . (2014). Restriction of Francisella novicida genetic diversity during infection of the vector midgut. PLoS Pathog 10: e1004499.
Reif KE, Palmer GH, Ueti MW, Scoles GA, Margolis JJ, Monack DM et al. (2011). Dermacentor andersoni transmission of Francisella tularensis subsp. novicida reflects bacterial colonization, dissemination, and replication coordinated with tick feeding. Infect Immun 79: 4941–4946.
Rochon K, Scoles GA, Lysyk TJ . (2012). Dispersion and sampling of Adult Dermacentor andersoni in rangeland in Western North America. J Med Entomol 49: 253–261.
Scoles GA, Ueti MW, Palmer GH . (2005). Variation among geographically separated populations of Dermacentor andersoni (Acari: Ixodidae) in midgut susceptibility to Anaplasma marginale (Rickettsiales: Anaplasmataceae). J Med Entomol 42: 153–162.
Telford SR III . (2009). Status of the ‘East Side Hypothesis’ (Transovarial Interference) 25 Years Later. Ann N Y Acad Sci 1166: 144–150.
Turner SW, Pryer KM, Miao VPW, Palmer JD . (1999). Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRND sequence analysis. J Eukaryot Microbiol 46: 327–338.
Ueti MW, Reagan JO, Knowles DP, Scoles GA, Shkap V, Palmer GH . (2007). Identification of midgut and salivary glands as specific and distinct barriers to efficient tick-borne transmission of Anaplasma marginale. Infect Immun 75: 2959–2964.
Weiss B, Aksoy S . (2011). Microbiome influence on insect host vector competence. Trends Parasitol 27: 514–522.
Ze'le F, Nicot A, Berthomieu A, Weill M, Duron O, Rivero A . (2014). Wolbachia increases susceptibility to Plasmodium infection in a natural system. Proc R Soc B 281: 20132837–20132837.
Zhong J, Jasinskas A, Barbour AG . (2007). Antibiotic treatment of the tick vector Amblyomma americanum reduced reproductive fitness. PLoS One 2: e405.
Acknowledgements
We thank Lisa Orfe, Mark Wildung, Derek Pouchnik, Ralph Horn, James Allison and the staff at the USDA-ARS in Moscow, ID, USA for excellent guidance and technical assistance. This research was supported by National Institutes of Health grants, AI044005, AI093524, 5T32GM008336-25, www.experiment.com (crowdsourcing grant), USDA-ARS-CRIS 5348-32000-033-00D and CVM intramural funds.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Gall, C., Reif, K., Scoles, G. et al. The bacterial microbiome of Dermacentor andersoni ticks influences pathogen susceptibility. ISME J 10, 1846–1855 (2016). https://doi.org/10.1038/ismej.2015.266
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2015.266
This article is cited by
-
Entomopathogenic fungus treatment changes the gut bacterial diversity of Rhipicephalus microplus ticks
Parasites & Vectors (2023)
-
Cross-alteration of murine skin and tick microbiome concomitant with pathogen transmission after Ixodes ricinus bite
Microbiome (2023)
-
Microbiota perturbation by anti-microbiota vaccine reduces the colonization of Borrelia afzelii in Ixodes ricinus
Microbiome (2023)
-
New insights into the impact of microbiome on horizontal and vertical transmission of a tick-borne pathogen
Microbiome (2023)
-
Tick microbial associations at the crossroad of horizontal and vertical transmission pathways
Parasites & Vectors (2022)
