
Abstract
Disruption of healthy microbial communities has been linked to numerous diseases, yet microbial interactions are little understood. This is due in part to the large number of bacteria, and the much larger number of interactions (easily in the millions), making experimental investigation very difficult at best and necessitating the nascent field of computational exploration through microbial correlation networks. We benchmark the performance of eight correlation techniques on simulated and real data in response to challenges specific to microbiome studies: fractional sampling of ribosomal RNA sequences, uneven sampling depths, rare microbes and a high proportion of zero counts. Also tested is the ability to distinguish signals from noise, and detect a range of ecological and time-series relationships. Finally, we provide specific recommendations for correlation technique usage. Although some methods perform better than others, there is still considerable need for improvement in current techniques.
Similar content being viewed by others

Introduction
Microbes interact with their hosts and their communities, and these interactions have been implicated in numerous human health conditions including obesity and metabolic syndrome (Ley et al., 2005; Turnbaugh et al., 2009; Vrieze et al., 2012; Ridaura et al., 2013), cardiovascular disease (Wang et al., 2011), Clostridium difficile colitis (Gough et al., 2011), inflammatory bowel diseases (Gevers et al., 2014) and HIV (Lozupone et al., 2013a). These communities are influenced by diet, culture, geography, age and antibiotic use, among other factors (Lozupone et al., 2013b), and are also very important in other systems, such as soils, lakes and oceans (Chaffron et al., 2010; Beman et al., 2011; Steele et al., 2011). An emerging approach to their study through sequencing is ‘correlation networks’. Broadly, correlation networks have individual microbes (operational taxonomic units (OTUs), or features) as nodes and feature–feature pairs as edges, where an edge may imply a biologically or biochemically meaningful relationship between features. For instance, one may expect that mutualistic microbes, or those that benefit each other, will positively correlate across samples. In contrast, microbes with antagonistic relationships such as competition for the same niche may negatively correlate. In practice, microbes also may positively or negatively correlate for indirect reasons, based on their environmental preferences. This notion is supported by the observation that phylogenetically related microbes have a tendency to positively co-occur (Lozupone et al., 2012). Recent studies suggest that the microbial relationships shown in correlation interaction networks can be used to determine drivers in environmental ecology (Ruan et al., 2006; Steele et al., 2011; Zhou et al., 2011; Lima-Mendez et al., 2015) or contribution to habitat niches or disease (Chaffron et al., 2010; Arumugam et al., 2011; Faust and Raes 2012; Faust et al., 2012; Greenblum et al., 2012; Oakley et al., 2013; Goodrich et al., 2014; Buffie et al., 2015). Correlation is also a powerful tool to help researchers with hypothesis generation, such as determining which interactions might be biologically relevant in their system, and should be given further study (for example, through co-culturing or whole-genome sequencing).
Unfortunately, measuring correlation networks is computationally challenging. One such challenge comes from the complexity of microbial communities: many microbial data sets easily have >5000 features. As the number of possible two-feature interactions for a data set with n features is (n*(n−1))/2, this implies almost 12.5 million possible two-feature correlations. Also, as microbes live in communities, there are likely three-feature interactions, four-feature interactions and more. An additional challenge is that microbial sequence data provide relative abundances based on a fixed total number of sequences rather than absolute abundances, which introduces the problem of compositions (Lovell et al., 2010; Friedman and Alm, 2012). Sparsity of the features and missing data owing to incomplete sampling further complicates statistical analysis (Reshef et al., 2011; Friedman and Alm, 2012). Finally, microbes may display diverse types of relationships, such as linear, exponential or periodic, and most tests are not general enough to detect them all; even those that do are unlikely to detect different functions with the same efficiency (Reshef et al., 2011).
There are many different approaches for computing these correlation networks. In theory, any method that measures relationships between features can be used: for example, metrics like Bray–Curtis (Bray and Curtis, 1957), which measures abundance similarity; the Pearson correlation coefficient, which assesses linear relationships; and the Spearman correlation coefficient, which measures rank relationships are all potentially applicable (Spearman, 1904; Pearson, 1909). Software programs have been developed and optimized specifically to correct for certain aspects of correlation analysis of natural populations. For example, CoNet (Faust et al., 2012) acknowledges that various techniques have different strengths and weaknesses and/or are designed to optimally detect different functional relationships, and thus uses an ensemble method with the ReBoot procedure for P-value computation to combine information from several different standard comparison metrics. Local Similarity Analysis (LSA) (Ruan et al., 2006; Beman et al., 2011; Steele et al., 2011; Xia et al., 2013) is optimized to detect non-linear, time-sensitive relationships and can be used to build correlation networks from time-series data. The Maximal Information Coefficient (MIC) (Reshef et al., 2011) is a non-parametric method designed to capture a wide range of associations without limitation to specific function types (such as linear or exponential) and to give similar scores to equally noisy relationships of different types. MENA (Zhou et al., 2011; Deng et al., 2012) adapts Random Matrix Theory (RMT) from physics to microbiome data, and attempts to be robust to noise and to arbitrary significance thresholds. Finally, SparCC (Friedman and Alm, 2012) is particularly designed to deal with compositional data, as it is based on Aitchison’s log-ratio analysis (Aitchison, 1986).
The performance and limitations of most of these computational methods for inferring correlation networks have not been comparatively evaluated using either real or theoretical data sets, leaving researchers to guess at important properties of their networks such as sensitivity, specificity, precision and—most importantly—ability to provide interpretable results. Counts of true positives (TP), false positives (FP), TN (true negatives), FN (false negatives), and calculations of sensitivity (true positive rate—TP/(TP+FN)), specificity (true negative rate—TN/(FP+TN)) and precision (TP/(TP+FP)) are among standard benchmark measures. Without an understanding of these important properties, correlation analysis risks diverting attention from meaningful interactions and leading to wasteful pursuit of expensive in vitro or in vivo validations of mechanisms. One previous effort in this area tested mainly basic correlation measures for one type of model system (Berry and Widder, 2014).
Here, we tested the ability of each of these widely used correlation measures and tools to detect a variety of dependent relationships in both simulated and real microbial data sets. Figure 1a outlines the general workflow. Supplementary Table 1 and the Methods section detail how mock data were generated, and all code, test-code and documentation is available at ftp.microbio.me/pub/cooccurrence_files.zip. In brief, our simulations comprised 91 different data tables (columns in microbiome data typically represent samples, whereas microbes/features represent rows) with the number of microbes per table ranging from 200 to 10 000, and generated from eight different sample data generation models: distribution/copula (Trivedi and Zimmer, 2007), experimental, normalization, feature filtering, null/random, linear and non-linear (Lotka–Volterra) ecological (Volterra, 1926) and time-series. Within some models, we also introduced the aforementioned compositional and sparsity challenges.

Overview and motivation of correlation network technique benchmarking. (a) Mathematical properties of microbial communities naturally present in the environment are simulated in different feature × sample tables. These tables are evaluated for significant feature correlation networks by different metrics and toolkits. The networks are then assessed for accuracy. (b) Correlation tools find very different significant pairs on the same data set. A blue (pink) line connects significant positively (negatively) correlated OTU pairs.
Materials and methods
Tools
CoNet
For each of five similarity measures ((Bray and Curtis, 1957), Kullback–Leibler dissimilarity, Pearson (1909) and Spearman (1904) correlation, and mutual information), a distribution of all pair-wise scores was computed (Faust et al., 2012). Given these distributions, initial thresholds were selected such that the initial network contained 2000 positive and 2000 negative edges supported by all five measures. For each measure and edge, 1000 permutation (with renormalization for correlation measures) and bootstrap scores were generated, following the ReBoot routine. The measure-specific P-value was then computed as the probability of the null value (represented by the mean of the null distribution) under a Gauss curve generated from the mean and s.d. of the bootstrap distribution. As a one-sided test was carried out, P-values close to one were considered indicative of mutual exclusion and converted into low P-values by subtraction from one. Next, measure-specific P-values were merged using Brown’s method (Volterra, 1926), which takes dependencies between measures into account. After applying Benjamini–Hochberg’s (Benjamini and Hochberg, 1995) false discovery rate correction, edges with merged P-values below 0.05 were kept. Any edge for which the five measures did not agree on the interaction type (that is positive or negative) or whose initial interaction type contradicted the interaction type determined with the P-value was also discarded. Edges with scores outside the 95% confidence interval defined by the bootstrap distribution or not supported by all five measures were discarded as well.
RMT
All RMT calculations were implemented through the Molecular Ecological Network Approach Pipeline at http://ieg2.ou.edu/MENA (Deng et al., 2012). Pearson correlation coefficient (r-value) was calculated between each pair of OTUs and a symmetric similarity matrix was formed after all r-values were calculated. Theoretically, the RMT approach is applicable to any similarity matrix (Deng et al., 2012), but here it was only used to automatically detect a reliable cutoff for the Pearson correlation matrix based on the χ2-test with Poisson distribution. The threshold for defining a network is mathematically determined by calculating the transition from Gaussian orthogonal ensemble to Poisson distribution of the nearest-neighbor eigenvalues, and hence the network is automatically defined based on the data structure itself. To control the FP rate, the most stringent thresholds (significance of χ2>0.05) were set for the tests.
MIC
MIC was calculated with default parameters in minerva, an R wrapper for the cmine implementation of Maximal Information-based Nonparametric Exploration statistics, to quantify the linear or non-linear association between pairs of OTUs (Reshef et al., 2011). An empirical approach was taken for P-value calculation; for example, with a P-value threshold of 0.001, the MIC threshold that made the top 0.001 (one-thousandths) of the edges significant was chosen. Bonferroni multiple hypothesis test correction was applied (Dunn, 1961).
LSA
The eLSA analysis was run with the program’s default parameters, that is, with no delay allowed (delayLimit=0), P-value calculated by theoretical approximation (P-valueMethod=theo), required precision of P-value as 1/1000 (precision=1000), and data rank-normalized and z-transformed (normMethod=robustZ) (Ruan et al., 2006; Xia et al., 2013). Multiple hypothesis correction was done using q-values (Storey, 2002).
SparCC
SparCC was run with default parameters and 500 bootstraps (Friedman and Alm, 2012). Pseudo P-values were calculated as the proportion of simulated bootstrapped data sets with a correlation at least as extreme as the one computed for the original data set.
Pearson and Spearman correlations
The Fisher z-transformation was used to calculate P-values (Fisher, 1915; Spearman, 1904; Pearson, 1909). Bonferroni multiple hypothesis test correction was applied (Dunn, 1961).
Bray–Curtis
An empirical approach was taken for P-value calculation; for example, with a P-value threshold of 0.001, a correlation threshold that made the top 0.001 (one-thousandth) of the edges significant was chosen (Bray and Curtis, 1957). Bonferroni multiple hypothesis test correction was applied (Dunn, 1961).
Models
Copula
This model enabled generation of random variables having a specified covariance matrix from a given distribution (Supplementary Methods) (Trivedi and Zimmer, 2007).
Null model
This model was used to generate data tables from null distributions of several types to support testing the false discovery rates of various tools. Three methods were implemented. In method 1, the OTU table was created by randomly drawing sample vectors from a given distribution and parameters. In method 2, the OTU table was created with compositions in mind and therefore the sum of each sample was constrained. Tables were either not sum-constrained (raw abundance) or sum-constrained (providing relative abundances by dividing each OTU by the total number of sequences in its sample) and were produced by the Dirichlet distribution. In method 3, the OTU table was created with compositional data in mind, similar to model 2, but with higher sparsity than is normally created with the Dirichlet procedure by subtracting the mean value of the table from all entries (entries<0=0).
Ecological
This model helped create tables with simple (ecologically based) relationships between OTUs to test if the tools can accurately recapture relationships that are defined by a mechanism rather than by a high correlation score. We chose this method to assess if relationships that exist in biological contexts can be revealed through correlation analysis as frequently reported. Amensal, commensal, mutual, parasitic, competitive and partial-obligate-syntrophic ecological models were tested. All interactions were linear and dependent on OTU abundance.
-
1
The amensal model depresses the abundance of OTU2 when OTU1 is present by strength*OTU1; OTU1 is unaffected by the presence of OTU2.
-
2
The commensal model increases abundance of OTU2 when OTU1 is present by strength*OTU1; OTU1 is unaffected by the presence of OTU2.
-
3
The mutualism relationship increases the abundance of OTU1 and OTU2 when both are present; the strength of increase in each OTU is proportional to the abundance of the other OTU.
-
4
The parasitism model increases the abundance of OTU1 and decreases abundance of OTU2 when both are present. Thus, OTU1 grows at the expense of OTU2 with strength proportional to the abundance of OTU2.
-
5
The competitive model depresses the abundance of both OTUs if both OTUs are present. This simulates OTU competition for some limiting resource with the strength of each OTU’s decrease proportional to the abundance of the other OTU.
-
6
The obligate syntrophy model allows OTU2 only when OTU1 is present at abundance proportional to strength. This mimics a relationship where OTU2 depends on the presence of OTU1 and cannot exist without it.
-
7
The partial-obligate-syntrophy model allows OTU2 only if and only if OTU1 is present. This is similar to obligate syntrophy except the presence of OTU1 does not necessarily mean OTU2 is also present.
Lotka–volterra
These are systems of n differential equations that model the dependencies and interactions of the abundances of n species. The most widely used are simple two-species system of equations modeling predator-prey (for example, fox and rabbit) abundances (Supplementary Figures 12a–f), developed by Volterra (1926). The behavior of the Lotka–Volterra equations is much less understood for systems larger than two-species; for example, starting with the three-species equations, chaotic behavior may occur, the system dynamics become much more complex (Idema, 2005). For the six-species equations in this paper, we used small variations of the six-species systems of equations explored by Idema (2005). Because of the system complexity, small variations in the interaction matrix lead to very different abundance patterns (Supplementary Figures 12g–i).
Time Series
This model creates OTU tables with simple time-series relationships. All signals take the form of: y_shift+alpha*signal_function(phi(theta+omega))+noise, where alpha is the amplitude, phi is the frequency, and omega is the phase shift. Options to subsample the waves at even/randomly selected indices, or add sparsity are included.
Table Sets
Details of table set construction and filtering are provided in Supplementary Table 1 and Supplementary Methods.
Results
Tools infer significantly different numbers of edges in most data sets
Different tools consistently produce very different numbers and types of significant edges for the same data (Figure 1b, Supplementary Figure 1). As a corollary, tools are generally dissimilar in which edges they detect; demonstrating an average of 31.5% shared edge inference for all pair-wise combinations of tools, and for all data sets/models tested. This discordance further underscores the need for benchmarking, and suggests that the techniques may have differing strengths and weaknesses in response to the diverse challenges presented by microbiome data.
Sampling significantly alters edge inferences
Compositions can be troublesome to sequencing data interpretation because if the abundance of one species increases, and the others do not change, there is less room in the fixed sample sum for the other species to be counted, thus inducing spurious correlations (Pearson, 1897; Lovell et al., 2010; Friedman and Alm, 2012). Theory suggests that lower numbers of species types should increase compositional effects (Friedman and Alm, 2012). We used a set of five copula tables with decreasing numbers of effective species (a measure of microbial diversity) to test how compositional data impacts each of the correlation measures (Figure 2, Supplementary Figure 4). We also tested different normalization approaches, which are applied to tables of OTU sequence counts (OTU tables) to correct for differences in sampling efforts (McMurdie and Holmes, 2014). Rarefying, or drawing without replacement from each sample’s distribution until all samples have the same total number of sequences, metagenomeSeq’s cumulative sum scaling (Paulson et al., 2013) and DESeq’s log-ratio-based variance stabilizing transformation (Anders and Huber, 2010) were examined.

The impact of compositional data and normalization strategy on reconstructing actual microbial interactions. Five tables with varying neff (36, 25, 19, 10, 4) were created by multiplication of the abundances of one OTU pair by a constant; all other OTU abundances remained the same for all tables. These ‘Abundance’ tables represent the actual OTU abundances in the environment. SparCC assumes the data table is compositional, and hence is not shown. Then, the ‘Abundance’ tables were sampled without replacement (rarefied), constraining the sum and inducing compositionality, mimicking the experimental sampling process. The rarefied (2000 library size) tables were then either rarefied further (rarefy 1000 library size), CSS normalized or DESeq normalized. From left to right: (a) The five circles within each normalization technique represent: of all the edges found in the five neff tables, the number of edges found 1 (red)—5 (blue) times. A technique less affected by the compositional nature of the data has a larger circle at point 5, as most tools do in the ‘Abundance’ tables. (b) Precision of a tool’s estimates on the compositional normalized tables as compared with the same tool’s predictions on the ‘Abundance’ tables for a given neff. A larger circle represents better reconstruction of the true ‘Abundance’ OTU correlations.
Although the correlations do well on the ‘Abundance’ tables, we see a marked shift in the number of correct edges for most tools as soon as the total sum of counts is constrained, which worsens with smaller neff. Many edge pairs vary between the same data set at different neff (Figure 2a), and deviate from the edge predictions based on absolute environmental OTU abundances (Figure 2b). Rank-based measures such as MIC and Spearman, as well as Bray–Curtis, are less affected by compositional data but still not immune. SparCC maintain high precision compared with predictions on ‘Abundance’ tables with low neff. However, if network overlap is measured, no technique does well (Supplementary Figure 9). We do not recommend DESeq normalization for correlations owing to the negative values it produces. Normalization is discussed more in the Supplementary Note, and Supplementary Figures 2 and 3. In general, across all tools and normalization techniques, the slope of the function describing the number of total edges for a given neff (Supplementary Figure 4) changes particularly quickly at low neff (Inverse Simpson neff<13), suggesting that the smaller the number of effective species, the larger the impact on edge inference results. Given these findings, promising work has been done on addressing compositional data as a significant challenge to co-occurrence network inference, but the problem is still not solved.
The number of FP in null data is within expectations but differs by tool/technique and in some cases distribution
Control of the number of FP is well established in traditional statistical analysis (Dunn, 1961; Hochberg and Benjamini, 1990; Storey and Tibshirani, 2003) but has not been standardized for correlation inference. RMT allows the method itself to set the correlation threshold, rather than employing an arbitrary user-imposed threshold. LSA, CoNet and SparCC calculate the P-value through permutation-based approaches, and q-value (Storey and Tibshirani, 2003) and Benjamini–Hochberg multiple hypothesis testing correction. MIC and Bray–Curtis calculate the P-value through distributional approaches, Pearson and Spearman calculate the P-value with Fisher z-transformation, and all apply stricter Bonferroni multiple hypothesis testing correction. Note that as the correlation techniques use different approaches for generating P-values and multiple hypothesis testing correction, they are not quite comparable. The impact of this is beyond the scope of the paper, but to lessen its effects we evaluate the techniques at multiple P-value thresholds.
To enable assessment of the relative performance of these methods, we created two ‘null’ data tables, one containing random draws from six different zero-heavy distributions and the other from a Dirichlet distribution modeled on real data. (The former simulates differently distributed non-compositional data in which vectors are independent and identically distributed within a distribution, whereas the latter simulates compositional data, which are not independent and identically distributed, but for which no correlation matrix is specified. Both of these data tables should have no true associations between features.) The performance of the tested tools on these data is generally excellent (Supplementary Figure 10), despite differences in P-value calculation and multiple hypothesis testing. RMT and CoNet have the lowest rate of FP. However, although the false-positive rates (FP/(FP+TN)) are in-line with specified P-values for tools that rely on them, the false discovery rates (FP/(FP+TP)) are not, as TP=0 for these tables. This suggests extremely low precision (below 0.2) for all tools.
All tools are sensitive to several distribution shapes, except for LSA, MIC, Spearman and SparCC. For example, RMT and CoNet demonstrate an unexpected tendency to preferentially select edges from certain distributions. RMT shows a preference for χ2-distributed OTUs, and CoNet prefers OTUs from the χ2-, Nakagami and lognormal distributions (Supplementary Figure 11). Bray–Curtis almost exclusively selects edges from the uniform distributions, whereas Pearson finds three times fewer edges from the uniform distribution compared with the other distributions. This means that these tools may preferentially select as correlated the OTUs exhibiting these distributions. For example, if uniform or χ2-distributed OTU correlations are preferred, parasitic relationships, where one species benefits and the other is harmed, may go undetected.
A subset of common linear ecological relationships is detectable by some tools
Correctly detecting ecologically meaningful relationships such as competition and mutualism is essential for a correlation tool. To test tools’ capacity to identify these relationships, we developed simple linear models of the amensal, commensal, competitive, mutual, obligate, parasitic and partial-obligate-syntrophic ecological relationships (Materials and methods). These ecological relationships manifest as a dependency between the species abundances for a given ecological relationship type. We built tables where the type, strength and number of OTUs in a linear relationship varied, and introduced compositions, sparsity or both. Mutualism and commensalism are well detected by most tools (Figure 3a,Supplementary Note), whereas amensalism and partial-obligate-syntrophy are undetectable. All tools detect parasitism as a co-presence rather than as mutual exclusion, but three tools (SparCC, Spearman and LSA) correctly identify competitive relationships as mutual exclusions. As expected, tool performance generally improves with increasing strength of a relationship (that is, increasing signal/noise ratio). Literature suggests that many biological interactions are mediated by more than two-species interactions (Shade et al., 2012). In tests of data with more than two members, detection profiles were similar to two-species relationships, but considerably attenuated (Figure 3b). SparCC and LSA are unique among the tested tools for their ability to correctly infer a competitive three-member relationship as having components of both co-presence and mutual exclusion. Nonetheless, our results suggest that microbial relationships having greater than three members are likely impossible to detect with current approaches.

Types of linear ecological relationships detected by each correlation technique. The columns represent the seven types of engineered ecological relationships, and the rows indicate the eight tools tested. Each cell contains three histograms with increasing ‘strength’ of relationship from left to right. The fill in each bar represents the fraction of engineered edges detected as significant when the relationships were composed of (a) pairs of features or (b) triples or more.
The features in these data sets were independent and identically distributed unless part of an engineered correlation, which allowed us to accurately assess tool sensitivity and specificity. ROC curves of the ecological data confirm that increasing the complexity of the ecological relationships by mixing three-species relationships with simpler two-species relationships (Supplementary Figure 12a) significantly decreases tool specificity and sensitivity. Although tool performance improves on only two-species ecological data even with the addition of compositional effects (Supplementary Figure 12b), increasing sparsity (Supplementary Figure 12c) to levels commonly seen in microbiome data sets drastically reduces tool performance to little better than random guessing.
In agreement with the above null data, precision of the tools is also extremely poor (close to or at zero) under realistic conditions (Figures 4a–c). We place more importance on precision and sensitivity, because although it is easy to create a large network, it is much more important to predict interactions that are true and can be investigated further. Tool performance above the 45-degree line, which represents random guessing, is useful. LSA, and at a few times, MIC and Spearman rise above the 45-degree line; however, not far above the line, which indicates large room for future improvement. Performance does improve for stronger ecological relationships (Supplementary Fig 13), but only slightly. In light of how drastically performance decreases with increasing OTU sparsity (Figure 4, Supplementary Figures 12 and 13a–c), we suggest removing rare OTU predictions from the network. Plots of TP and FP predictions show that the ratio of TP to FP decreases markedly at ~50% OTU sparsity (Supplementary Figure 14). This 50% threshold could be adjusted depending on the technique, data set, and user preferences. Although OTU removal destroys network structure, we found that a high rate of FP is likely more destructive.

Tool precision is extremely low under realistic microbiome data set conditions. Precision vs recall (sensitivity) curves for linear ecological relationships (a–c) and non-linear/Lotka–Volterra ecological relationships (d–h). All tables were ~40% sparse, except (c) and (h), which were 70% sparse. The CoNet ROC curve does not extend from the bottom left corner to the top right corner of the ROC curves because of the filtering procedure CoNet uses prior to inferring any correlations. RMT is only a single point since the algorithm sets the P-value, instead of the user imposing a P-value. Although the dots are connected by interpolation, only the dots themselves have been measured.
Non-linear ecological relationships are harder to detect than linear ecological relationships
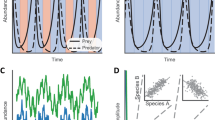
Lotka–Volterra models are a set of classic ecological models for interacting species based on coupled first-order differential equations (Volterra, 1926) that are applicable in a wide range of macro-scale ecological relationships (Shade et al., 2012). Evidence is emerging for their applicability at the micro scale as well—for example, in describing the microbial dynamics in a cheese model community (Mounier et al., 2008) and within individuals (Gerber 2014), as well as their shifts in response to environmental perturbations (Pepper and Rosenfeld, 2012). Previous investigation in this area mostly tested standard correlation metrics not developed for microbiome data (Berry and Widder, 2014). We created two- and six-species Lotka–Volterra interactions (Supplementary Figure 15) and tested whether tools accurately capture these relationships when they are embedded in random noisy signals.
The irregularity of the Lotka–Volterra equations proves difficult for all measures, with an average 10% drop in sensitivity compared with the linear ecological relationships. For the two-species edges, MIC, SparCC, LSA, CoNet and Spearman all perform strongly for both count and compositional tables (Figures 4d and e, Supplementary Figure 12d and e,Supplementary Table 2), whereas SparCC consistently performs well on the six-species Lotka–Volterra tables (Figures 4f and g). Pearson also performs well on the six-species tables because some of the dissipative relationships display linear correlations. However, again under realistic conditions, when sparsity is boosted from 40 to 70%, performance drops to little better (or even worse) than random guessing (Supplementary Figure 12h). The same is true for precision (Figure 4h).
Time-dependent relationships vary based on signal, sampling frequency and time shift
Correlations in time-series data are well studied in other fields, but microbiological studies are just beginning to show predictable shifts in microbial communities over time (Caporaso et al., 2011; Gonzalez et al., 2012; Shade et al., 2013). For example, in Caporaso et al., the fluctuations appear sinusoidal (Caporaso et al., 2011). Generally, detected edges varied depending upon at which point in time/how many samples were taken of the fluctuating OTUs (Figure 5). More details can be found in the Supplementary Note, and Supplementary Figures 16 and 17. Together, the time-series results indicate an important area of future research, as researchers take discrete samples, and therefore cannot know the abundance of each OTU at every point in time.

The time, or point in the feature signal cycle, at which a sample is taken introduces variability in detected correlations. The number of samples is also a large influence in reconstructing the correct signal, and therefore correlation. The number of co-occurring feature pairs found in 26, 50 and 76 points randomly sampled from a 100 time point time-series of features composed of signals with varying noise, amplitude offset, phase shift, frequency and coupling. These mixture model tables had signals composed of sine, cosine, sawtooth and logarithmic patterns.
Ensemble approaches boost precision and the F1 score
Because tools detect different edges in the same data, we hypothesized that combining tools for detection purposes might improve precision. We treat the CoNet approach (Materials and methods), which is an ensemble approach of the standard metrics in itself and implements renormalization and permutation (ReBoot) for P-value calculation (Faust et al., 2012), as one tool. The ensemble approach tested included the toolkits, for example, SparCC, and simply calculated the intersection of the edges below a certain P-value, here 0.001, yielded by each technique (Figure 6a). In our tests on the linearly ecologically modeled data where engineered correlations are known, the increase in precision for the ensemble approach is marked compared with most tools alone—with many combinations finding zero FP—at a cost to sensitivity (Supplementary Table 3). Although the ensemble shows little gain against MIC or LSA (Figure 6b) in theoretical data, the gains become larger when sparsity is increased from 40% to a more realistic 70%, although all tools still suffer from drastically decreased sensitivity or hit rate. Our results suggest that an ensemble approach including CoNet, SparCC, Spearman and Pearson, should be used when precision is required, for example, for developing biological hypotheses on species interactions to test with co-culturing. If low FP rates are not critically important, and the OTU table is over half zeroes, we recommend using an ensemble of CoNet and Pearson for increased F1 score. For Lotka–Volterra 70% sparse ecological relationships, LSA also has high precision/F1 score (Supplementary Table 2).

Ensemble approach increases precision and the harmonic mean of precision and sensitivity. (a) Simple two-tool explanation of ensemble approach. Edges in green are found to be significant by tool one in left network and tool two in middle network. Blue edges in the right network are those edges found by both tool one and tool two. The ensemble approach tested all 28 possible one to eight member combinations. (b) The top three ensemble approaches ranked by F1 score (harmonic mean of precision and sensitivity, Supplementary Table 4) on each linear ecological table type (tables 1.6, 1.7—two- and three-species abundance tables—45% sparse, table 2.16 compositional—40% sparse, table 2.17 counts—70% sparsity, table 2.18 compositional—70% sparse) compared with the tools alone. LSA is hidden beneath the ensemble approaches for the tables 1.6 and 1.7.
Discussion
Correlation detection is an emerging analytical technique that can select biochemically or ecologically relevant feature pairs in microbial sequencing data. At the highest level, there is much disagreement between inferred networks generated from different tools on the same data (Figure 1b,Supplementary Figure 1), necessitating benchmarking. Although the potential of this approach is clear, our work shows that current tools have significant limitations that must be accounted for when performing correlation analyses. More specifically, the usual corrected P-value threshold of 0.05 is too lenient to allow high-precision detection with almost all tools; a threshold such as 0.001 is more useful. Also, processing choices such as sequencing technology type and normalization (Supplementary Notes) have a great impact on which network edges are detected. New strategies must be explored and validated to mitigate the impact of preprocessing on network topology. It is noteworthy that the RMT approach, which in this study is paired with Pearson correlation, significantly improves the precision and F1 score of Pearson correlation alone. Hence, future investigation of RMT paired with other correlation measures, such as Spearman, is promising. Our results confirm that progress, as measured by precision, has been made on addressing previously published compositional effects in the context of low numbers of effective species (Friedman and Alm, 2012) (meaning that when a few microbes are highly abundant, fluctuations in these dominant abundances changed the resulting correlation networks dramatically owing to the sum constraint on the total number of sequences per sample).
Encouragingly, all tools have reasonable false-positive rates. However, detection of ecological relationships (manifested as abundance dependencies) is poor for relationships other than commensalism and mutualism (Figure 3), and sparsity is perhaps the most significant unaddressed challenge of all (Figures 4c and h). Hence, we recommend filtering out extremely rare OTUs prior to network construction. Tool performance degraded significantly for OTUs containing >50% zeroes. Nonetheless, the best options depending upon input data set characteristics are summarized in Figure 7 and Table 1, and tool computational time in the Supplementary Note. If associations between sparse OTUs are to be predicted, a reality in many data sets, an ensemble approach is best for high-precision detection of linear relationships in, for example, situations where explicit tests of all hypothesized interactions are prohibitively inefficient. For sparse Lotka–Volterra relationships, LSA alone yields the highest precision (0.2). Also, tools robust to noise (for example, assessed by multiple rarefactions on experimental data—see Supplementary Figures 2 and 3)—are likely to perform better on real-world data sets. Finally, although the tools may accurately identify certain overall biological relationships, researchers should be aware of which relationships a given tool is actually capable of detecting: for instance, concluding that a particular microbial community shows no signs of amensal interactions on the basis of a correlation analysis is likely incorrect, as none of the tested tools could accurately identify engineered amensal correlations.

Workflow diagram summary indicating the best correlation technique depending upon data set characteristics and desired ecological relationship discovery.
Thus, we have identified the strengths and weaknesses of the main microbial correlation analysis techniques, and provided many recommendations for future study and toolkit use.
Despite their weaknesses, the correlation techniques have proved useful in a number of biological and experimental settings, as mentioned in the introduction. Study of correlation network analysis will likely continue to grow, given its significance. Supplementation of the data sets utilized here with new data sets containing experimentally verified microbial interactions would be invaluable to progress in this area.
References
Aitchison J . (1986) The Statistical Analysis of Compositional Data. Chapman and Hall: London; New York, NY, USA.
Anders S, Huber W . (2010). Differential expression analysis for sequence count data. Genome Biol 11: R106.
Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR et al. (2011). Enterotypes of the human gut microbiome. Nature 473: 174–180.
Beman JM, Steele JA, Fuhrman JA . (2011). Co-occurrence patterns for abundant marine archaeal and bacterial lineages in the deep chlorophyll maximum of coastal California. ISME J 5: 1077–1085.
Benjamini Y, Hochberg Y . (1995). Controlling the false discovery rate - a practical and powerful approach to multiple testing. J Roy Stat Soc B Met 57: 289–300.
Berry D, Widder S . (2014). Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front Microbiol 5: 219.
Bray JR, Curtis JT . (1957). An ordination of upland forest communities of southern Wisconsin. Ecol Monographs 27: 325–349.
Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A et al. (2015). Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517: 205–208.
Caporaso JG, Lauber CL, Costello EK, Berg-Lyons D, Gonzalez A, Stombaugh J et al. (2011). Moving pictures of the human microbiome. Genome Biol 12: R50.
Chaffron S, Rehrauer H, Pernthaler J, von Mering C . (2010). A global network of coexisting microbes from environmental and whole-genome sequence data. Genome Res 20: 947–959.
Deng Y, Jiang YH, Yang Y, He Z, Luo F, Zhou J . (2012). Molecular ecological network analyses. BMC Bioinformatics 13: 113.
Dunn OJ . (1961). Multiple comparisons among means. J Am Stat Assoc 56: 52–64.
Faust K, Raes J . (2012). Microbial interactions: from networks to models. Nat Rev Microbiol 10: 538–550.
Faust K, Sathirapongsasuti JF, Izard J, Segata N, Gevers D, Raes J et al. (2012). Microbial co-occurrence relationships in the human microbiome. PLoS Comput Biol 8: e1002606.
Fisher RA . (1915). Frequency distribution of the values of the correlation coefficient in samples from an indefinitely large population. Biometrika 10: 507–521.
Friedman J, Alm EJ . (2012). Inferring correlation networks from genomic survey data. PLoS Comput Biol 8: e1002687.
Gerber GK . (2014). The dynamic microbiome. FEBS Lett 588: 4131–4139.
Gevers D, Kugathasan S, Denson LA, Vazquez-Baeza Y, Van Treuren W, Ren B et al. (2014). The treatment-naive microbiome in new-onset Crohn's disease. Cell Host Microbe 15: 382–392.
Gonzalez A, King A, Robeson MS 2nd, Song S, Shade A, Metcalf JL et al. (2012). Characterizing microbial communities through space and time. Curr Opin Biotechnol 23: 431–436.
Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R et al. (2014). Human genetics shape the gut microbiome. Cell 159: 789–799.
Gough E, Shaikh H, Manges AR . (2011). Systematic review of intestinal microbiota transplantation (fecal bacteriotherapy) for recurrent Clostridium difficile infection. Clin Infect Dis 53: 994–1002.
Greenblum S, Turnbaugh PJ, Borenstein E . (2012). Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proc Natl Acad Sci USA 109: 594–599.
Hochberg Y, Benjamini Y . (1990). More powerful procedures for multiple significance testing. Stat Med 9: 811–818.
Idema T . (2005), The behaviour and attractiveness of the Lotka-Volterra equations. Doctorate thesis, Leiden University.
Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI . (2005). Obesity alters gut microbial ecology. Proc Natl Acad Sci USA 102: 11070–11075.
Lima-Mendez G, Faust K, Henry N, Decelle J, Colin S, Carcillo F et al. (2015). Ocean plankton. Determinants of community structure in the global plankton interactome. Science 348: 1262073.
Lovell D, Müller W, Taylor J, Zwart A, Helliwell C . (2010). Caution! compositions! technical report and companion software (publication–technical). Technical Report EP10994, CSIRO.
Lozupone C, Faust K, Raes J, Faith JJ, Frank DN, Zaneveld J et al. (2012). Identifying genomic and metabolic features that can underline early successional and opportunistic lifestyles of human gut symbionts. Genome Res 22: 1974–1984.
Lozupone CA, Li M, Campbell TB, Flores SC, Linderman D, Gebert MJ et al. (2013a). Alterations in the gut microbiota associated with HIV-1 infection. Cell Host Microbe 14: 329–339.
Lozupone CA, Stombaugh J, Gonzalez A, Ackermann G, Wendel D, Vazquez-Baeza Y et al. (2013b). Meta-analyses of studies of the human microbiota. Genome Res 23: 1704–1714.
McMurdie PJ, Holmes S . (2014). Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput Biol 10: e1003531.
Mounier J, Monnet C, Vallaeys T, Arditi R, Sarthou AS, Helias A et al. (2008). Microbial interactions within a cheese microbial community. Appl Environ Microbiol 74: 172–181.
Oakley BB, Morales CA, Line J, Berrang ME, Meinersmann RJ, Tillman GE et al. (2013). The poultry-associated microbiome: network analysis and farm-to-fork characterizations. PloS One 8: e57190.
Paulson JN, Stine OC, Bravo HC, Pop M . (2013). Differential abundance analysis for microbial marker-gene surveys. Nat Methods 10: 1200–1202.
Pearson K . (1897). On a form of spurious correlation which may arise when indices are used in the measurement of organs. Proc R Soc London 60: 489–502.
Pearson K . (1909). Determination of the coefficient of correlation. Science 30: 23–25.
Pepper JW, Rosenfeld S . (2012). The emerging medical ecology of the human gut microbiome. Trends Ecol Evol 27: 381–384.
Reshef DN, Reshef YA, Finucane HK, Grossman SR, McVean G, Turnbaugh PJ et al. (2011). Detecting novel associations in large data sets. Science 334: 1518–1524.
Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL et al. (2013). Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341: 1241214.
Ruan Q, Dutta D, Schwalbach MS, Steele JA, Fuhrman JA, Sun F . (2006). Local similarity analysis reveals unique associations among marine bacterioplankton species and environmental factors. Bioinformatics 22: 2532–2538.
Shade A, Peter H, Allison SD, Baho DL, Berga M, Burgmann H et al. (2012). Fundamentals of microbial community resistance and resilience. Front Microbiol 3: 417.
Shade A, Caporaso JG, Handelsman J, Knight R, Fierer N . (2013). A meta-analysis of changes in bacterial and archaeal communities with time. ISME J 7: 1493–1506.
Spearman C . (1904). The proof and measurement of association between two things. Am J Psychol 15: 72–101.
Steele JA, Countway PD, Xia L, Vigil PD, Beman JM, Kim DY et al. (2011). Marine bacterial, archaeal and protistan association networks reveal ecological linkages. ISME J 5: 1414–1425.
Storey JD . (2002). A direct approach to false discovery rates. J Roy Stat Soc B 64: 479–498.
Storey JD, Tibshirani R . (2003). Statistical significance for genomewide studies. Proc Natl Acad Sci USA 100: 9440–9445.
Trivedi PK, Zimmer DM . (2007) Copula Modeling: an Introduction for Practitioners. Now publishers inc.: Boston, UK.
Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE et al. (2009). A core gut microbiome in obese and lean twins. Nature 457: 480–484.
Volterra V . (1926). Variazioni e fluttuazioni del numero d’individui in specie animali conviventi. Mem Acad Lincei Roma 2: 31–113.
Vrieze A, Van Nood E, Holleman F, Salojarvi J, Kootte RS, Bartelsman JF et al. (2012). Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 143: 913–916 e917.
Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B et al. (2011). Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472: 57–63.
Xia LC, Ai D, Cram J, Fuhrman JA, Sun F . (2013). Efficient statistical significance approximation for local similarity analysis of high-throughput time series data. Bioinformatics 29: 230–237.
Zhou J, Deng Y, Luo F, He Z, Yang Y . (2011). Phylogenetic molecular ecological network of soil microbial communities in response to elevated CO2. mBio 2: doi: 10.1128/mBio.00122-11.
Acknowledgements
WVT and SJW were supported by the National Human Genome Research Institute Grant# 3 R01 HG004872-03S2, and the National Institute of Health Grant# 5 U01 HG004866-04. JAF and JAC were supported by the Gordon and Betty Moore Foundation Grant# GBMF3779 and NSF Grant# 1136818. This work was supported in part by the Howard Hughes Medical Institute (RK was an HHMI Early Career Scientist). The National Human Genome Research Institute Grant# 3 R01 HG004872-03S2, the National Institute of Health Grant# 5 U01 HG004866-04, the Gordon and Betty Moore Foundation Grant# GBMF3779, NSF Grant# 1136818 and the Howard Hughes Medical Institute.
Author contributions
WWVT, SJW, CL and RK designed and conceived analyses. WVT and SJW performed data analysis and wrote the manuscript. KF, JF, YD, LCX and ZX ran the CoNet, SparCC, RMT, LSA and MIC correlation network techniques, respectively. All authors provided invaluable feedback and insights into analyses and the manuscript. All authors approved the final version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
About this article

Cite this article
Weiss, S., Van Treuren, W., Lozupone, C. et al. Correlation detection strategies in microbial data sets vary widely in sensitivity and precision. ISME J 10, 1669–1681 (2016). https://doi.org/10.1038/ismej.2015.235
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2015.235
This article is cited by
-
Ecological insights into soil health according to the genomic traits and environment-wide associations of bacteria in agricultural soils
ISME Communications (2023)
-
The potential to produce tropodithietic acid by Phaeobacter inhibens affects the assembly of microbial biofilm communities in natural seawater
npj Biofilms and Microbiomes (2023)
-
Succession of bacterial biofilm communities following removal of chloramine from a full-scale drinking water distribution system
npj Clean Water (2023)
-
Deciphering microeukaryotic–bacterial co-occurrence networks in coastal aquaculture ponds
Marine Life Science & Technology (2023)
-
Deciphering Interactions Within a 4-Strain Riverine Bacterial Community
Current Microbiology (2023)
