
Abstract
Archaea domain is comprised of many versatile taxa that often colonize extreme habitats. Here, we report the discovery of strictly anaerobic extremely halophilic euryarchaeon, capable of obtaining energy by dissimilatory reduction of elemental sulfur using acetate as the only electron donor and forming sulfide and CO2 as the only products. This type of respiration has never been observed in hypersaline anoxic habitats and is the first example of such metabolic capability in the entire Archaea domain. We isolated and cultivated these unusual organisms, selecting one representative strain, HSR2, for detailed characterization. Our studies including physiological tests, genome sequencing, gene expression, metabolomics and [14C]-bicarbonate assimilation assays revealed that HSR2 oxidized acetate completely via the tricarboxylic acid cycle. Anabolic assimilation of acetate occurred via activated glyoxylate bypass and anaplerotic carboxylation. HSR2 possessed sulfurtransferase and an array of membrane-bound polysulfide reductase genes, all of which were expressed during the growth. Our findings suggest the biogeochemical contribution of haloarchaea in hypersaline anoxic environments must be reconsidered.
Similar content being viewed by others
Introduction
Hypersaline terrestrial and marine brines host extremely halophilic euryarchaea, a unique group of Archaea thriving at salt saturation conditions. According to physiological and genomic studies, cultivated haloarchaea are predominantly aerobic heterotrophs (Andrei et al., 2012). There are only a few known examples of facultative anaerobic haloarchaea species capable of growth by either fermentation, or anaerobic respiration using nitrate, fumarate, dimethyl sulfoxide or trimethylamine N-oxide as terminal electron acceptors (Oren and Trüper, 1990; Oren, 1991; Antunes et al., 2008; Bonete et al., 2008; Werner et al., 2014). As these electron acceptors are present in hypersaline anoxic habitats at very low concentrations (Oren, 2011), it might appear that these facultative anaerobes cannot play a significant role in anaerobic environments. Nonetheless, molecular ecology studies demonstrated that highly reduced sulfur-rich hypersaline sediments and salt deposits are inhabited by haloarchaea with no cultured representatives (Walsh et al., 2005; Youssef et al., 2011). Can these organisms use elemental sulfur, which may be very abundant, as the alternative terminal electron acceptor? This question was addressed several times in the past 30 years (Grant and Ross, 1986; Tindall and Trüper, 1986; Elshahed et al., 2004a, 2004b), but no conclusive answer has been found. Acetate is an abundant electron donor and carbon source in anoxic habitats, which accumulates in the absence of high-potential electron acceptors, such as nitrate or ferric ion (Afshar et al., 1998; Kashevi et al., 2002; Slobodkina et al., 2009). Therefore, anaerobic oxidation of acetate with low potential electron acceptors, such as partially oxidized sulfur compounds, can be an important cross-point of microbiologically mediated carbon and sulfur cycles. Until now, practically nothing was known about these processes in hypersaline habitats.
Here, we describe novel strictly anaerobic haloarchaea that grow with 3–5 M NaCl and reduce sulfur using acetate or pyruvate as the only electron donors and forming sulfide (HS−) and CO2 as the only products. Our genomic and functional studies point at unique mechanisms that support this type of anaerobic respiration. We successfully isolated these organisms from different sites around the world, which suggests their ubiquity and ecological significance. In addition, our study indicates that the anaerobic oxidation of acetate is a rather common phenomenon in anoxic salt-saturated habitats.
Materials and methods
Origin of samples, enrichments and cultivation conditions
The top 10 cm layer of sediment cores and brine samples were collected from several hypersaline chloride-sulfate lakes located in the Kulunda Steppe (southwestern Siberia, Altai region, Russia) (Sorokin et al., 2012). Superficial anoxic sediments were also collected from sulfidic hypersaline lake Lagoona Fuente de Piedra (coordinates: 37°05.717N; 04°47.483E, Malaga, Spain) and from the deep-sea hypersaline lake Medee (coordinates: 34°26.250N; 22°19.783E, Ionian Sea, Eastern Mediterranean, 3105 m below sea level) (Yakimov et al., 2013). The mineral medium used for enrichment and further cultivation of extremely halophilic sulfur reducers contained the following (g l−1): 240 NaCl; 3 K2HPO4; 0.5 NH4Cl; 10 HEPES. The pH was adjusted to 7.0 by 1 M KOH. After sterilization, the medium was supplemented with 5 mmol l−1 MgCl2 × 6H2O, 1 ml l−1 of trace metal solution (Pfennig and Lippert, 1966) and 10 mg l−1 of yeast extract. Elemental sulfur flour was sterilized at 110 °C for 30 min as a wet paste and added to approximately 2 g l−1. Cultivation was performed in serum bottles with butyl rubber stoppers. The medium was made anoxic first by ‘cold boiling’ upon evacuation followed by three cycles of flushing with argon and evacuation. Anaerobic conditions were achieved by the final addition of 0.2 mmol l−1 HS−. After five cycles of evacuation-flushing, the residual HS− concentration in the medium was less than 5 μmol l−1. The cultures were incubated at 30–37 °C with periodic shaking. The ability for aerobic growth was examined in the absence of sulfur and sulfide, and in the presence of 1.0 mmol l−1 magnesium sulfate. Air was added by syringe to anoxic medium to the final concentration of 1% vol. of headspace. Sulfide (HS−) formation was measured using the standard methylene blue method (Trüper and Schlegel, 1964). The values were obtained by subtraction of the measurements in control (no cells) from the data in the culture. When sulfide reached the concentration of several mmol l−1, the enrichments were sub-cultured four to six times after 1:100 dilution. Pure cultures were obtained by serial dilutions and the final isolates were checked microscopically and by 16S rRNA sequencing.
Chemical analyses
After the biomass removal and acidification of the supernatant to pH 4.0, acetate concentrations were measured by gas chromatography (Chromoteq-Crystall 5000.2 (Chromateq, Joshkar-Ola, Russian Federation); column Sovpol-5, 1 m, detector PID in the range of temperatures between 180 and 230 °C). The cells from 1 to 4 ml culture samples were pelleted by centrifugation in 2 ml Eppendorf tubes at 13 000 r.p.m. for 20 min and washed with 4 M NaCl solution (pH 5.0) to remove the cell-bound FeS. After final centrifugation, the pellets were stored frozen until further analysis. Cell protein was measured using the Lowry method. Polysulfides were analyzed after methylation, in the form of dimethyl polysulfide as described elsewhere (Roman et al., 2014). Dimethyl disulfide, dimethyl trisulfide and higher dimethyl polysulfanes (Me2S4 to Me2S8) were subjected to high-performance liquid chromatography on Zorbax Extend-C18 column (1.8 μm, 2.1 × 50 mm, Agilent Technologies, Santa Clara, CA, USA) at 20 °C; the UV detector (Dionex UltiMate 3000RS, Thermo Fisher Scientific, Sunnyvale, CA, USA) was set to 210 nm. The mobile phase was 15 vol.% of methanol in water, which was switched at 0.72 min to a linear gradient of the methanol reaching 85% at 10 min. The flow rate was maintained at 0.371 ml min−1 and the injection volume was 1.25 μl. The purities of the standards were above 98% for dimethyl disulfide and dimethyl trisulfide (Sigma-Aldrich, Zwijndrecht, the Netherlands). The sample preparation procedure and derivatization protocol are described elsewhere (Roman et al., 2014). The samples were analyzed for their total sulfur content with an inductively coupled plasma optical emission spectrometer (ICP-OES) (Perkin Elmer, Optima 5300 DV, Waltham, MA, USA). Commercially available dimethyl trisulfide (Sigma-Aldrich) was used to validate the method for determination of dimethyl polysulfane concentration. The difference between the dimethyl trisulfide concentration of prepared solutions and the results obtained from ICP-OES analysis was below 1.5% in each of the five replicates. A 3-point calibration with known concentrations of dimethyl polysulfanes was performed every time for ever new sample series, as described previously (Roman et al., 2014).
RNA isolation and quantitative reverse transcription PCR analysis (Q-RT-PCR)
Q-RT-PCR was used to estimate the abundance of polysulfide reductases, pyruvate:ferredoxin oxidoreductase, isocitrate lyase and malate synthase transcripts. HSR2 cultures were obtained with acetate or pyruvate. Cells were collected by filtering (0.22 μm, Millipore, Billerica, MA, USA) 15–25 ml culture samples and total RNA was immediately purified using miRVANA kit (Ambion, Austin, TX, USA). RNA samples were treated with Turbo DNA-free kit (Ambion) and cDNA synthesis was performed with SuperScript II Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instruction. The RT reaction was carried out with 100 ng of total RNA. All Q-RT-PCR experiments were performed using an ABI 7500 Fast Real-Time PCR System thermocycler (Applied Biosystems, Foster City, CA, USA). Specific primers and TaqMan probes were designed using Primer Express software v.2.0 (Applied Biosystems) and are presented in Supplementary Tables S7A and S7B. TaqMan probes were created only for psrA genes and obtained from Biomers (Ulm, Germany). RNA samples were tested in triplicates along with ‘No Template Control’. The reaction mixtures for Taqman Q-RT-PCR were as follows: 0.8 μM final concentration of each primer, 0.2 μM TaqMan probe, 20 ng of template, 12.5 μl of 2 × TaqMan 5 Universal PCR Master Mix (PE Applied Biosystems) and ultrapure water added to the final volume of 25 μl. The reactions were performed under the following conditions: 2 min at 50 °C followed by 10 min at 95 °C, followed by 40 cycles of 15 s at 95 °C and 1 min at 60 °C. For SYBR Green Q-RT-PCR, initial denaturation was for 5 min at 95 °C, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. Each 25 μl reaction contained 20 ng of template, 12.5 ml of 2X SYBR Green PCR Master Mix (Applied Biosystems) and 0.1 μM of each primer. A dissociation protocol was run at the end of each SYBR Green real-time PCR reaction to verify that only the expected amplification product was generated. Q-RT-PCR amplification was analyzed using an automatic setting for the baseline and threshold values and using the relative standard curve method. Standards for all amplifications were prepared using known amounts of cloned target templates. Amplicons were generated by PCR amplification of the target genes from genomic DNA. The resulting amplicons were then purified using the Wizard SV Gel and PCR Clean–up System kit (Promega, Madison, WI, USA), and cloned in pGEM-T Easy Vector System I (Promega). After cloning, plasmids were extracted using the QIAprep Spin Miniprep kit (Qiagen, Hilden, Germany) and DNA concentrations were measured using a Nanodrop ND-1000 spectrophotometer (Wilmington, DE, USA). Standard curves were based on serial dilution ranging between 107 and 101 gene copies.
[14C]-bicarbonate assimilation
Two replicate gas-tight 100 ml serum vials were filled with 10 ml of acetate- or pyruvate-grown HSR2 cultures (1.1–1.5 × 107 cells ml−1) and 10 ml of fresh medium. Thirty microCi of [14C]-bicarbonate (56.0 mCi mmol l−1, Amersham Italia, Milan, Italy) were then added. Anaerobic conditions were achieved by final addition of 0.2 mmol l−1 HS−. The cultures, accordingly supplemented with acetate or pyruvate, were incubated for 10 days at 37 °C, then fixed by adding formaldehyde to the final concentration of 2% (vol/vol). Samples were filtered through 0.1 μm polycarbonate filters (Millipore), washed three times with 10 ml of prefiltered seawater, acidified in an HCl fume hood for 12 h to remove inorganic carbon and air-dried before addition of scintillation cocktail for [14C]-counting in a Wallac 1414 analyzer (Perkin Elmer, Monza, Italy). The disintegrations per minute values were calculated by subtracting the values detected in the [14C]-bicarbonate control with no biomass from the absolute counts obtained in the culture samples (Yakimov et al., 2014).
Genome sequencing and assembly
Sequencing was performed with a Roche 454 GS FLX Ti sequencer (454 Life Sciences, Branford, CT, USA) at Lifesequencing S.L. (Valencia, Spain). In parallel, sequencing was carried out on Illumina HiSeq 2000 platform at BGI (Hong Kong, China), yielding 5 736 282 paired reads with an average read length of 101 bp, totalling 579.36 Mbp. 454 assemblies were performed with Roche Newbler assembler v. 2.5.3 using the default parameters and Velvet v.1.1. The number of reads accounted to 527 470 sequences with a mean read length of 463.97 bp that provided a total of 244.72 Mbp. The assembly provided a total of 23 721 contigs, where 837 were longer than 500 bp and the N_50 10 833 with the largest contig of 32 349. These reads were further assembled using both Velvet 1.2.10 (Zerbino and Birney, 2008) and Geneious 7.1 software (http://www.geneious.com/). Gaps between contigs were closed in a conventional PCR-based approach and supported by manual refining with Geneious 7.1 embedded tools, resulting in a fully closed circular chromosome of 2 085 482 bp, and one circular plasmid of 124 256 bp. Together, all sequences provided 151 × coverage for HSR2 chromosome and 62 × for the plasmid.
Genome annotation
Genes were annotated by Glimmer 3.02 (Delcher et al., 2007), Geneious 7.1 software, NCBI online tool for rRNA and by tRNAScan-SE online tool for tRNA. Operon predictions were performed by the FgenesB online tools (Tyson et al., 2004). For each predicted gene, similarity searches were performed by BLAST in Geneious 7.1 against public sequence databases (nr, SwissProt. protein family databases (Pfam) and clusters of orthologous groups (COGs)). Finally, annotations were manually curated using the Artemis 15.0 program (Rutherford et al., 2000) and refined for each gene with NCBI blastx against nr database (Altschul et al., 1997). Secreted proteins were predicted with SignalP 4.1 server (http://www.cbs.dtu.dk/services/SignalP/index.php (Petersen et al., 2011)). Transmembrane helices were predicted using TMHMM Server v. 2.0 (http://www.cbs.dtu.dk/services/TMHMM/).
Phylogenetic analysis and general predictions
16S rRNA gene phylogeny of the HSR strains was inferred from a 16S rRNA gene sequence alignment with PAUP*4.b10 using a LogDet/paralinear distance method. Support for nodes in the tree corresponds to bootstrap values for 1000 pseudoreplicates. The tree has been arbitrarily rooted on the closest haloarchaea Salt Deposit Clade II (Walsh et al., 2005). The complete genomes of 29 haloarchaeal isolates were obtained from NCBI genome database (http://www.ncbi.nlm.nih.gov/sites/genome, see Supplementary Table S1). We selected six single copy genes identified in most archaea genomes (Lloyd et al., 2013) to form a group of orthologous proteins, in the following order: COG0052, Ribosomal protein S2; COG0092, Ribosomal protein S3; COG0081, Ribosomal protein L1; COG0090, Ribosomal protein L2; COG0087, Ribosomal protein L3; COG0088, Ribosomal protein L4. We concatenated them maintaining the same order for each haloarchaeon to form sequences ready to be aligned with the Clustal W (Larkin et al., 2007) plugin inside Geneious 7.1 software using the IUB DNA weight matrix. The resulted alignment was used for construction of the maximum likelihood phylogenetic tree using PhyML program version 3.0 (Guindon and Gascuel, 2003) with the Tamura-Nei substitution model. All positions containing gaps and missing data were eliminated.
The dataset of molybdopterin reductases was chosen as follows: 4 HSR2 molybdopterin oxidoreductases subunits A (HLASFs); 10 best BLAST hits of all 4 HLASFs were obtained via Uniprot BLAST utility (http://www.uniprot.org/blast/) and 2 databases for each: UniProtKB and UniProtKB-SwissProt. The results were filtered by ‘complete proteomes only’ option. All best hits were then filtered through 0.99 filter using CD-hit utility (http://weizhong-lab.ucsd.edu/cdhit_suite/cgi-bin/index.cgi?cmd=h-cd-hit) (Huang et al., 2010) to eliminate duplicates and reduce redundancy. Biochemically characterized sulfur reductase SreA from Acidianus ambivalens (Laska et al., 2003) was not detected among these best hits as it was still found in UniProt (but not in SwissProt) database and was more distant than the first 10 HLASFs UniProtKB best hits. Thus, the above SreA was added manually along with its top 10 UniProtKB hits. The homologous sequences were included to the previous dataset (Duval et al., 2008) after removal of partial sequences (ArrA_DesDC; ArrA_DesY5; ArrA_SulBa; ArrA_BacSe; Psr/PhsA_DesDe; AroA_AlcFa). Upon manual removal of duplicates, the final dataset consisted of 147 sequences. The dataset was aligned in MEGA6 (Tamura et al., 2013) using Muscle (Edgar, 2004). The evolutionary history was inferred by using the Maximum Likelihood method based on the Whelan And Goldman+Freq. model (Whelan and Goldman, 2001). All positions containing gaps and missing data were eliminated. There were a total of 361 positions in the final dataset. The tree with the highest log likelihood (−77939.2454) is shown. The percentages of trees in which the associated taxa clustered together (bootstrap values, 1000 replicates) are shown next to the branches. Evolutionary analyses were conducted in MEGA6. Final processing was carried out in MacVector (v. 11.1.2).
Analysis of clustered regularly interspaced short palindromic repeats (CRISPR)
Pilercr v1.02 with default parameters (Edgar, 2007) was used to identify short palindromic repeats and spacers in the genome of strain HSR2, using default parameters. Also, the CRISPRfinder online tool was used for control (Grissa et al., 2007). CRISPR and Cas homologs were identified with BLAST (Altschul et al., 1997). Spacer sequences detected in CRISPR system were analyzed to find similarity against plasmid, virus and prophage. Using the ACLAME database (Leplae et al., 2010), nucleotide sequences were blasted against Plasmid, Virus and Prophages databases separately inside the ALCAME web site (http://aclame.ulb.ac.be/) with default parameter. Moreover, spacer sequences detected by Pilercr v1.02 were blasted against env_nt blast using Geneious 7.1 software. A portion of the best hit sequences (max bit score) containing the spacers of at least 100 bp was manually compared against nr and nt NCBI databases using blastx and blastn, respectively. Only hits with bit score higher than 30 were considered for analysis.
Targeted metabolomic analysis
Next, we studied the chemical species in central metabolism of HSR2 including the pyruvate metabolism and the TCA cycle. The total of 31 chemicals was quantified in cell cultures grown on either acetate or pyruvate (Supplementary Table S8). Metabolites were extracted as previously described (Kubacka et al., 2014) and analyzed on three complementary platforms including mass spectrometry (MS) with gas and liquid chromatography (LC) and capillary electrophoresis (CE) separations (gas chromatography-quadrupole time-of-flight (QTOF)-MS, LC-QTOF-MS and CE-TOF-MS). As indicated in Supplementary Table S8, these analytical platforms matched the nature of the chemical species. Complete descriptions of the sample preparation protocols prior to analysis and analytical methods are extensively described in Supplementary Methods. To ensure the analytical reproducibility in LC-MS and CE-MS, quality controls samples were analyzed periodically throughout the analytical runs to monitor the drift of the signal (Dunn et al., 2011). Quality controls samples were independently prepared for LC-MS and CE-MS by pooling and mixing equal volume of each sample (see Supplementary Methods for full details). The resulting data files (GC-QTOF-MS, LC-ESI-QTOF-MS, CE-TOF-MS) files were cleaned of background noise and unrelated ions by Mass Hunter Qualitative Analysis software (B.05.00, Agilent Technologies). This tool was used to identify and quantify the target list of compounds. The peak area corresponding to each target compound was used to calculate the average and the coefficient of variation between the two technical replicates.
Data and strains deposition
16S rRNA gene sequences were deposited in the GenBank database (accession no. from KM875607 to KM875612). The genome sequence of strain HSR2 has been submitted to the GenBank (accession numbers CP008874 (circular chromosome) and CP008875 (circular plasmid)). All HSR acetate-oxidizing isolates have been deposited in the UNIQEM culture collection (Collection of Unique Cultures, Russian Academy of Sciences, Moscow, Russia) under the numbers: strain HSR2 (=Uniqem U935), strain HSR3 (=Uniqem U936), strain HSR4 (=Uniqem U937) and strain HSR5 (=Uniqem U938). Type strain HSR2T was additionally sent to the Japan Collection of Microorganisms (JCM) and the deposition procedure is actually in progress. The authors also guarantee the availability of the type strain HSR2T from personal collections of Dr Sorokin (d.sorokin@tudelft.nl, soroc@inmi.ru), maintained at Technical University of Delft (The Netherlands) and at Winogradsky Institute of Microbiology (Russian Academy of Sciences, Moscow, Russia).
Results
Isolation of haloarchaeal strains HSR2, HSR3, HSR4, HSR5, HSR7 and M27-SA2 and the global distribution of related organisms
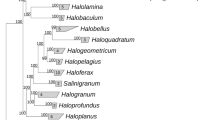
Active sulfur reduction was repeatedly observed in mixed anaerobic sulfur-rich sediment samples from the Kulunda Steppe hypersaline lakes (Altai, Russia) (Sorokin et al., 2012). To explore the microbiology of these samples, we isolated four axenic sulfidogenic acetate-oxidizing haloarchaeal cultures designated HSR2, HSR3, HSR4 and HSR5. The 16S rRNA gene sequences of these strains were nearly identical demonstrating that they belong to a single genetic species. Comparative phylogenetic analysis placed them within the family Halobacteriaceae as a novel lineage with the 92–93% sequence identity to members of genera Halarchaeum and Halobacterium (Figure 1 and Supplementary Figure S1). Together with uncultivated members of the ancient salt deposit cluster SD-II (Walsh et al., 2005), these isolates form a new monophyletic group. We constructed the phylogenetic tree using six conserved proteins from 29 currently available haloarchae genomes (Supplementary Figure S2). In this tree, HSR2 positioned in the Clade III of the Class Halobacteria, which previously was known to contain only two members, Halobacterium sp. NRC-1 and Halobacterium sp. DL1. The HSR isolates exhibited 97–98% sequence identity with the cloned 16S rRNA gene sequences of uncultured haloarchaea obtained from hypersaline anoxic soils, brines and sediments around the world (Supplementary Figure S3). To test that this type of haloarchaea is indeed widely distributed, we isolated similar acetate-oxidizing S0-reducing haloarchaea from anoxic hypersaline sediments of Lagoona Fuente de Piedra (Malaga, Spain) and of the deep-sea hypersaline lake Medee (Ionian Sea, Eastern Mediterranean, water depth 3105 m) designated HSR7 and M27-SA2, respectively. Together with the Kulunda isolates, all these strains form a distinct lineage within the family Halobacteriales and represent a novel genus with the proposed name ‘Halanaeroarchaeum sulfurireducens’.

Phylogenetic position of the proposed genus ‘Halanaeroarchaeum’ within the family Halobacteriaceae. Complete haloarchaeal genome sequences available in 2014 are denoted by red circles, scaffolds by green squares and contigs by blue triangles. Haloarchaeal genera known only by 16S rRNA gene sequences are shown in grey. For the genera included in the Group I and Group II, see Supplementary Table S1. Insert: The detailed phylogenetic tree of ‘Halanaeroarchaeum’-related sequences from SILVA database and those produced in this study. Similar clones are indicated in parentheses. Support for nodes in this tree corresponds to bootstrap values for 1000 pseudo-replicates. Only bootstrap values at nodes greater than 75% are displayed as solid circles. The tree has been arbitrarily rooted with the closest sequences from haloarchaea of the Salt Deposit Clade II (SD-II) (7). Sequence of Natronomonas pharaonis (D87971) was used for outgrouping. Sequences from cultured organisms are denoted in red.
General physiological description of the strain HSR2
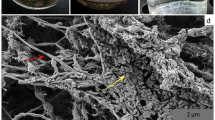
We performed a detailed characterization of the HSR2 isolate, which was the first to be isolated in pure culture, and which we chose as the type strain of this new group of Halobacteria. This organism grew between pH 6.7 and 8.0 (with the optimum at pH 7.2–7.5), salinity between 3.0 and 5.0 M NaCl and with the optimum growth temperature of 40 °C. The cells had very thin cell walls, a large nucleoid and a typical haloarchaeal morphology mostly in the form of flat irregular cocci and rods (Figures 2a and b). The biomass visually lacked any of light-scavenging pigments present in most of the Halobacteria. Fermentative growth on lactate and sugars was not observed. No anaerobic growth was observed on peptone and yeast extract either in absence or in the presence of elemental sulfur. The ability to grow anaerobically by arginine fermentation was checked in basal 4.0 M NaCl medium supplemented with 20 mM arginine and no growth was observed with any of four HSR strains tested. The isolate has a very limited metabolic profile restricted to acetate and pyruvate as the only available sources of carbon and energy. Among various electron acceptors tested either with acetate or pyruvate (anthraquinone-2,6-disulfonate, arsenate, arsenite, dimethyl sulfoxide, ferrihydrite, fumarate, manganese [IV] oxide, nitrate, nitrite, nitrous oxide, selenate, selenite, sulfate, sulfite, tetrathionate, thiosulfate and trimethylamine N-oxide), the growth occurred only with elemental sulfur. The overall reaction of S0-dependent acetate oxidation is a low energy-yielding conversion with the following stoichiometry and standard Gibbs energy yield (Thauer, 1988):

Morphology, cell ultrastructure and growth dynamics of strain HSR2. Phase contrast micrographs show the cells grown on acetate (a), pyruvate (b) and S0 and electron micrographs—the cells the cells grown on acetate and S0 (c). Growth dynamic with acetate (d) or pyruvate (e) as sole energy and carbon source. Error bars show the standard deviations for the mean values obtained from five independent cultures.

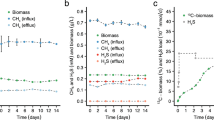
Growth of HSR2 on acetate and S0 resulted in production of approximately 9.10±0.92 mmol l−1 HS− at the expense of 2.50±0.34 mmol l−1 consumed acetate (Figure 2c), which is a near-stoichiometric oxidation. Part of the cell carbon might have also originated from the yeast extract present in the medium at the low concentration (10 mg l−1). Compared with acetate, growth with pyruvate as the electron donor resulted in lower HS− formation but higher biomass yield (Figure 2d).
General features of the HSR2 genome
We determined complete genome sequence of the HSR2 strain and discussed its general features in more detail in the supplement (Supplementary Tables S2-S4 and Supplementary Figures S4–S8). The genome is composed of two circular replicons: a chromosome of 2 085 482 base pairs (bp) and a 124 256-bp plasmid correspondingly containing 2111 and 117 predicted protein-encoding open reading frames, respectively. Out of all these coding sequences, 1336 chromosomal (63.3%) and only 23 plasmid genes (19.7%) were affiliated with known COGs. The genome harbors 47 tRNA genes and one complete rRNA operon. One gene cluster encoding CRISPRs and CRISPR-associated proteins was found in the HSR2 genome. This system is known to be responsible for prokaryotic immunity to phage infection (Deveau et al., 2010). Detailed comparisons revealed particularly high similarity between the HSR2 strain and Halorhabdus utahensis with regards to both the CRISPR-associated proteins and the number of repeats and their consensus sequence (Supplementary Figure S7). Remarkably, these clusters showed no homology between the spacer sequences, likely implying a different history of phage interaction for the isolated strains in their respective environments, regardless of the genomic and structural similarity of these two CRISPR systems.
Genetic determinants of acetate oxidation and respiratory chain
Consistent with the narrow specialization of HSR2 in metabolizing acetate, its genome contains multiple genes for acetate import, activation, oxidation and assimilation. These include a sodium-acetate symporter (HLASF_0396) of the 2.A.21 family (Saier et al., 2014), two ADP-forming acetyl-CoA synthases (operon HLASF_0398-0400), one putative succinyl-CoA:acetate CoA-transferase (HLASF_2086), one pyruvate ferredoxin oxidoreductase (PFOR, HLASF_0492-0493), all enzymes of the TCA cycle, and a complete set of glyoxylate bypass enzymes (Figure 3).

Acetate activation and central metabolism in HSR2. Pathways shown in the model were deduced on the basis of genome annotation, gene expression, metabolic profiling and [14C]-bicarbonate assimilation assay. Enzymes involved: (A) acetyl-CoA synthase (HLASF_0398, HLASF_0400, HLASF_1340); (B) putative succinyl-CoA:acetate CoA transferase (HLASF_2086); (C) citrate(si)-synthase (HLASF_0233, HLASF_1099); (D) aconitate hydratase (HLASF_0248); (E) NADP-specific isocitrate dehydrogenase (HLASF_1921); (F) 2-oxoglutarate:ferredoxin oxidoreductase (HLASF_0747-0748, HLASF_1469-1470); (G) succinyl-CoA synthetase (HLASF_1699-1700); (H) putative membrane-bound succinate:menaquinone oxidoreductase (HLASF_1576-1579); (I) fumarate hydratase (HLASF_1088), (J) NAD-dependent malate dehydrogenase (HLASF_0041); (K) isocitrate lyase (HLASF_2006); (L) malate synthase (HLASF_2007); (M) malic enzyme (HLASF_1723); (N) pyruvate:ferredoxin oxidoreductase (HLASF_0492-0495). Pyruvate and metabolites of the TCA cycle detected in acetate-grown cells are shown in red. Their concentrations, normalized as mV μg−1 cell protein, are shown in brackets. The details on targeted metabolomics analysis are given in the Supplementary Table S8. Metabolites relevant to the carbon assimilation pathways are underlined. Gene transcripts, detected and quantified by Q-RT-PCR gene expression analysis (Supplementary Table S5), are marked with blue circles.
In addition to the membrane-bound succinate dehydrogenase (Complex II; HLASF_1576-1579) of the TCA cycle, the Complex I oxidoreductase provides another electron entry point in the HSR2 respiratory chain. Complex I genes were found in many genomes of anaerobic Archaea including Archaeoglobus, Thermoproteus and several methanogens. Only 11 of the 14 Complex I subunits are observed in most of these organisms (Siebers et al., 2011). Three other subunits, catalyzing NADH binding and oxidation in Complex I of aerobes, are replaced by nonhomologous ferredoxin- or F420-oxidizing subunits. Similarly, HSR2 has a complete set of ‘anaerobic’ Complex I genes, located at a single site in the genome (HLASF_1225-1235). The F420-oxidizing NuoF subunit is missing indicating that this organism uses ferredoxins, rather than this cofactor, to funnel electrons into the menaquinone pool. The HSR2 strain has only one ATP synthase operon (HLASF_0205-0213) belonging to H+-translocating V-type ATPases (Mulkidjanian et al., 2008), thus resembling all known haloarchaeal homologues.
The HSR2 genome lacks genes encoding cytochrome c-oxidases found in aerobic prokaryotes and any atypical variants of cytochromes found in anaerobic bacteria. Furthermore, the genes coding for another crucial complex of aerobic respiratory electron transfer chain—the cytochrome bc1 complex—are absent, along with homologues of the alternative Complex III (Refojo et al., 2010), confirming the inability of this organism for oxygen respiration. Cytochrome bd ubiquinol oxidase, present in HSR2 (HLASF_0492-0493), likely serves for oxygen detoxification, similarly to oxidases of other strict anaerobes, such as acetogens and sulfate reducers (Jünemann, 1997). Other candidate proteins involved in oxidative stress protection in HSR2 include a protein homologous to the bifunctional enzyme catalase/peroxidase from Haloarcula marismortui (HLASF_0191) and a putative manganese superoxide dismutase (HLASF_1272).
HSR2 genetic determinants and biochemical challenges of anaerobic respiration on elemental sulfur
With regard to electron acceptor utilization, four operons encoding oxidoreductases of the molybdopterin superfamily were identified in the HSR2 genome (Supplementary Figure S5). According to the entire operon structures, the phylogeny of catalytic subunits A and the number of transmembrane regions of subunits C, three of these oxidoreductases were annotated as membrane-bound polysulfide reductases PsrABC (HLASF_0052-0054, HLASF_0694-0696 and HLASF_1287-1285). Concatenated sequences of core subunits of these operons have a strong similarity, suggesting recent duplication events that led to their formation. Phylogenetic analysis of catalytic molybdenum subunits placed HSR2 polysulfide reductases in a distinct haloarchaea-specific cluster, adjacent to the Psr/Phs family (Duval et al., 2008) (Figure 4,Supplementary Figure S6). Catalytic subunit A phylogeny and core genes organization attribute a fourth operon of molybdopterin oxidoreductase to the clade of tetrathionate reductases. However, as we mentioned above, the HSR2 strain did not grow with tetrathionate.

Phylogenetic tree of molybdopterin oxidoreductase catalytic subunits PsrA. Polysulfide reductase/thiosulfate reductase (Psr/Phs) clade is marked in yellow. Other abbreviations: arsenite oxidases (Aro); arsenate reductases (Arr); dimethylsulfoxide/trimethylamine-N-oxide reductases (DMSO/TMAO); formate dehydrogenases (Fdh); periplasmic nitrate reductases (Nap); membrane-bound nitrate/perchlorate reductases (Nar/Pcr); assimilatory nitrate reductases (Nas); tetrathionate reductases (Ttr); unknown reductases (Unk). Bacteria are highlighted in black, Crenarchaeota in blue, Euryarchaeota in green and Halobacteria in pink (see Supplementary Figure S6 for details).
The analysis of the supernatant from HSR2 culture at a stationary phase grown with acetate demonstrated the presence of inorganic polysulfides at a total sulfur concentration of 1.3 mmol l−1 with a domination of S42− (that is, 0.65 mmol l−1 mobilized zero-valent sulfur). The formation of these soluble inorganic polysulfides likely occurred because of the spontaneous reaction of elemental sulfur with HS− (Hedderich et al., 1999). Although the initial concentration of HS− measured in the HSR2 growth medium was very low (5 μmol l−1), its amount increased up to 9.0 mmol l−1 HS− in the stationary phase (Figure 2c). However, the concentration of polysulfides in neutral condition is low because of their chemical instability and therefore their utilization as an electron acceptor should still pose biochemical challenges to the sulfur-respiring microorganisms. We propose that this process may rely on the utilization of a specific polysulfide-binding carrier protein that presents the substrate to the catalytic molybdenum (Mo)-containing PsrA subunit of polysulfide reductases. Indeed, one of the three HSR2 PsrABC operons contains a gene encoding a protein (HLASF_0693) belonging to sulfurtransferase family (Supplementary Figure S5), which may play a role in binding, stabilizing and transferring inorganic polysulfide molecules to PsrA subunit (Klimmek et al., 1998) (Figure 5).

Proposed pathways for sulfur metabolism and energy generation in HSR2 enzymes are labeled with locus tag numbers. The central intermediate in the sulfur pathway is polysulfide. This process would require a polysulfide carrier (HLASF_0693) similar to the sulfurtransferase of Wolinella succinogenes. Enzymes involved in the sulfur metabolism are shown in yellow, Complex I (HLASF_1576-1579) and Complex II (HLASF_1225-1235) are pink.
Gene expression, metabolism and [14C]-bicarbonate assimilation during growth of the HSR2 cells
Transcription analysis of genes for catalytic subunits of all four oxidoreductases demonstrated that during the growth on acetate and elemental sulfur, psrA (HLASF_0694) of the sulfurtransferase-containing operon had the highest expression levels (21.05±3.59 copy per cell, Supplementary Table S5). Synthesis of pyruvate from acetyl-CoA through a carboxylation reaction catalyzed by PFOR was confirmed by gene expression analysis for this enzyme (1.58±0.23 copy per cell), [14C]-bicarbonate assimilation assay (40±10 ag C per cell per day) (Figure 3, Supplementary Tables S5–S7), and by the analysis of the intracellular metabolites, linked with the pyruvate metabolism and the TCA cycle (Supplementary Table S8). Metabolomic analysis confirmed that in addition to puryvate, HSR2 uses oxaloacetate and 2-oxoglutarate as precursors for biosynthesis (Supplementary Table S8). To avoid the depletion of metabolites of the TCA cycle metabolites, acetyl-CoA and glyoxylate are likely converted into malate via the glyoxylate bypass (Figure 3). The expression of isocitrate lyase (10.45±2.42 copy per cell) and malate synthase (4.75±1.65 copy per cell) genes during the growth on acetate supported the likelihood of this reaction (Supplementary Table S5). As shown by [14C]-labeling experiments, during the growth on pyruvate, the bicarbonate assimilation was also observed at a much higher extent (360±10 ag C per cell per day, Supplementary Table S6).
Discussion
Hypersaline aquatic environments are widespread on our planet and store nearly as much water as freshwater lakes (Vallentyne, 1972; Hammer, 1986). These hydrological formations, including inland salt lakes, marine coastal marshes, solar salterns and deep-sea brine lakes are often densely populated by the unique group of extremely halophilic euryarchaea (haloarchaea). These predominantly aerobic heterotrophs have developed a low energy-demanding (so called ‘salt-in’) strategy to thrive under extremely high salinity. The present work provided new insights into the molecular mechanisms underlying energy conservation and metabolism of this group of prokaryotes. By isolation, genome and physiology analysis of strictly anaerobic sulfur-respiring acetate-oxidizing haloarchaea, we demonstrated that these organisms have a much broader functional capability than known haloarchaea, which further expands their colonization abilities towards anoxic niches.
Phylogenetically, the strains described here had nearly identical 16S rRNA gene sequences and formed a novel monophyletic cluster within the family Halobacteriaceae, with a maximum of 93% sequence identity with the nearest cultured members. This suggested their affiliation with a novel genus. Closely related strains were numerically dominant microbes in various terrestrial and deep-sea hypersaline anoxic sites around the globe. We considered that these strains were highly relevant to understanding the functional contribution of haloarchaea to anaerobic carbon and sulfur cycling in salt-saturated environments.
Analysis of the HSR2 genome provided remarkable insights into its unique metabolic capabilities and strategies for survival of this strain under such habitat-specific stress factors as extreme salinity and low solubility of electron acceptor. The two main factors that determine whether a microorganism can live anaerobically at extreme salinity (>250 g l−1) are (i) the amount of energy it can generate through dissimilatory metabolism and (ii) the mode of maintaining osmotically balanced functional cytoplasm (haloadaptation) (Oren, 2011; 2013; Andrei et al., 2012). Haloadaptation has high energetic costs, thus limiting the number of prokaryotes that can thrive in these environments. In the dark, anoxic, nitrate-impoverished and salt-saturated environments, highly exergonic processes such as photosynthesis, aerobic and nitrate respiration cannot be operative. This is why under such conditions, the final oxidation of acetate to CO2 by secondary anaerobes with low-potential electron acceptors, such as sulfur or sulfate, is problematic. Haloarchaea have an advantage over other organisms, because they use the ‘energetically cheap’ mode of haloadaptation based on potassium ions import (Oren, 2011). Central to the HSR2 metabolism is its ability to anaerobically oxidize acetate, which is the main recalcitrant intermediate of anaerobic degradation of organic matter. Using elemental sulfur as terminal electron acceptor, HSR2 oxidizes acetate completely, converting it to CO2 and H2O. The only alternative to this type of respiration would be disproportionation of elemental sulfur with assimilation of acetate as carbon source. However, we ruled out this possibility by showing the near 4:1 stoichiometry of sulfide produced to acetate consumed and the absence of detectable sulfate production. Hitherto, this type of sulfidogenesis was described for a very limited number of non-halophilic anaerobic acetate-oxidizing bacterial species (Rabus et al., 2006). In all organisms of this functional group characterized to date, acetate is activated via ATP-dependent acetyl-CoA synthase and oxidized via the tricarboxylic acid (TCA) cycle. HSR2 appears to be no exception, as its genome contains all enzymes of the classical TCA cycle. HSR2 has developed multiple anabolic strategies to grow on acetate as the sole electron donor and carbon source. Specifically, acetate assimilation in HSR2 requires the synthesis of acetyl-CoA and the simultaneous activity of PFOR complex to produce pyruvate and the enzymes of glyoxylate bypass to produce malate. As shown by our [14C]-labeling experiments (Figure 3 and Supplementary Tables S5–S8), anaplerotic fixation of CO2 through its carboxylation is catalyzed by PFOR (HLASF_0492-0495) at the rates 0.04±0.01 fg C per cell per day. During the growth on pyruvate the anaplerotic assimilation of bicarbonate by HSR2 is likely catalyzed by a NADP-malic enzyme (HLASF_1723), working in the direction of carboxylation (Hong and Lee, 2001; Zelle et al., 2011).
Our studies revealed that oxidation of acetate by HSR2 is accompanied by a near stoichiometric production of sulfide. Utilization of elemental sulfur as the terminal electron acceptor must require a sophisticated suite of energy-conserving enzymes. Indeed, HSR2 genome harbors three operons encoding membrane-bound polysulfide reductases PsrABC and, according with the gene expression studies, all of them are expressed during this type of respiration. Solubility of elemental sulfur in water at neutral pH is extremely low (less than 160 nmol l−1) and, therefore, S0 can hardly be used as the terminal electron acceptor without any adaptive implementations (Klimmek et al., 1998). However, in the presence of sulfide, a significant amount of S0 is transformed into soluble inorganic polysulfide (Hedderich et al., 1999). One of the mechanisms of its utilization as a terminal acceptor was described for bacterial sulfur reducers Wolinella succinogenes and Nautilia profundicola (Klimmek et al., 1998; Campbell et al., 2009). The process relies on the activity of a periplasmic sulfurtransferase/rhodanese-like protein (Sud), which acts as a polysulfide-binding carrier and represents the actual substrate for the catalytic molybdenum (Mo)-containing subunit PsrA. The Sud of W. succinogenes binds up to 10 mol of polysulfide sulfur per 1 mol of enzyme and drastically increases the activity of polysulfide reductase (Klimmek et al., 1998; Hedderich et al., 1999). As the supernatant of a stationary phase HSR2 culture contained 1.3 mmol l−1 of polysulfide (S42−) sulfur, we suggested that HSR2 could use a similar strategy. Because about 50% of sulfur in the S42− species pool is reduced, the actual zero-valent linear sulfur concentration must be around 0.65 mmol l−1. Indeed, we detected this polysulfide concentration at the final point of HSR2 growth when 9 mmol l−1 sulfur was reduced to HS−. As at neutral pH and low sulfide concentrations polysulfide (S42−) rapidly decomposes back to sulfur and HS−, polysulfide can reach high concentrations only when HS− is in a millimolar range. However, accumulation of sulfide inhibits the further growth of HSR2. Therefore, this organism can grow within the certain range of sulfide concentrations. On one hand, sulfide concentration must be sufficient to support the availability of polysulfides for the initial growth of HSR2. On the other hand, the further growth of HSR2 is limited by the toxicity of sulfide at concentrations above 9 mmol.
The HSR2 genome contains the gene HLASF_0693 encoding an extracellular sulfurtransferase, that is likely to be responsible for mobilization of insoluble sulfur. Compared with the Sud protein of W. succinogenes, which has one sulfur-binding domain and needs dimerization to achieve high substrate affinity (Klimmek et al., 1998; Rabus et al., 2006), the HSR2 sulfurtransferase contains two homologous domains. This feature might be especially important during the early growth phase of the HSR2 cells when sulfide concentration is in the micromolar range. The role of this enzyme may decrease as sulfide/polysulfide concentrations rise at the later stages of HSR2 growth.
In summary, our discovery of the strictly anaerobic sulfur-respiring haloarchaea significantly extended our understanding of Halobacteria physiology and their contribution to energy fluxes and elements cycling in hypersaline anoxic ecosystems. Thermodynamically, anaerobic oxidation of acetate with sulfur as terminal electron acceptor is an exceptionally unfavorable conversion, and for a long time, scientists have been searching for this type of metabolism. As sulfur-rich anoxic saline habitats were a commonplace on early Earth, this mechanism of elemental sulfur utilization suggests new scenarios in the evolution of Biosphere.
References
Afshar S, Kim C, Monbouquette HG, Schröder I . (1998). Effect of tungstate on nitrate reduction by the hyperthermophilic archaeon Pyrobaculum aerophilum. Appl Environ Microbiol 64: 3004–3008.
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402.
Andrei AS, Banciu HL, Oren A . (2012). Living with salt: metabolic and phylogenetic diversity of archaea inhabiting saline ecosystems. FEMS Microbiol Lett 330: 1–9.
Antunes A, Taborda M, Huber R, Moissl C, Nobre MF, da Costa MS . (2008). Halorhabdus tiamatea sp. nov., a non-pigmented extremely halophilic archaeon from a deep-sea, hypersaline anoxic basin of the Red Sea, and emended description of the genus Halorhabdus. Int J Syst Evol Microbiol 58: 215–220.
Bonete MJ, Martínez-Espinosa RM, Pire C, Zafrilla B, Richardson DJ . (2008). Nitrogen metabolism in haloarchaea. Saline Systems 4: 9.
Campbell BJ, Smith JL, Hanson TE, Klotz MG, Stein LY, Lee CK et al. (2009). Adaptations to submarine hydrothermal environments exemplified by the genome of Nautilia profundicola. PLoS Genet 5: e1000362.
Delcher AL, Bratke KA, Powers EC, Salzberg SL . (2007). Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23: 673–679.
Deveau H, Garneau JE, Moineau S . (2010). CRISPR/Cas system and its role in phage-bacteria interactions. Annu Rev Microbiol 64: 475–493.
Dunn WB, Broadhurst D, Begley P, Zelena E, Francis-McIntyre S, Anderson N et al. (2011). Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat Protoc 6: 1060–1083.
Duval S, Ducluzeau AL, Nitschke W, Schoepp-Cothenet B . (2008). Enzyme phylogenies as markers for the oxidation state of the environment: the case of respiratory arsenate reductase and related enzymes. BMC Evol Biol 8: 206.
Edgar RC . (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797.
Edgar RC . (2007). PILER-CR: fast and accurate identification of CRISPR repeats. BMC Bioinformatics 8: 18.
Elshahed MS, Najar FZ, Roe BA, Oren A, Dewers TA, Krumholz LR et al. (2004a). Survey of archaeal diversity reveals an abundance of halophilic Archaea in a low-salt, sulfide- and sulfur-rich spring. Appl Environ Microbiol 70: 2230–2239.
Elshahed MS, Savage KN, Oren A, Gutierrez MC, Ventosa A, Krumholz LR et al. (2004b). Haloferax sulfurifontis sp. nov., a halophilic archaeon isolated from a sulfide- and sulfur-rich spring. Int J Syst Evol Microbiol 54: 2275–2279.
Grant WD, Ross HNM . (1986). The ecology and taxonomy of Halobacteria. FEMS Microbiol Rev 39: 9–15.
Grissa I, Vergnaud G, Pourcel C . (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res 35: W52–W57.
Guindon S, Gascuel O . (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52: 696–704.
Hammer UT . (1986) Saline lake ecosystems of the world. Dr. W. Junk Publishers, P.O. Box 163, 3300 ADDordrecht, The Netherlands, p616.
Hedderich R, Klimmek O, Kröger A, Dirmeier R, Keller M, Stetter KO et al. (1999). Anaerobic respiration with elemental sulfur and with disulfides. FEMS Microbiol Rev 22: 353–381.
Hong SH, Lee SY . (2001). Metabolic flux analysis for succinic acid production by recombinant Escherichia coli with amplified malic enzyme activity. Biotechnol Bioeng 74: 89–95.
Huang Y, Niu B, Gao Y, Fu L, Li W . (2010). CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 26: 680–682.
Jünemann S . (1997). Cytochrome bd terminal oxidase. Biochim Biophys Acta 1321: 107–127.
Kashefi K, Tor JM, Holmes DE, Gaw Van Praagh CV, Reysenbach AL, Lovley DR et al. (2002). Geoglobus ahangari gen. nov., sp. nov., a novel hyperthermophilic archaeon capable of oxidizing organic acids and growing autotrophically on hydrogen with Fe(III) serving as the sole electron acceptor. Int J Syst Evol Microbiol 52: 719–728.
Klimmek O, Kreis V, Klein C, Simon J, Wittershagen A, Kröger A et al. (1998). The function of the periplasmic Sud protein in polysulfide respiration of Wolinella succinogenes. Eur J Biochem 253: 263–269.
Kubacka A, Diez MS, Rojo D, Bargiela R, Ciordia S, Zapico I et al. (2014). Understanding the antimicrobial mechanism of TiO2-based nanocomposite films in a pathogenic bacterium. Sci Rep 4: 4134.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948.
Laska S, Lottspeich F, Kletzin A . (2003). Membrane-bound hydrogenase and sulfur reductase of the hyperthermophilic and acidophilic archaeon Acidianus ambivalens. Microbiology 149: 2357–2371.
Leplae R, Lima-Mendez G, Toussaint A . (2010). ACLAME: a CLAssification of Mobile genetic Elements, update 2010. Nucleic Acids Res 38: D57–D61.
Lloyd KG, Schreiber L, Petersen DG, Kjeldsen KU, Lever MA, Steen AD et al. (2013). Predominant archaea in marine sediments degrade detrital proteins. Nature 496: 215–218.
Mulkidjanian AY, Galperin MY, Makarova KS, Wolf YI, Koonin EV . (2008). Evolutionary primacy of sodium bioenergetics. Biol Direct 3: 13.
Oren A, Trüper HG . (1990). Anaerobic growth of halophilic archaeobacteria by reduction of dimethylsulfoxide and trimethylamine N-oxide. FEMS Microbiol Lett 70: 33–36.
Oren A . (1991). Anaerobic growth of archaeobacteria by reduction of fumarate. J Gen Microbiol 137: 1387–1390.
Oren A . (2011). Thermodynamic limits to microbial life at high salt concentrations. Environ Microbiol 13: 1908–1923.
Oren A . (2013). Life at high salt concentration. In: Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F (eds). The Prokaryotes—Prokaryotic Communities and Ecophysiology. Springer-Verlag: Berlin, Heidelberg, Germany, pp 421–440.
Petersen TN, Brunak S, von Heijne G, Nielsen H . (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8: 785–786.
Pfennig N, Lippert KD . (1966). Über das Vitamin B12-Bedürfnis phototropher Schwefelbakterien. Arch Mikrobiol 55: 245–256.
Rabus R, Hansen TA, Widdel F . (2006). Dissimilatory sulfate- and sulfur-reducingprokaryotes. Dworkin M (ed.). The Prokaryotes—A Handbook on the Biology of Bacteria, vol. 2. Springer Science+Business Media: Singapore, pp 659–768.
Refojo PN, Sousa FL, Teixeira M, Pereira MM . (2010). The alternative complex III: a different architecture using known building modules. Biochim Biophys Acta 1797: 1869–1876.
Roman P, Bijmans MF, Janssen AJ . (2014). Quantification of individual polysulfides in lab-scale and full-scale desulfurisation bioreactors. Environ Chem 11: 702–708.
Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA et al. (2000). Artemis: sequence visualization and annotation. Bioinformatics 16: 944–945.
Saier MH Jr, Reddy VS, Tamang DG, Västermark A . (2014). The transporter classification database. Nucleic Acids Res 42: D251–D258.
Siebers B, Zaparty M, Raddatz G, Tjaden B, Albers SV, Bell SD et al. (2011). The complete genome sequence of Thermoproteus tenax: a physiologically versatile member of the Crenarchaeota. PLoS One 6: e24222.
Slobodkina GB, Kolganova TV, Querellou J, Bonch-Osmolovskaya EA, Slobodkin AI . (2009). Geoglous acetivorans sp. nov., an iron(III)-reducing archaeon from a deep-sea hydrothermal vent. Int J Syst Bacteriol 59: 2880–2883.
Sorokin DY, Zacharova EE, Pimenov NV, Tourova TP, Panteleeva AN, Muyzer G et al. (2012). Sulfidogenesis in hypersaline chloride-sulfate lakes of Kulunda Steppe (Altai, Russia). FEMS Microbiol Ecol 79: 445–453.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S . (2013). MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30: 2725–2729.
Thauer RK . (1988). Citric-acid cycle, 50 years on. Modifications and an alternative pathway in anaerobic bacteria. Eur J Biochem 176: 497–508.
Tindall BJ, Trüper HG . (1986). Ecophysiology of the aerobic halophilic archaebacteria. Syst Appl Microbiol 7: 202–212.
Trüper HG, Schlegel HG . (1964). Sulfur metabolism in Thiorhodaceae. 1. Quantitative measurements on growing cells of Chromatium okenii. Ant van Leeuwenhoek 30: 225–238.
Tyson GW, Chapman J, Hugenholtz P, Allen EE, Ram RJ, Richardson PM et al. (2004). Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 428: 37–43.
Vallentyne JR . (1972) Freshwater supplies and pollution: Effects of the demophoric explosion on water and manIn: Polunin N (Ed.) The Environmental Future. Macmillan: London, pp 181–211.
Walsh DA, Papke RT, Doolittle WF . (2005). Archaeal diversity along a soil salinity gradient prone to disturbance. Environ Microbiol 7: 1655–1666.
Werner J, Ferrer M, Michel G, Mann AJ, Huang S, Juarez S et al. (2014). Halorhabdus tiamatea: proteogenomics and glycosidase activity measurements identify the first cultivated euryarchaeon from a deep-sea anoxic brine lake as potential polysaccharide degrader. Environ Microbiol 16: 2525–2537.
Whelan S, Goldman N . (2001). A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol Biol Evol 18: 691–699.
Yakimov MM, La Cono V, Slepak VZ, La Spada G, Arcadi E, Messina E et al. (2013). Microbial life in the Lake Medee, the largest deep-sea salt-saturated formation. Sci Rep 3: 3554.
Yakimov MM, La Cono V, Smedile F, Crisafi F, Arcadi E, Leonardi M et al. (2014). Heterotrophic bicarbonate assimilation is the main process of de novo organic carbon synthesis in hadal zone of the Hellenic Trench, the deepest part of Mediterranean Sea. Environm Microb Rep 6: 709–722.
Youssef NH, Ashlock-Savage KN, Elshahed MS . (2011). Phylogenetic diversities and community structure of members of the extremely halophilic Archaea (order Halobacteriales in multiple saline sediment habitats. Appl Environ Microbiol 78: 1332–1344.
Zelle RM, Harrison JC, Pronk JT, van Maris AJ . (2011). Anaplerotic role for cytosolic malic enzyme in engineered Saccharomyces cerevisiae strains. Appl Environm Microbiol 77: 732–738.
Zerbino DR, Birney E . (2008). Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18: 821–829.
Acknowledgements
This work was supported by research fund from European Commission’s Program under MicroB3 Project (Contract FP7-OCEAN.2011-2-287589), the Russian Foundation of Basic Research (13-04-00049, 13-04-40205-N) and the Russian Academy of Science Program ‘Molecular and Cellular Biology’. This work was further funded by grant BIO2011-25012 from the Spanish Ministry of the Economy and Competitiveness’. David Rojo would like to acknowledge funding from the Ministry of Science and Technology (CTQ2014-55279-R). We thank Prof. Coral Barbas (CEU San Pablo University) for her excellent support in relation to the metabolomics analyses. We sincerely thank Erika Arcadi, Gina LaSpada for technical assistance and Douglas Boettner and Helen Glanville for critical reading the manuscript and useful comments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Sorokin, D., Kublanov, I., Gavrilov, S. et al. Elemental sulfur and acetate can support life of a novel strictly anaerobic haloarchaeon. ISME J 10, 240–252 (2016). https://doi.org/10.1038/ismej.2015.79
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2015.79
This article is cited by
-
Functional differentiation determines the molecular basis of the symbiotic lifestyle of Ca. Nanohaloarchaeota
Microbiome (2022)
-
Role of organic/sulfide ratios on competition of DNRA and denitrification in a co-driven sequencing biofilm batch reactor
Environmental Science and Pollution Research (2022)
-
New approaches for archaeal genome-guided cultivation
Science China Earth Sciences (2021)
-
Halo(natrono)archaea from hypersaline lakes can utilize sulfoxides other than DMSO as electron acceptors for anaerobic respiration
Extremophiles (2021)
-
The human gut archaeome: identification of diverse haloarchaea in Korean subjects
Microbiome (2020)
