
Abstract
Marine microbes have a pivotal role in the marine biogeochemical cycle of carbon, because they regulate the turnover of dissolved organic matter (DOM), one of the largest carbon reservoirs on Earth. Microbial communities and DOM are both highly diverse components of the ocean system, yet the role of microbial diversity for carbon processing remains thus far poorly understood. We report here results from an exploration of a mosaic of phytoplankton blooms induced by large-scale natural iron fertilization in the Southern Ocean. We show that in this unique ecosystem where concentrations of DOM are lowest in the global ocean, a patchwork of blooms is associated with diverse and distinct bacterial communities. By using on-board continuous cultures, we identify preferences in the degradation of DOM of different reactivity for taxa associated with contrasting blooms. We used the spatial and temporal variability provided by this natural laboratory to demonstrate that the magnitude of bacterial production is linked to the extent of compositional changes. Our results suggest that partitioning of the DOM resource could be a mechanism that structures bacterial communities with a positive feedback on carbon cycling. Our study, focused on bacterial carbon processing, highlights the potential role of diversity as a driving force for the cycling of biogeochemical elements.
Similar content being viewed by others


Introduction
Marine microbial communities contain an immense taxonomic and functional diversity (Pedrós-Alió, 2006; Sogin et al., 2006; Moran, 2008). The exploration of microbial diversity provided insight into distinct temporal dynamics (Fuhrman et al., 2006; Gilbert et al., 2012; Vergin et al., 2013) and spatial distributions of dominant and rare taxa across ocean regimes (Martiny et al., 2006; Pommier et al., 2007; Fuhrman et al., 2008; Ghiglione et al., 2012). Patterns in diversity are paralleled by changes in the repertoire and expression of genes of microbial communities (Rusch et al., 2007; Frias-Lopez et al., 2008; Poretsky et al., 2009) or individual taxa (Moran et al., 2004; Bauer et al., 2006; Grzymski et al., 2006; Gifford et al., 2011). How the ensemble of phylogenetic and functional diversity in a given ecosystem affect bacterial processes and thereby energy and nutrient cycling remains, however, poorly understood. This is a fundamental issue for a better comprehension of the ocean system and its response to perturbations that are predicted to increase with global change.
The role of species diversity in sustaining ecosystem processes was demonstrated by positive productivity–diversity relationships in terrestrial plant and soil communities (Hooper and Vitousek, 1997; Naeem and Li, 1997; Hector et al., 1999; Cadotte et al., 2008). The presence of more functional traits at higher richness allowing a larger range of resources to be exploited due to niche differentiation or positive interactions are mechanisms to explain this relationship (Loreau et al., 2001). To test the validity of this relationship for microbes in aquatic systems, experimental studies with assembled multi-level food webs (McGrady-Steed et al., 1997), reconstituted or manipulated bacterial communities (Bell et al., 2005; Gravel et al., 2011; Peter et al., 2011) were performed. These experimental results revealed positive relationships between microbial diversity, productivity and predictability of ecosystem function. However, these experimental observations could thus far not be confirmed in the ocean (Reinthaler et al., 2005; Obernosterer et al., 2010).
A present major challenge is to decipher how the identity and diversity of marine microbes affect the turnover of the globally important pool of marine dissolved organic matter (DOM). DOM is composed of a multitude of organic substrates, with distinct chemical structures and bioavailability to microbes (Benner, 2002; Hertkorn et al., 2006). The coupling between community composition and DOM cycling differs largely among studies, pointing to varying degrees of functional redundancy with respect to the degradation of different organic matter (Langenheder et al., 2006; Teira et al., 2008; Sjöstedt et al., 2013). The recent finding that a single bacterial strain can degrade labile marine DOM as rapidly as a natural bacterial community (Pedler et al., 2014) further challenges the question on the importance of diversity for bacterially mediated carbon processing in the ocean.
The Southern Ocean combines unique features that strongly affect bacterial heterotrophic metabolism, and likely also diversity. The permanent upwelling around Antarctica transports old and highly refractory DOM to the surface. Concurrent low primary production, due to iron limitation, leads to concentrations of dissolved organic carbon (DOC) in surface waters that are the lowest of the world oceans (~50 μm; Hansell, 2013). As a consequence, the availability of organic carbon strongly constrains bacterial heterotrophic metabolism (Church et al., 2000). To date, however, this remote open ocean remains poorly studied (Selje et al., 2004; Giebel et al., 2009; Manganelli et al., 2009).
Natural iron fertilization in the wake of islands stimulates phytoplankton primary production and the related release of DOM (Blain et al., 2007; Pollard et al., 2009). Heterotrophic bacteria rapidly respond to these blooms and process a substantial fraction of primary production (Zubkov et al., 2007; Obernosterer et al., 2008). The largest naturally iron-fertilized region is located in the vicinity of the Kerguelen Islands, where massive spring phytoplankton blooms occur annually (Blain et al., 2007). This original environmental context allowed us to address the questions on whether different blooms harbor distinct bacterial communities and whether changes in bacterial diversity affect the processing of organic matter.
Materials and methods
Site description
We investigated the composition of the bacterial communities associated with spring phytoplankton blooms in the naturally iron-fertilized region east of Kerguelen Islands and in high-nutrient low-chlorophyll (HNLC) waters (Station R-2) west of the islands during the KEOPS2 cruise (Kerguelen Ocean and Plateau Compared Study, October–November 2011; Figure 1). For the southeastern bloom above the Kerguelen plateau (Station A3) samples were also collected during its decline in late summer (KEOPS1 cruise; January–February 2005; Stations A3-3 and A3-4; Table 1). The hydrographic context of the study region is described in detail in Park et al. (2014) and basic parameters are provided in Table 1. The sites were intensively sampled to characterize the nutrient regime, the composition of plankton communities and the associated fluxes (Supplementary Tables S1 and S2).

Composite satellite images provided by GlobColour illustrating the temporal and spatial evolution of the phytoplankton blooms induced by natural iron fertilization in the region of Kerguelen Island. Surface ocean chlorophyll is derived from ocean color satellite observations. Stations are indicated on the satellite images that were available for corresponding sampling dates. See Table 1 for the exact dates of sampling of the stations.
Sampling for bacterial diversity analysis
Seawater for bacterial diversity analysis was collected at 20 m depth with 12 l Niskin bottles (General Oceanics, Miami, FL, USA) mounted on a CTD Rosette. Seawater was pre-filtered with a Nylon screen (25-μm pore size) and collected in 10 l polycarbonate (PC) carboys (Nalgene, Thermo Fisher Scientific, Waltham, MA, USA) rinsed three times with sample water. The samples were immediately sequentially filtered through 3 μm and 0.8 μm PC filters (47-mm diameter, Nuclepore, Whatman, Sigma Aldrich, St Louis, MO, USA) and cells were concentrated on a 0.2-μm cartridge (Sterivex, Millipore, EMD, Billerica, MA, USA). To each cartridge, 500 μl of lysis buffer (40 mm EDTA, 50 mm Tris, 0.75 m sucrose) was added and the samples were stored at −80 °C until analysis.
The on-board continuous-cultures experiment
To investigate the response of a winter-water bacterial community to phytoplankton DOM, we set up a continuous-culture experiment during our first visit to Station A3 (A3-1). The set-up of the continuous cultures followed the design previously published with some modifications (Landa et al., 2013). The experiment consisted of two treatments, a control and a diatom-DOM treatment. Triplicate cultures (2 l final volume) were run for each treatment. In the control treatment, bacteria were supplied with 0.2 μm-filtered seawater, and in the diatom-DOM treatment, bacteria were supplied with 0.2 μm-filtered seawater mixed with phytoplankton-derived DOM. To obtain phytoplankton-derived DOM, the diatom Chaetoceros debilis isolated from the Southern Ocean (strain L41-B1, Alfred Wegener Institute, Germany) was cultivated in low DOC (15 μm) artificial seawater amended with nutrients according to Guillard’s F/2 medium in a light:dark cycle of 16 h:8 h at 4 °C. This diatom species accounted for 10–50% of the diatom biomass at 5 out of the 9 bloom stations (Lasbleiz, 2014). The diatom exudate was collected by sequential filtration as described previously (Landa et al., 2013). The DOC concentration of the phytoplankton exudate was identical to the DOC concentration in 0.2 μm-filtered seawater. Therefore, despite the considerable contribution of diatom-DOM to the total DOC (20%), the bulk DOC concentration in the diatom-DOM cultures (51±2 μm, n=8) were similar to those in the control cultures (54±2 μm, n=8). The roughly 3-μm difference in DOC concentration indicates the preferential uptake of diatom-DOM. Previous experiments have shown that this approach yields differences in the chemical composition of DOM (Landa et al., 2014). Concentrations of inorganic phosphate (2.1±0.3 μm) and nitrate (41.2±1.7 μm) were identical in the cultures of both treatments throughout the experiment. A natural bacterial community collected at Station A3-1 was used as inoculum for both treatments.
For the preparation of media for the entire duration of the experiment, 60 l of seawater from the upper 80 m were sampled at the same station (A3-1) with 10 l Teflon-lined Niskin-1010X bottles (General Oceanics, Miami, FL, USA) mounted on a 1018 rosette system (General Oceanics) adapted for trace-metal clean work. The 60 l of seawater were collected in acid-washed (10% HCl), Milli-Q water-rinsed 20 l PC carboys and they were immediately sequentially filtered through 0.4-μm and 0.2-μm filters using a Sartobran cartridge (150 cm2, Sartorius, Goettingen, Germany). Seawater for the bacterial inoculum was collected at 40 m and 4 l were filtered through a 0.8-μm PC filter (47-mm diameter, Nuclepore). The 0.8 μm filtrate was stored at 6 °C for a few hours until the beginning of the experiment. Concurrently, 4.8 l of the <0.8 μm filtrate were collected on a 0.2-μm cartridge (Sterivex, Millipore) and treated, as described above, for further diversity analyses.
The cultures were conducted in a cold room (6 °C) in the dark. Each culture was bubbled with filter-sterilized air (Sartorious). A Milli-Q water purge was placed between the source and the filters to avoid potential DOC contamination by air. Cultures were homogenized by stirring. The imposed flow rate was 0.2 day−1 and resulted in a generation time of 5.6 days. This flow rate was chosen based on the in situ growth rates determined during the KEOPS1 cruise. The experiment was run for 19 days, during which the cultures were sampled every 2 to 3 days. The air purge was renewed every day and the input media were prepared every 2 days. Samples for bacterial diversity were collected in acid-washed, Milli-Q water-rinsed PC flasks every 5 days from the outflow of the cultures. The tubes were placed in the flasks at night and between 135 ml and 216 ml of collected culture were filtered the next morning on 0.2-μm PC filters (Nuclepore), placed in tubes containing 500 μl of lysis buffer and stored at −80 °C until further analysis. The last day of the experiment, the volumes filtered for bacterial diversity were ~500 ml for each culture.
Chemical, biological and molecular analyses
For the samples collected in situ or in the continuous cultures, bacterial abundance was determined by flow cytometry and bacterial heterotrophic production was measured by 3H-leucine incorporation (Christaki et al., 2014). Concentrations of DOC and inorganic nutrients were measured as described previously (Landa et al., 2014). For DNA extraction and pyrosequencing, samples collected at the different bloom stations and from the continuous-culture experiment were further analyzed together. Briefly, the 0.2-μm fractions were lysed using lysis buffer (40 mm EDTA, 50 mm Tris and 0.75 m sucrose) and performing 3 freeze–thaw cycles with liquid nitrogen and a water bath at 65 °C. After denaturation and degradation of proteins using sodium dodecyl sulfate (1%) and proteinase K (0.2 mg ml−1), DNA was purified using a Qiagen AllPrep DNA/RNA extraction kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The molecular size and purity of the DNA were analyzed using agarose gel electrophoresis (1%). In total, 16 samples of genomic DNA were sent to the Molecular Research DNA laboratory (Shallowater, TX, USA) for 454 pyrosequencing. The sequencing was performed using an FLX-titanium platform (Roche, Indianapolis, IN, USA; Wolcott et al., 2009). Universal bacterial primers 27 F (5′-AGRGTTTGATCMTGGCTCAG-3′) and 530 R (5′-CCGCNGCNGCTGGCAC-3′) were used, targeting hypervariable regions V1 to V3 of the 16 S rRNA gene. The raw sequences for this data set were deposited in the Sequence Read Archive (SRA) database under accession number SRP041580. Sequences from the continuous cultures and from stations R-2, A3-2 and F-L can be found on HUFUDAT01.sff, with the following barcodes: ACTGGTGT (continuous culture, control, replicate 1), ACTGTCAG (continuous culture, control, replicate 2), ACTGTCTC (continuous culture, control, replicate 3), ACTGGACT (continuous culture, diatom-DOM treatment, replicate 1), ACTGGAGA (continuous culture, diatom-DOM treatment, replicate 2), ACTGGTCA (continuous culture, diatom-DOM treatment, replicate 3), ACTGTGAC (R-2), AGACAGAC (A3-2) and AGACCTCA (F-L). Sequences from stations A3-1, A3-3, A3-4, E-1, E-3, E-4E and E-4W can be found on HUFUDAT02.sff, with the following barcodes: ACCGCTAC (A3-1), ACCGGCTT (A3-3), ACCGTAGA (A3-4), AGACGTCT (E-1), AGACTGAG (E-3), ACAGTGAA (E-4E) and AGAGAGAG (E-4W).
Bioinformatics
Analyses were conducted in QIIME (Caporaso et al., 2010) as described previously (Landa et al., 2013) with the following modifications. Raw data was denoised with AmpliconNoise (Quince et al., 2011) run through Mothur (Schloss et al., 2009). The denoised reads were clustered into operational taxonomic units (OTUs) with a 97% cutoff using Usearch 5.2.326 (Edgar, 2010). At this stage potential chimeras were removed using de novo and reference-based chimera detection in Uchime. Taxonomy was assigned as described using the rdp classifier against the Greengenes database released in August 2013, and the Silva database (release 119). For each identified phylogenetic group, we further searched the literature for sequences found in marine polar environments. On the basis of all these sequences, we constructed phylogenetic trees using Mr Bayes3, and kept the most relevant sequences to build three final trees.
Data processing
In total, 748 OTUs were obtained for the 16 samples, with a total number of sequences per sample comprised between 3957 and 12 172. To enable comparison between samples, the data set was randomly subsampled to 3957 sequences per sample. Bacterial communities from the HNLC and the various bloom sites and stages were compared by calculating a weighted UNIFRAC dissimilarity matrix (Lozupone and Knight, 2005) that considers both the phylogeny and the relative abundances of the OTUs to calculate the distance between each pair of samples. This matrix was used to construct a UPGMA dendrogram, whose nodes were further tested using a bootstrap analysis.
To identify the OTUs most enhanced in the bloom conditions compared with the reference HNLC site, the relative abundances of each individual OTU was divided by its relative abundance in the R-2 station when detected at R-2. Only OTUs for which the obtained ratio was ⩾3 and whose relative abundance in the bloom site was ⩾1% were considered for further analysis. The same criteria were applied to identify the OTUs enhanced by either condition in the continuous-culture experiment after four bacterial-generation times.
Results and discussion
Natural iron fertilization induces a mosaic of phytoplankton blooms
The region east of the Kerguelen Islands is characterized by annually occurring spring phytoplankton blooms (⩾0.6 μg l−1 chlorophyll-a (Chl-a)) in response to large-scale iron fertilization of the Southern Ocean (Figure 1). The southeastern bloom above the plateau (overall depth 527 m, Station A3) is constrained by bathymetry and is maintained over roughly 3 months by the continuous supply of low quantities of iron and major nutrients from below (Blain et al., 2007). During early spring (October–November), station A3 had a deep mixed layer (roughly 150 m) and concentrations of Chl-a increased by threefold between our first (A3-1; 0.6 μg l−1; Table 1) and second visit (A3-2; 28 days apart; 2 μg l−1). During the decline of the bloom (Stations A3-3 and A3-4, January–February) Chl-a concentrations were overall lower, whereas bacterial abundances and growth rates were about twofold higher. The offshore stations were located in a region where, at the seasonal scale, horizontal transport is the main mechanism of iron fertilization (d’Ovidio et al., 2015). Stations F-L and E-4 W, located, respectively, north and south of the Polar Front were characterized by intermediate (E-4 W, 1.3 μg l−1) and high (F-L, 4.0 μg l−1) Chl-a concentrations in overall shallow mixed layers (61–38 m). Stations E-1, E-3 and E-4E were sampled in a quasi-Lagrangian manner within a meander south of the Polar Front.
This environmental context provided access to iron-fertilized sites that varied with respect to hydrographic and biogeochemical conditions. Each of the sampled sites harbored a distinct phytoplankton community (Supplementary Tables S3 and S4). Above the Kerguelen plateau, the dominant phytoplankton species were Chaetoceros debilis (A3-2), Chaetoceros hyalochaete (A3-3) and Eucampia ant v. antarctica (A3-4), each accounting for roughly 50% of total phytoplankton-carbon biomass at the respective sites (Armand et al., 2008; Lasbleiz, 2014). At the off-plateau stations, small centrics (Thalassiosira-like; F-L), Chaetoceros debilis (E-4W) and Corethron pennatum (E-1 and E-3) accounted each for roughly 50% of phytoplankton-carbon biomass, and Eucampia antarctica and Chaetoceros debilis were the dominant phytoplankton species (each roughly 20%) at Station E-4E (Lasbleiz, 2014). The dominance of varying diatom species reflects the different biogeochemical features across stations and it indicates that we investigated a variety of phytoplankton blooms each with specific characteristics.
Diverse bacterial responses to iron-induced phytoplankton blooms
We sampled 1 reference site in HNLC waters and 9 bloom stations, and obtained 600 OTUs, defined at 97% identity of the 16 S rRNA gene. A clustering of the samples indicated that the communities in the various bloom stations were different from the community at the reference Station R-2 (Figure 2). The dissimilarity to the reference station varied from 7% to 22%, indicating the intensity and possibly the nature of the community changes were different among the bloom stations (Supplementary Figure S1). The clustering of the bacterial communities could not be explained based on the occurrence of different water parcels characterized by their physical properties. This is clearly demonstrated using the temperature and salinity diagram (Supplementary Figure S2) where the stations clustered in a different way than the bacterial communities, suggesting that factors other than physical processes were the main drivers of changes in community composition. To explore which OTUs contributed most to the observed dissimilarities, we identified the dominant OTUs (⩾1% of total sequences) that were enhanced in each of the bloom stations as compared with the reference station. Dominant OTUs with relative abundances at least three times higher in one of the bloom stations than at the reference Station R-2 were considered enhanced. OTUs that were dominant at a bloom station, but not detectable at the reference station were also considered enhanced.

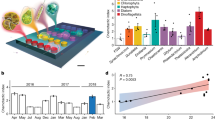
Heatmap of the OTUs identified as enhanced in at least one bloom station compared with the reference Station R-2. Each of the OTUs is identified by a number and the most detailed phylogenetic assignment obtained. Blue-colored OTUs were not detected at the reference Station R-2, and red-colored OTUs were detected at station R-2. The different shades represent the relative abundance (Rel. Ab.) of the OTUs. White indicates absence of detection. The dashes indicate the OTU was detected in the sample but did not meet the criteria to be considered as enhanced (see Results and Discussion section). The stations are ordered according to a UPGMA clustering built from a weighted UNIFRAC distance matrix. The numbers at each node of the cluster indicate the percent of dissimilarity. Blue nodes correspond to a bootstrap support of 50% to 75% and red nodes correspond to a bootstrap support >75%. OTUs that were specifically associated to one of the two treatments in the continuous-culture experiment are noted with an asterisk (*). See Figure 3 and Results and Discussion section for details.
Including all bloom stations, a total of 42 enhanced OTUs were identified (Figure 2). Twenty-seven OTUs had relative abundances at least three times higher in one of the bloom stations than at the reference Station R-2, and 15 OTUs were not detected at the reference Station R-2, but had relative abundances ⩾1% of total sequences at one of the bloom stations. The number of enhanced OTUs at a bloom station varied between 3 and 21 (mean 10±6, n=9; Figures 2 and Supplementary Figure S3), and their cumulative relative abundances accounted for 16–68% (mean 38±19%, n=9) of the total communities at the various bloom stations (Supplementary Figure S3). Only few enhanced OTUs were identical across all stations. Each pair of stations shared 0 to 12 enhanced OTUs (mean 2.33±2.31; n=36), 14 OTUs were enhanced in at least 3 stations, and only 2, OTUs 1663 (SAR86) and OTU 17 (Ulvibacter), were enhanced in at least 5 stations (Figure 2).
The increased abundance of these various OTUs in the bloom stations resulted in changes in the overall phylogenetic composition of the total communities (Supplementary Figure S1). Though the 42 enhanced OTUs belonged to all major phylogenetic groups found in marine environments, 22 of them belonged to the Roseobacter clade (7 OTUs) and the Flavobacteriaceae (Figure 2; Supplementary Figure S4 and Supplementary Table S5). These groups are known for their ability to thrive in bloom conditions in polar systems, as well as in temperate waters (see Buchan et al. (2014) for review). In the Roseobacter clade, four OTUs were identified as members of the RCA cluster, which has been found abundant in polar systems and particularly the Southern Ocean (Selje et al., 2004). The low abundance of these 4 OTUs (OTU 7, 1714, 2048 and 2049 in Figure 2) at the HNLC reference station (<1% of total sequences) and their increased contribution to the bloom stations F-L, A3-3 and A3-4 (range 1–10% of total sequences) point to a key role of the RCA members in the processing of organic matter during phytoplankton blooms as suggested by previous studies (Giebel et al., 2009, 2011; Teeling et al., 2012; Voget et al. 2015).
Flavobacteria and in particular Polaribacter are frequently found in polar waters, especially during phytoplankton blooms (Abell and Bowman, 2005; Straza et al., 2010), including blooms resulting from artificial iron-fertilization experiments (Agawin et al., 2006; Kataoka et al., 2009). Flavobacteria were described to be efficient polymer degraders (Cottrell and Kirchman, 2000), an idea that is supported from recent genomic and proteomic analyses (Williams et al., 2013; Xing et al., 2014). Overall, our data confirm the dominance of fast-growing, traditionally bloom-associated bacterial groups in the pool of enhanced OTUs. Interestingly, some SAR11 were also detected as enhanced in bloom stations (Figure 2), though as a whole this clade was not competitive under bloom conditions (Supplementary Figure S1). Our results also revealed OTUs enhanced under bloom conditions belonging to other phylogenetic groups such as Gammaproteobacteria, Actinobacteria and Planctomycetes.
The number and the variability in the identity and phylogeny of the enhanced OTUs among stations suggest that naturally iron-induced phytoplankton blooms in the Southern Ocean stimulate a large diversity of bacterial groups. Our study extends previous investigations of the southeastern bloom above the Kerguelen plateau (Station A3) that have demonstrated major differences in the bacterial community composition during the late stage of the bloom as compared with HNLC waters (West et al., 2008; Obernosterer et al., 2011). Our results from the Kerguelen region are, however, in contrast to the artificial iron-enrichment experiments, EisenEx and LOHAFEX, performed in the Southern Ocean where minor or no changes in bacterial community composition were observed in the iron-fertilized patch (Arrieta et al., 2004; Thiele et al., 2012). This suggests that the bacterial response is in part driven by the mode of fertilization that differs drastically between these studies. In contrast to the Southern Ocean, bacterial community composition associated with phytoplankton blooms has been extensively studied in other ocean regions, however, with focus on a single bloom event (González et al., 2000; Fandino et al., 2001; Riemann and Winding, 2001; Larsen et al., 2004; Rink et al., 2007). Our study brings novel perspectives on the subject, through the concurrent exploration of a patchwork of blooms provided by a large-scale natural laboratory in the Southern Ocean. The overall low overlap of the enhanced OTUs between distinct stations or bloom stages suggests that the conditions set by each site or stage favored a given community, dominated by OTUs adapted to the environmental conditions.
Is DOM a driver of the changes in bacterial community composition?
DOC in surface waters of the Southern Ocean is present at ~50 μm and dominated by refractory material originating from the deep ocean. As a consequence, accessing carbon substrates is a major constraint for bacterial growth. In that context, the supply of phytoplankton DOM is key in regulating bacterial heterotrophic metabolism (Christaki et al., 2014). In our study, features that are intimately linked to DOM production, such as phytoplankton primary production and taxonomic composition, revealed pronounced differences among stations, most likely resulting from physical setting, varying iron inputs and bloom age. Furthermore, multivariate analyses including parameters that describe the nutrient regime, the composition of the phytoplankton community and major fluxes did not identify one or a set of parameters that significantly explained the bacterial community composition in the study region (Supplementary Figure S5). This leads to the hypothesis that the nature of DOM could be a major force driving bacterial community composition at the different bloom sites. To explore this question we used continuous cultures run with an ambient bacterial community aboard the research vessel during the KEOPS2 cruise. Continuous cultures allow investigation of the response of a microbial community to a given limitation over the time scale of days to weeks, because of the maintenance of the same growth-limiting factor over several generation times (Landa et al., 2013, 2014; Beier et al., 2014).
The composition of the bacterial communities growing on seawater or on seawater mixed with diatom DOM was strikingly different after four bacterial-generation times. A comparative analysis of the major OTUs (⩾1% of total sequences), performed with the same criteria used to identify in situ-enhanced OTUs, revealed that 6 and 8 OTUs markedly responded to the seawater and to the diatom-DOM treatment, respectively (Figure 3a). These 14 OTUs accounted for a large fraction of the respective treatments (58±6% and 73±7% of total sequences in the seawater and diatom-DOM triplicate cultures, respectively). This experiment provides evidence that DOM properties were a strong factor shaping bacterial community composition, each tested condition sustaining diverse but comparatively distinct OTUs. We propose that the OTUs responsive in the seawater treatment are competitive when most of the carbon pool is constituted of semi-labile and refractory compounds. By contrast, the OTUs responsive to the diatom-DOM treatment are probably competitive at increased concentrations of labile compounds of phytoplankton origin.

Relative abundances of the OTUs associated to the seawater cultures (blue) and of the OTUs associated to the diatom-DOM cultures (red) in the continuous-culture experiment after four bacterial-generation times (a) and relative abundance in surface waters of the visited stations of the OTUs identified in the continuous-culture experiment (b). The relative abundances in individual triplicate cultures are shown. OTUs that were also detected enhanced at a bloom site are noted with an asterisk (*). See Figure 3 and Results and Discussion section for details.
The majority of the OTUs responsive to either of the two experimentally set DOM resource regimes (13 out of 14) were also detected in the bloom sites, and their cumulative abundances were particularly high at 3 sites (9–19% of total sequences; Figure 3b). Common features of these 3 stations were the slightly enhanced temperatures and the high growth rates of the bulk community (about 0.2 d−1; Table 1). This was the minimum growth rate to be maintained in the continuous cultures. It was particularly interesting to note that all OTUs responsive to the diatom-DOM treatment were present at Station F-L. This site represents the early phase of a well-developed spring bloom, characterized by highest concentrations of Chl-a constrained in a shallow mixed layer, likely leading to a pronounced quantitative and compositional signature of freshly produced, labile phytoplankton DOM. More surprisingly, OTU 1 and 6 (Polaribacter) responsive to the seawater treatment had high relative abundances at Stations A3-3 and A3-4. These stations are distinct from all other sites in that they represent the declining phase of the 3-month bloom above the Kerguelen plateau with much lower supply of recent phytoplankton DOM and a predominance of more degraded and background DOM. Thus, our experimental approach defined by two DOM resource regimes was able to identify part of the fast-growing members of the bacterial community developing in contrasting situations.
The combined experimental and in situ observations suggest that at least some of the abundant bacterial taxa exhibit distinct metabolic preferences with respect to degradation of DOM of variable origin and reactivity. These preferences could lead to partitioning of the available resource between active members of the community at a given site. The co-existence at Station F-L of OTUs that are adapted to distinct DOM conditions strongly supports this idea. Resource partitioning through the existence of complementary traits involved in carbon metabolism could decrease competition for substrate and allow positive interactions among bacterial taxa, which ultimately could benefit the bacterial community by optimizing the resource usage.
Linking composition changes to bacterially mediated carbon cycling
We established positive relationships (P⩾0.01, r2⩾0.60) between the changes observed in community structure at the various bloom sites, and bacterial heterotrophic production. First, the number of enhanced OTUs in each station was positively related with bacterial heterotrophic production in the surface mixed layer (Figure 4a). This suggests that an increase in bacterial production, driven by an increased supply of phytoplankton DOM, was associated with a higher number of responding taxa. If, as suggested by our continuous-culture experiment, DOM pools of different reactivities are exploited by distinct and diverse taxa, the higher number of responding taxa could reflect an increase in niche availability, with a positive feedback on bulk performance due to resource partitioning.

Regression analysis between bacterial heterotrophic production (mean value±s.d. in the mixed layer) and the number of enhanced OTUs identified at each bloom site (a) and between bacterial heterotrophic production and the extent of change, determined as the sum of the relative abundances of the enhanced OTUs identified at each bloom site (b). For bacterial heterotrophic production, the mean value±s.d. in the mixed layer is given. The gray data point corresponds to station E-3, which was excluded for the calculation of P and r2 values.
Second, we used the sum of the relative abundances of the enhanced OTUs at each bloom site as an estimate of the degree of change in community composition (Supplementary Figure S3). This extent of change in community composition revealed an even stronger positive relation with bacterial heterotrophic production (Figure 4b). There is, however, an interesting feature to note. The correlation coefficient was particularly high when Station E-3 was excluded from the data set, though it was still significant when included (P=0.01, r2=0.60). A specificity of Station E-3 was the low number of enhanced OTUs, 3, and the high relative abundance of one of them, an Actinomycetale (OTU 1853), which accounted for 36% of the total community at this station. For comparison, the relative abundances of enhanced OTUs at the other bloom sites did not exceed 11% of the total community, which is indicative of a higher evenness among responding taxa in these sites.
These results suggest that rates of bacterial production were tightly linked to the compositional changes induced by the growth of these 42 enhanced OTUs. Bacterial production also increased as a function of temperature (P=0.006, r2=0.68) across the study sites. Interestingly, only the extent of change in community composition revealed a significant correlation with temperature (P=0.04, r2=0.46), whereas the number of enhanced OTUs did not. This suggests that higher temperatures acted as an additional factor, rendering the changes in bacterial community composition and bacterial production more pronounced by stimulating the metabolic activity of the OTUs that responded to the bloom conditions.
We present here relationships based on a sub set of the bacterial community, composed of 42 taxa that through their capacity to respond to an environmental change, were major drivers of composition shifts and were associated with up to 40-fold variability in bacterial production. Within this community, the diversity of the enhanced OTUs, driven by both their number and their relative abundances, were key ecological features sustaining the relationship. By contrast, the diversity of the total communities was rather consistent among sites (Shannon index 5 to 5.7, except for Station E-3: 4) and not related to bacterial production (Supplementary Figure S6). This result is not surprising as total communities comprise both active and dormant cells (Lennon and Jones, 2011), rendering it difficult to study the link between community diversity and productivity. The lack of evidence for other factors structuring the bacterial communities supports our assumption that the observed bacterial response is mainly bottom–up driven, which is also consistent with in situ and experimental observations of fast-growing taxa that are likely to contribute substantially to bulk bacterial production. Grazing by heterotrophic nanoflagellates removed roughly 1% of the bacterial standing stock per day and viral lysis was negligible during the early bloom phase (Christaki et al., 2014, 2008). This further suggests that top–down control was not a dominant removal process at any of the investigated sites.
Our exploration of a mosaic of phytoplankton blooms extending over several thousands of km2 in the Southern Ocean provides novel insights on the potential role of bacterial community structure in carbon cycling. The coupling between changes in the bacterial community composition and bulk bacterial productivity across concurrently occurring spring phytoplankton blooms point to the importance of taxonomic identity and diversity for the functional outcome of the community. The combined laboratory and in situ observations demonstrate that DOM shapes the bacterial community, and they suggest that substrate preferences might lead to resource partitioning with consequences on biogeochemical fluxes. We provide mechanisms of biodiversity effects that could serve as working hypotheses to be explored in other microbial ecosystems at different spatial scales.
Accession codes
References
Abell GCJ, Bowman JP . (2005). Ecological and biogeographic relationships of class Flavobacteria in the Southern Ocean. FEMS Microbiol Ecol 51: 265–277.
Agawin NSR, Hale MS, Rivkin RB, Matthews P, Li WKW . (2006). Microbial response to a mesoscale iron enrichment in the NE Subarctic Pacific: bacterial community composition. Deep Sea Res II 53: 2248–2267.
Armand LK, Cornet-Barthaux V, Mosseri J, Quéguiner B . (2008). Late summer diatom biomass and community structure on and around the naturally iron-fertilised Kerguelen Plateau in the Southern Ocean. Deep Sea Res II 55: 653–676.
Arrieta JM, Weinbauer MG, Lute C, Herndl GJ . (2004). Response of bacterioplankton to iron fertilization in the Southern Ocean. Limnol Oceanogr 49: 799–808.
Bauer M, Kube M, Teeling H, Richter M, Lombardot T, Allers E et al. (2006). Whole genome analysis of the marine Bacteroidetes ‘Gramella forsetii’ reveals adaptations to degradation of polymeric organic matter. Environ Microbiol 8: 2201–2213.
Beier S, Rivers AR, Moran MA, Obernosterer I . (2014). The transcriptional response of prokaryotes to phytoplankton-derived dissolved organic matter in seawater. Environ Microbiol e-pub ahead of print; doi:10.1111/1462-2920.12434.
Bell T, Newman J a, Silverman BW, Turner SL, Lilley AK . (2005). The contribution of species richness and composition to bacterial services. Nature 436: 1157–1160.
Benner R Chemical composition and reactivity. Hansell DA, Carlson CA . (2002) Biogeochemistry of Marine Dissolved Organic Matter. Elsevier Science, 59–90.
Blain S, Quéguiner B, Armand L, Belviso S, Bombled B, Bopp L et al. (2007). Effect of natural iron fertilization on carbon sequestration in the Southern Ocean. Nature 446: 1070–1074.
Buchan A, LeCleir GR, Gulvik CA, González JM . (2014). Master recyclers: features and functions of bacteria associated with phytoplankton blooms. Nat Rev Micro 12: 686–698.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336.
Cadotte MW, Cardinale BJ, Oakley TH . (2008). Evolutionary history and the effect of biodiversity on plant productivity. Proc Natl Acad Sci USA 105: 17012–17017.
Cavagna A-J, Fripiat F, Elskens M, Dehairs F, Mangion PL et al. (2014). Biological productivity regime and associated N cycling in the vicinity of Kerguelen Island area, Southern Ocean. Biogeosci Discuss 11: 18073–18104.
Christaki U, Lefèvre D, Georges C, Colombet J, Catala P, Courties C et al. (2014). Microbial food web dynamics during spring phytoplankton blooms in the naturally iron-fertilized Kerguelen area (Southern Ocean). Biogeosciences 11: 6739–6753.
Christaki U, Obernosterer I, Wambeke F, Van, Veldhuis M, Garcia N, Catala P . (2008). Microbial food web structure in a naturally iron-fertilized area in the Southern Ocean (Kerguelen Plateau). Deep Sea Res II 55: 706–719.
Church MJ, Hutchins DA, Ducklow HW . (2000). Limitation of bacterial growth by dissolved organic matter and iron in the Southern Ocean. Appl Environ Microbiol 66: 455–466.
Cottrell MT, Kirchman DL . (2000). Natural assemblages of marine proteobacteria and members of the cytophaga-flavobacter cluster consuming low- and high-molecular-weight dissolved organic matter. Appl Environ Microbiol 66: 1692–1697.
Edgar RC . (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460–2461.
Fandino LB, Riemann L, Steward GF, Long RA, Azam F . (2001). Variations in bacterial community structure during a dinoflagellate bloom analyzed by DGGE and 16 S rDNA sequencing. Aquat Microb Ecol 23: 119.
Frias-Lopez J, Shi Y, Tyson GW, Coleman ML, Schuster SC, Chisholm SW et al. (2008). Microbial community gene expression in ocean surface waters. Proc Natl Acad Sci USA 105: 3805–3810.
Fuhrman J, Hewson I, Schwalbach MS, Steele JA, Brown M V, Naeem S . (2006). Annually reoccurring bacterial communities are predictable from ocean conditions. Proc Natl Acad Sci USA 103: 13104–13109.
Fuhrman J, Steele J, Hewson I, Schwalbach MS, Brown M V, Green JL et al. (2008). A latitudinal diversity gradient in planktonic marine bacteria. Proc Natl Acad Sci USA 105: 7774–7778.
Fukuda R, Ogawa H, Nagata T, Koike I . (1998). Direct determination of carbon and nitrogen contents of natural bacterial assemblages in marine environments. Appl Environ Microbiol 64: 3352–3358.
Ghiglione J-F, Galand PE, Pommier T, Pedrós-Alió C, Maas EW, Bakker K et al. (2012). Pole-to-pole biogeography of surface and deep marine bacterial communities. Proc Natl Acad Sci USA 109: 17633–17638.
Giebel H-A, Brinkhoff T, Zwisler W, Selje N, Simon M . (2009). Distribution of Roseobacter RCA and SAR11 lineages and distinct bacterial communities from the subtropics to the Southern Ocean. Environ Microbiol 11: 2164–2178.
Giebel H-A, Kalhoefer D, Lemke A, Thole S, Gahl-Janssen R, Simon M et al. (2011). Distribution of Roseobacter RCA and SAR11 lineages in the North Sea and characteristics of an abundant RCA isolate. ISME J 5: 8–19.
Gifford SM, Sharma S, Rinta-Kanto JM, Moran MA . (2011). Quantitative analysis of a deeply sequenced marine microbial metatranscriptome. ISME J 5: 461–472.
Gilbert JA, Steele JA, Caporaso JG, Steinbrück L, Reeder J, Temperton B et al. (2012). Defining seasonal marine microbial community dynamics. ISME J 6: 298–308.
González JM, Simó R, Massana R, Covert JS, Casamayor EO, Pedrós-Alió C et al. (2000). Bacterial community structure associated with a dimethylsulfoniopropionate-producing North Atlantic algal bloom. Appl Environ Microbiol 66: 4237–4246.
Gravel D, Bell T, Barbera C, Bouvier T, Pommier T, Venail P et al. (2011). Experimental niche evolution alters the strength of the diversity-productivity relationship. Nature 469: 89–92.
Grzymski JJ, Carter BJ, DeLong EF, Feldman RA, Ghadiri A, Murray AE . (2006). Comparative genomics of DNA fragments from six Antarctic marine planktonic bacteria. Appl Environ Microbiol 72: 1532–1541.
Hansell D . (2013). Recalcitrant dissolved organic carbon fractions. Ann Rev Mar Sci 5: 421–445.
Hector A, Schmid B, Beierkuhnlein C, Caldeira MC, Diemer M, Dimitrakopoulos PG et al. (1999). Plant diversity and productivity experiments in European grasslands. Science 286: 1123–1127.
Hertkorn N, Benner R, Frommberger M, Schmitt-Kopplin P, Witt M, Kaiser K et al. (2006). Characterization of a major refractory component of marine dissolved organic matter. Geochim Cosmochim Ac 70: 2990–3010.
Hooper DU, Vitousek PM . (1997). The effects of plant composition and diversity on ecosystem processes. Science 277: 1302–1305.
Kataoka T, Suzuki K, Hayakawa M, Kudo I, Higashi S, Tsuda A . (2009). Temporal changes in community composition of heterotrophic bacteria during in situ iron enrichment in the western subarctic Pacific (SEEDS-II). Deep Sea Res II 56: 2779–2787.
Landa M, Cottrell MT, Kirchman DL, Blain S, Obernosterer I . (2013). Changes in bacterial diversity in response to dissolved organic matter supply in a continuous culture experiment. Aquat Microb Ecol 69: 157–168.
Landa M, Cottrell MT, Kirchman DL, Kaiser K, Medeiros PM, Tremblay L et al. (2014). Phylogenetic and structural response of heterotrophic bacteria to dissolved organic matter of different chemical composition in a continuous culture study. Environ Microbiol 16: 1668–1681.
Langenheder S, Lindstro ES, Tranvik LJ . (2006). Structure and function of bacterial communities emerging from different sources under identical conditions. Appl Environ Microbiol 72: 212–220.
Larsen A, Flaten GF, Sandaa R-A, Castberg T, Thyrhaug R, Erga SR et al. (2004). Spring phytoplankton bloom dynamics in Norwegian coastal waters: microbial community succession and diversity. Limnol Oceanogr 49: 180–190.
Lasbleiz M . (2014). Cycles biogéochimiques (Si, C, N, P) en lien avec la dynamique nutritionnelle du phytoplancton dans la région naturellement fertilisée des Kerguelen, PhD thesis. Aix-Marseille Université: Marseille, France, p 313..
Lasbleiz M, Leblanc K, Blain S, Ras J, Cornet-Barthaux V, Helias Nunige S et al. (2014). Pigments, elemental composition (C, N, P, Si) and stoichiometry of particulate matter, in the naturally iron fertilized region of Kerguelen in the Southern Ocean. Biogeosciences 11: 8259–8324.
Lennon JT, Jones SE . (2011). Microbial seed banks: the ecological and evolutionary implications of dormancy. Nat Rev Microbiol 9: 119–130.
Loreau M, Naeem S, Inchausti P, Bengtsson J, Grime JP, Hector A et al. (2001). Biodiversity and ecosystem functioning: current knowledge and future challenges. Science 294: 804–808.
Lozupone C, Knight R . (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71: 8228–8235.
Manganelli M, Malfatti F, Samo TJ, Mitchell BG, Wang H, Azam F . (2009). Major role of microbes in carbon fluxes during Austral winter in the Southern Drake passage. PLoS One 4: e6941.
Martiny JBH, Bohannan BJM, Brown JH, Colwell RK, Fuhrman J, Green JL et al. (2006). Microbial biogeography: putting microorganisms on the map. Nat Rev Microbiol 4: 102–112.
McGrady-Steed J, Harris PM, Morin PJ . (1997). Biodiversity regulates ecosystem predictability. Nature 390: 162–165.
Moran MA Genomics and metagenomics of marine prokaryotes. Kirchman DL . (2008) Microbial Ecology of the Oceans. John Wiley and Sons Inc:: Hoboken, NJ, USA, 91–129.
Moran MA, Buchan A, González JM, Heidelberg JF, Whitman WB, Kiene RP et al. (2004). Genome sequence of Silicibacter pomeroyi reveals adaptations to the marine environment. Nature 432: 910–913.
Naeem S, Li S . (1997). Biodiversity enhances ecosystem reliability. Nature 390: 507–509.
Obernosterer I, Catala P, Lebaron P, West NJ . (2011). Distinct bacterial groups contribute to carbon cycling during a naturally iron fertilized phytoplankton bloom in the Southern Ocean. Limnol Oceanogr 56: 2391–2401.
Obernosterer I, Christaki U, Lefèvre D, Catala P, Van Wambeke F, Lebaron P . (2008). Rapid bacterial mineralization of organic carbon produced during a phytoplankton bloom induced by natural iron fertilization in the Southern Ocean. Deep Sea Res II 55: 777–789.
Obernosterer I, Lami R, Larcher M, Batailler N, Catala P, Lebaron P . (2010). Linkage between bacterial carbon processing and the structure of the active bacterial community at a coastal site in the NW Mediterranean Sea. Microb Ecol 59: 428–435.
d’Ovidio F, Della Penna A, Trull TW, Nencioli F, Pujol I, Rio MH et al. (2015). The biogeochemical structuring role of horizontal stirring: Lagrangian perspectives on iron delivery downstream of the Kerguelen plateau. Biogeosci Discuss 12: 779–814.
Park Y-H, Durand I, Kestenare E, Rougier G, Zhou M, d’ Ovidio F et al. (2014). Polar Front around the Kerguelen Islands: an up-to-date determination and associated circulation of surface/subsurface waters. J Geophys Res Oceans 119: 6575–6592.
Pedler BE, Aluwihare LI, Azam F . (2014). Single bacterial strain capable of significant contribution to carbon cycling in the surface ocean. Proc Natl Acad Sci USA 111: 7202–7207.
Pedrós-Alió C . (2006). Marine microbial diversity: can it be determined? Trends Microbiol 14: 257–263.
Peter H, Beier S, Bertilsson S, Lindström ES, Langenheder S, Tranvik LJ . (2011). Function-specific response to depletion of microbial diversity. ISME J 5: 351–361.
Pollard RT, Salter I, Sanders RJ, Lucas MI, Moore CM, Mills RA et al. (2009). Southern Ocean deep-water carbon export enhanced by natural iron fertilization. Nature 457: 577–580.
Pommier T, Canbäck B, Riemann L, Boström KH, Simu K, Lundberg P et al. (2007). Global patterns of diversity and community structure in marine bacterioplankton. Mol Ecol 16: 867–880.
Poretsky RS, Hewson I, Sun S, Allen AE, Zehr JP, Moran MA . (2009). Comparative day/night metatranscriptomic analysis of microbial communities in the North Pacific subtropical gyre. Environ Microbiol 11: 1358–1375.
Quince C, Lanzen A, Davenport RJ, Turnbaugh PJ . (2011). Removing noise from pyrosequenced amplicons. BMC Bioinformatics 12: 38.
Reinthaler T, Winter C, Herndl GJ . (2005). Relationship between bacterioplankton richness, respiration, and production in the Southern North Sea. Appl Environ Microbiol 71: 2260–2266.
Riemann L, Winding A . (2001). Community dynamics of free-living and particle-associated bacterial assemblages during a freshwater phytoplankton bloom. Microb Ecol 42: 274–285.
Rink B, Seeberger S, Martens T, Duerselen C-D, Simon M, Brinkhoff T . (2007). Effects of phytoplankton bloom in a coastal ecosystem on the composition of bacterial communities. Aquat Microb Ecol 48: 47–60.
Rusch DB, Halpern AL, Sutton G, Heidelberg KB, Williamson S, Yooseph S et al. (2007). The Sorcerer II Global Ocean Sampling Expedition: Northwest Atlantic through Eastern Tropical Pacific. PLoS Biol 5: e77.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537–7541.
Selje N, Simon M, Brinkhoff T . (2004). A newly discovered Roseobacter cluster in temperate and polar oceans. Nature 427: 445–448.
Sjöstedt J, Pontarp M, Tinta T, Alfredsson H, Turk V, Lundberg P et al. (2013). Reduced diversity and changed bacterioplankton community composition do not affect utilization of dissolved organic matter in the Adriatic Sea. Aquat Microb Ecol 71: 15–24.
Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, Neal PR et al. (2006). Microbial diversity in the deep sea and the underexplored ‘rare biosphere’. Proc Natl Acad Sci USA 103: 12115–12120.
Straza TRA, Ducklow HW, Murray AE, Kirchman DL . (2010). Abundance and single-cell activity of bacterial groups in Antarctic coastal waters. Limnol Oceanogr 55: 2526–2536.
Teeling H, Fuchs BM, Becher D, Klockow C, Gardebrecht A, Bennke CM et al. (2012). Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 336: 608–611.
Teira E, Gasol JM, Aranguren-Gassis M, Fernández A, González J, Lekunberri I et al. (2008). Linkages between bacterioplankton community composition, heterotrophic carbon cycling and environmental conditions in a highly dynamic coastal ecosystem. Environ Microbiol 10: 906–917.
Thiele S, Fuchs BM, Ramaiah N, Amann R . (2012). Microbial community response during the iron fertilization experiment LOHAFEX. Appl Environ Microbiol 78: 8803–8812.
Vergin K, Done B, Carlson C, Giovannoni S . (2013). Spatiotemporal distributions of rare bacterioplankton populations indicate adaptive strategies in the oligotrophic ocean. Aquat Microb Ecol 71: 1–13.
Voget S, Wemheuer B, Brinkhoff T, Vollmers J, Dietrich S, Giebel H-A et al. (2015). Adaptation of an abundant Roseobacter RCA organism to pelagic systems revealed by genomic and transcriptomic analyses. ISME J 9: 371–384.
West NJ, Obernosterer I, Zemb O, Lebaron P . (2008). Major differences of bacterial diversity and activity inside and outside of a natural iron-fertilized phytoplankton bloom in the Southern Ocean. Environ Microbiol 10: 738–756.
Williams TJ, Wilkins D, Long E, Evans F, DeMaere MZ, Raftery MJ et al. (2013). The role of planktonic Flavobacteria in processing algal organic matter in coastal East Antarctica revealed using metagenomics and metaproteomics. Environ Microbiol 15: 1302–1317.
Wolcott RD, Gontcharova V, Sun Y, Dowd SE . (2009). Evaluation of the bacterial diversity among and within individual venous leg ulcers using bacterial tag-encoded FLX and Titanium amplicon pyrosequencing and metagenomic approaches. BMC Microbiol 9: 226.
Xing P, Hahnke RL, Unfried F, Markert S, Huang S, Barbeyron T et al. (2014). Niches of two polysaccharide-degrading Polaribacter isolates from the North Sea during a spring diatom bloom. ISME J 9: 1410–1422.
Zubkov M V, Holland RJ, Burkill PH, Croudace IW, Warwick PE . (2007). Microbial abundance, activity and iron uptake in vicinity of the Crozet Isles in November 2004–January 2005. Deep Sea Res II 54: 2126–2137.
Acknowledgements
We thank the captain and the crew of the ‘R/V Marion Dufresne’ and the chief scientist B Quéguiner for their help and support aboard. We thank L Oriol for inorganic nutrient analyses, J Caparros for DOC analyses and the trace-metal team for providing large volumes of filtered seawater. N Fuchs (Alfred Wegener Institute, Germany) kindly provided the strain Chaetoceros debilis. We thank M Rembauville for providing Figure 1. The ocean color products for the Kerguelen area were obtained from GlobColour. We thank M-A Moran and R Lami for insightful comments on a previous version of the manuscript. Three anonymous reviewers improved a previous version of the manuscript. The KEOPS2 project received financial support from the CNRS-INSU-LEFE-CYBER, the ANR-10-BLAN-0614 and the IPEV. ML was supported by a PhD fellowship from the French Ministry of Science.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Landa, M., Blain, S., Christaki, U. et al. Shifts in bacterial community composition associated with increased carbon cycling in a mosaic of phytoplankton blooms. ISME J 10, 39–50 (2016). https://doi.org/10.1038/ismej.2015.105
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2015.105
This article is cited by
-
The functional diversity of microbial communities in the multi-compartment biofilters of shrimp mariculture effluents using 16S rRNA metabarcoding
Aquaculture International (2024)
-
Metagenome-assembled genomes reveal greatly expanded taxonomic and functional diversification of the abundant marine Roseobacter RCA cluster
Microbiome (2023)
-
First report on the bacterial community composition, diversity, and functions in Ramsar site of Central Himalayas, Nepal
Environmental Monitoring and Assessment (2023)
-
Vertical microbial profiling of water column reveals prokaryotic communities and distribution features of Antarctic Peninsula
Acta Oceanologica Sinica (2023)
-
Role of Bacterial Community Composition as a Driver of the Small-Sized Phytoplankton Community Structure in a Productive Coastal System
Microbial Ecology (2023)
