
Abstract
Objective:
Renin–angiotensin system (RAS) regulates adipogenic response with adipocyte hypertrophy by increasing oxidative stress. Recent studies have shown the role of peroxisome proliferator-activated receptor-δ (PPARδ) agonist in attenuation of angiotensin II-induced oxidative stress. The aim of this study was to explore a potential mechanistic link between PPARδ and the cytoprotective enzyme heme oxygenase-1 (HO-1) and to elucidate the contribution of HO-1 to the adipocyte regulatory effects of PPARδ agonism in an animal model of enhanced RAS, the Goldblatt 2 kidney 1 clip (2K1C) model.
Method:
We first established a direct stimulatory effect of the PPARδ agonist (GW 501516) on the HO-1 gene by demonstrating increased luciferase activity in COS-7 cells transfected with a luciferase-HO-1 promoter construct. Sprague-Dawley rats were divided into four groups: sham-operated animals, 2K1C rats and 2K1C rats treated with GW 501516, in the absence or presence of the HO activity inhibitor, stannous mesoporphyrin (SnMP).
Results:
2K1C animals had increased visceral adiposity, adipocyte hypertrophy, increased inflammatory cytokines, increased circulatory and adipose tisssue levels of renin and Ang II along with increased adipose tissue gp91 phox expression (P<0.05) when compared with sham-operated animals. Treatment with GW 501516 increased adipose tissue HO-1 and adiponectin levels (P<0.01) along with enhancement of Wnt10b and β-catenin expression. HO-1 induction was accompanied by the decreased expression of Wnt5b, mesoderm specific transcript (mest) and C/EBPα levels and an increased number of small adipocytes (P<0.05). These effects of GW501516 were reversed in 2K1C animals exposed to SnMP (P<0.05).
Conclusion:
Taken together, our study demonstrates, for the first time, that increased levels of Ang II contribute towards adipose tissue dysregulation, which is abated by PPARδ-mediated upregulation of the heme-HO system. These findings highlight the pivotal role and symbiotic relationship of HO-1, adiponectin and PPARδ in the regulation of metabolic homeostasis in adipose tissues.
Similar content being viewed by others

Introduction
Adipose tissues are a site for energy storage and also functions as an endocrine organ that secretes various biologically active molecules called adipocytokines. Hyperplasia and hypertrophy of adipocytes is central to pathological conditions such as obesity and metabolic syndrome,1, 2, 3 conditions that are frequently associated with chronic redox imbalances. Reactive oxygen species (ROS) regulate the adipogenic process where ROS-induced adipocyte hypertrophy has been observed.4, 5, 6 In this regard, angiotensin II activates cellular oxidases and precipitates redox imbalance.7 Angiotensin II has also been reported to regulate adipocyte growth and differentiation along with modulation of adipocytokine expression and release.8 Increased Ang II levels in adipocytes induce oxidative stress and attenuate adiponectin release.4 The Goldblatt 2 kidney 1 clip (2K1C) model is a model for reno-vascular hypertension characterized by activation of the renin–angiotensin system (RAS)9 whose local adipose tissue effects have recently been reported.10 However, precise regulatory effect of Ang II on adipose tissue is still unclear and needs further examination.
Peroxisome proliferator-activated receptor-δ (PPARδ), a member of the PPAR nuclear receptor family, is a ligand-activated transcriptional factor with a multifunctional role in lipid metabolism, namely, to increase lipolysis, promote fatty acid catabolism and energy expenditure in adipose tissues.11, 12 PPARδ forms obligate heterodimers with the retinoid X receptor and bind to defined PPAR elements in the promoter region of target genes. PPARδ is expressed in many tissues, including metabolically active sites such as the liver, muscle and fat, and its role in the metabolic syndrome is currently elucidated.12 PPARδ is activated by oxidative stress and has an important role in decreasing inflammation, apoptosis and leukocyte adhesion. Recent studies have shown the role of PPARδ agonist in the attenuation of angiotensin II-induced oxidative stress in vascular smooth muscles.13 Additionally, upregulation of the antioxidant enzyme system, that is, the heme–heme oxygenase system (HO) has also been shown to reduce Ang II-induced oxidative stress, with abatement of associated cardiovascular complications. HO system comprises of two isoforms, a constitutively expressed isoform called HO-2 and an inducible HO-1.14 HO-1 is stimulated by a variety of stimuli, including heavy metals, ROS, NO, Ang II and cytokines.15, 16, 17 Induction of the HO-1 gene, both in vivo and in cell cultures, reduces adipocyte hypertrophy with an increase in adiponectin levels and the number of small adipocytes, which are regarded as ‘healthy’ insulin-sensitive adipocytes.18, 19
This study examines adipose tissue effects of increased Ang II, both in vitro and in vivo (Goldblatt’s 2K1C model). In addition, we use this model to evaluate a possible interplay between PPARδ and HO-1 in regulating adipocyte morphology and function, as both HO-1 and PPARδ have been shown to attenuate Ang II-induced redox imbalance and associated pathologies. In contextual light, the present study aims to explore the effectiveness of a PPARδ agonist in the prevention of Ang II-induced adipocyte dysfunction and the possible interaction between PPARδ and HO-1 system in a model of enhanced oxidative stress. This novel study corroborates the existence of a PPARδ-HO-1 axis whose direct stimulation via the administration of an exogenous PPARδ agonist induces HO-1 expression, abates RAS-associated adipocyte dysfunction and induces adipocyte remodeling with smaller adipocytes while decreasing inflammation and increasing adiponectin secretion.
Materials and methods
Animal treatment
All animal studies were approved by the New York Medical College Animal Care and Use Committee in accordance with the National Institutes of Health Guidelines for Care and Use of Laboratory Animals. Forty, 8-week-old Sprague-Dawley rats were used in the studies. Sprague-Dawley rats were divided into four groups: sham-operated animals, 2K1C rats and 2K1C rats treated with GW 501516, in the absence or presence of the HO activity inhibitor, stannous mesoporphyrin (SnMP). All surgically altered rats were fitted with a 0.25-mm U-shaped silver clip with an internal gap of 0.25 mm around the left renal artery. GW501516 (Enzo Life Sciences Inc., Plymouth Meeting, PA, USA) was dissolved in dimethyl sulfoxide and suspended in 0.5% carboxymethylcellulose. Immediately after surgery, GW 501516 was administered via oral gavage daily for 3 weeks at a concentration of 3 mg kg−1 body weight. SnMP was injected intraperitoneally, three times a week at a dose of 20 mg kg−1 of body weight, for 2 weeks. Blood pressure was measured by the tail cuff method immediately before surgery and then 3 weeks after surgery. Before the experiment, rats were all acclimatised to the tail cuff method. Rats were placed in a heat-controlled box (36–38 °C) for approximately 10 min before applying the tail cuff. The mean of a minimum of five measurements was obtained from each rat. After a 6-h fast, rats were anesthetized with sodium pentobarbital (65 mg kg−1, intraperitoneally) and blood was obtained from a tail vein for glucose measurement using a glucometer (Lifescan Inc., Miligitas, CA, USA). At the time of killing, body weight, liver weight and visceral fat content of all rats was measured. Blood samples were collected in K3EDTA tubes at killing and the plasma was separated. Samples were flash frozen in liquid nitrogen and maintained at −80 °C until assayed.
Gene reporter assay
Cos7 cells were grown to 90% confluence in 24-well plates and then transiently transfected with expression vectors for PPARδ and RXR using Lipofectamine 2000, according to the manufacturer’s protocol. A luciferase plasmid construct containing 15 kb of the HO-1 promoter (HO-1–15 kb-Luc reporter; courtesy of Dr Jawed Alam) was co-transfected, as appropriate, along with a pRL-CMV renilla reporter to normalize for transfection efficiency. Twenty-four hours post transfection, cells were treated with either vehicle or GW501516 (PPARδ agonist) for an additional 24 h until harvest. Cell lysates were prepared by repeated freezing and thawing in Promega lysis buffer (Promega, Madison, WI, USA), and luciferase activity was measured using Promega’s luciferase assay system in a Dual luciferase plate reader (Promega).
Cell culture and differentiation into adipocytes
Frozen mouse preadipocytes (3T3L-1) were purchased from ATTC (ATTC, Manassas, VA, USA). After thawing, 3T3L cells were resuspended in a Dulbecco's Modified Eagle Medium (DMEM; Invitrogen, Carlsbad, CA, USA) supplemented with 10% heat inactivated fetal bovine serum (FBS; Invitrogen) and 1% antibiotic/antimycotic solution (Invitrogen). The cells were plated at a density of 1–5 × 106 cells per 100 cm2 dish. The cultures were maintained at 37 °C in a 5% CO2 incubator and the medium was changed after 48 h and every 3∼4 days thereafter. When the 3T3L cells were confluent, the cells were recovered by the addition of 0.25% trypsin/EDTA (Invitrogen). 3T3L cells (Passages 2 and 3) were plated in a 75-cm2 flask at a density of 1–2 × 104 cells and cultured in DMEM with 10% FBS until 80% confluence was achieved. The medium was replaced with adipogenic medium, and the cells were cultured for an additional 7 days as described previously.5
Cells were treated every alternate day with GW501516 (10 μM), Ang II (10 μM) and SnMP (5 μM) for 7 days in mouse preadipocytes.
Effect of Ang II on adipogenesis
To measure the effect of increasing Ang II concentration on MSC-derived adipocyte differentiation, cells were treated with 0.1, 1, 10, 100 and 1000 μ M of Ang II every alternate day. After 7 days, cells were stained with Oil Red O solution to study adipogenesis.
Oil Red O staining
Lipid droplets were detected by Oil red O staining. For Oil Red O staining, 0.21% Oil Red O in 100% isopropanol (Sigma-Aldrich, St Louis, MO, USA) was used. Briefly, mouse preadipocytes were fixed in 10% formaldehyde, washed in Oil-red O for 10 min, rinsed with 60% isopropanol (Sigma-Aldrich), and the Oil red O eluted by adding 100% isopropanol for 10 min and optical density measured at 490 nm, for a 0.5-sec reading. Mouseb preadipocytes were measured by Oil red O staining (optical density=490 nm) after day 7.
Determination of adipocyte size and number
Visceral adipose tissue was collected from all the groups and prepared for morphological analysis, as previously described.20 Samples were fixed in 4% paraformaldehyde for 24 h, cut into small pieces and embedded in paraffin for histological analysis. The samples were cut by microtome (5 μM thick), mounted on d-polylisinated glass slides, deparaffinized in xylene and stained with haematoxylin and eosin for the evaluation of adipocyte size. Each sample was examined and evaluated for the adipocyte size and number using an optical microscopy ( × 200 magnification). Specifically, for each animal 100 adipocytes from different hematoxylin–eosin-stained sections were analyzed by determining individual adipocyte areas (μM2) using an image analyzer (Image Pro Plus, Milan, Italy). The data were pooled to represent a mean value and a statistical analysis was applied to compare the results obtained from the different experimental groups with a statistical significance set at P<0.05. The data were presented as the mean±s.d.
Western blot analysis of HO-1, adiponectin, gp phox91 and adipogenic markers
Frozen adipose tissue was pulverized under liquid nitrogen and placed in a homogenization buffer comprising (mmol l−1): 10 phosphate buffer, 250 sucrose, 1 EDTA, 0.1 phenylmethanesulfonylfluoride, and 0.1% v/v tergitol, pH 7.5. Homogenates were centrifuged at 27 000 g for 10 min at 4 °C. The supernatant was isolated, and protein levels were assayed (Bradford Method). The supernatant was used for the determination of HO-1, gp phox91, adiponectin, Pref-1, fatty acid synthase, Wnt10b, Wnt5b, β-catenin, mest and CEBPα. Immunoblotting was performed in visceral adipose tissue as previously described.19, 21 β-Actin was used to ensure adequate sample loading for all western blots.
Cytokines, adiponectin, Ang II, renin and lipid profile measurements
The plasma levels of interleukin (IL)-6, monocyte chemotactic protein-1 (MCP-1) and the high molecular weight (HMW) form of adiponectin were determined using an enzyme-linked immunosorbent assay (ELISA) as previously described.4, 21 Angiotensin II and renin was measured in plasma and visceral adipose tissues using an ELISA assay (Assay Gate, Inc., Ijamsville, MD, USA). Triglycerides and total low-density lipoprotein levels are measured in plasma using an ELISA assay (Assay Gate, Inc.).
Quantitative real-time PCR analysis
Total RNA was extracted from liver tissues using 5-Prime PerfectPure RNA Tissue Kit (Fisher Scientific Company, LLC, Waltham, MA, USA). Total RNA was read on a NanoDrop 2000 Spectrophotometer (Thermo Scientific, Wilmington, DE, USA) and cDNA was synthesized using iScript cDNA Synthesis kit (Bio-Rad, Hercules, CA, USA). PCR amplification of the cDNA was performed by quantitative real-time PCR using qPCR Core kit for SYBR Green I (Applied Biosystems, Grand Island, NY, USA). The thermocycling protocol consisted of 10 min at 95 °C, 40 cycles of 15 s at 95 °C, 30 s at 60 °C and finished with a melting curve ranging from 60 to 95 °C to allow distinction of specific products. Primers were designed specific to each gene using Primer Express 3.0 software (Applied Biosystems). Normalization was performed in separate reactions with primers to 18S mRNA (5′-TTCGAACGTCTGCCCTATCAA-3′ and 5′-ATGGTAGGCACGGCGACTA-3′).
Statistical analysis
Data are expressed as means±s.e.m. Significance of difference in mean values was determined using one-way analysis of variance followed by the Newman–Keul’s post hoc test. P<0.05 was considered to be significant.
Results
Effect of the PPARδ agonist on adipogenesis in mouse preadipocytes
To study the effects of Ang II on adipogenesis, in vitro, 3T3L1 cells were exposed to Ang II in a dose-dependent manner. At 10 μM, Ang II increased adipogenesis in mouse preadipocytes, measured as the relative absorbance of Oil Red O on day 7 of the experiment (Figure 1a; P<0.05); follow-up in vitro experiments were performed with 10 μM of Ang II. We then studied the effects of the PPARδ agonist on adipogenesis in mouse preadipocytes, in the absence or in the presence of Ang II (Figure 1b). Our results show that concurrent administration of PPARδ agonist significantly (P<0.05) decreased adipogenesis in preadipocytes treated with Ang II; this effect was not seen in experiments conducted with SnMP, the HO inhibitor (Figure 1b; P<0.05).

(a) Effect of increasing Ang II concentrations on adipogenesis in mouse preadipocytes. Lipid droplet area was measured as the relative absorbance of Oil Red O at day 7 after inducing adipogenesis as described in Materials and Methods (mean±s.e., *P<0.05 vs vehicle; n=6). (b) Effect of the PPARδ-agonist (GW501516) and Ang II on adipogenesis in mouse preadipocytes. Adipogenesis was measured as the relative absorbance of Oil Red O at day 7 after inducing adipogenesis as described in Methods and procedures (mean±s.e., *P<0.05 vs vehicle; n=6).
Effect of the PPARδ agonist on body weight and the metabolic profile
As shown in Table 1, animals with clipped kidneys have body weight similar to sham-operated animals. Liver weight, plasma glucose, serum triglycerides and total low-density lipoprotein levels were also not dissimilar between the two groups. Treatment with the PPARδ agonist, alone or in combination with SnMP, did not affect these anthropometric and general metabolic parameters in Goldblatt’s hypertensive rats.
Effect of the PPARδ agonist on plasma renin and Ang II levels
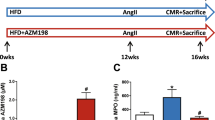
Quantitative analysis, performed on the plasma from Goldblatt’s 2K1C and sham-operated animals, showed significant elevation of plasma renin (P<0.05) and Ang II levels in rats with a clipped kidney (Figures 2a and b). Treatment with the PPARδ agonist significantly decreased renin (P<0.05) and Ang II levels (P<0.05) in these rats, an effect reversed by the administration of SnMP (P<0.05). We measured blood pressure in this animal model of renovascular hypertension and, as expected, systolic blood pressure was increased in 2K1C rats as compared with sham animals (167.3±10.8 mm Hg vs 124.1±9.4 mm Hg, respectively; P<0.05). Treatment with the PPARδ agonist significantly reduced the levels of systolic blood pressure in the 2K1C rats (135±8.9 mm Hg; P<0.05 vs 2K1C); this was abolished in animals also treated with SnMP (159±8.4 mm Hg; P<0.05 vs 2K1C+GW).

(a, b) Effects of the PPARδ-agonist in 2K1C animals on plasma renin and Ang II levels; results are means±s.e., n=6/group. (a) Plasma renin; *P<0.05 vs control, #P<0.05 vs 2K1C,+P<0.05 vs 2K1C+GW. (b) Plasma Ang II; *P<0.05 vs control, #P<0.05 vs 2K1C,+P<0.05 vs 2K1C+GW. (c–e) Effects of the PPARδ-agonist in 2K1C animals on plasma cytokines and adiponectin levels; results are means±s.e., n=6/group. (c) Plasma IL-6; *P<0.05 vs control, #P<0.05 vs 2K1C,+P<0.05 vs 2K1C+GW. (d) Plasma MCP-1; *P<0.05 vs control, #P<0.05 vs 2K1C,+P<0.05 vs 2K1C+GW. (e) Plasma adiponectin levels; *P<0.05 vs control, #P<0.05 vs 2K1C,+P<0.05 vs 2K1C+GW.
Effect of the PPARδ agonist on plasma inflammatory cytokines and adiponectin levels
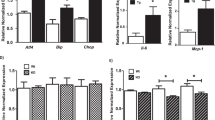
The Goldblatt 2K1C model is associated with systemic inflammation, and this was examined in the current study. Plasma IL-6 and MCP-1 levels were greater in the 2K1C rats when compared with sham-operated rats (Figures 2c and d; P<0.05). The PPARδ agonist (P<0.05) decreased plasma IL-6 and MCP-1 levels and this was prevented in animals also treated with SnMP (P<0.05; Figures 2c and d). One of the established markers of dysfunctional adipose tissues is the attenuation of plasma adiponectin levels. Goldblatt’s 2K1C rats displayed reduced plasma adiponectin levels when compared with sham animals (Figure 2e; P<0.05). Plasma adiponectin levels were increased in 2K1C rats treated with the PPARδ agonist (P<0.05). SnMP prevented the increase in adiponectin levels when compared with animals administered the PPARδ agonist alone (Figure 2e).
Effect of the PPARδ agonist on adipose tissue renin and Ang II levels and on the marker of oxidative stress
Complementary results show that visceral adipose tissue renin was increased in Goldblatt 2K1C model, a model of enhanced RAS, as compared with control animal (Figure 3a; P<0.05). Treatment with the PPARδ agonist decreased renin levels (P<0.05) and this effect was reversed by administration of PPARδ agonist and SnMP. Similarly, Ang II levels in visceral adipose tissue were increased in 2K1C1 rats as compared with control animals (P<0.05). The PPARδ agonist decreased Ang II levels, and this effect was reversed by the concurrent administration of SnMP (Figure 3b). Oxidative stress was assessed in visceral adipose tissues by the measurement of gp phox 91 protein expression level, which was increased in 2K1C rats when compared with control animals (Figure 3c; P<0.05). PPARδ agonist decreased the expression of gp phox 91 (P<0.05) while SnMP prevented this decrease (P<0.05).

Effects of the PPARδ-agonist on adipose tissue renin and Ang II levels, and on the marker of oxidative stress; results are means±s.e., n=6/group. (a) Adipose tissue renin; *P<0.05 vs control, #P<0.05 vs 2K1C,+P<0.05 vs 2K1C+GW. (b) Adipose tissue Ang II; *P<0.05 vs control, #P<0.05 vs 2K1C,+P<0.05 vs 2K1C+GW. (c) Western blot and densitometry analysis of gp phox 91expression; *P<0.05 vs control, #P<0.05 vs 2K1C,+P<0.05 vs 2K1C+GW. Data are shown as mean band density normalized to β-actin.
Effect of the PPARδ agonist on adipogenesis and visceral fat content
Histological examination of the visceral adipose tissues revealed that cross-sectional area of the adipocytes from 2K1C rats was significantly higher when compared with control rats (Figure 4a; P<0.05). The adipocyte area is reduced in rats treated with the PPARδ agonist and SnMP reverses this effect, indicating the interactive role of PPARδ-HO-1 in regulating adipocyte hypertrophy and lipid accumulation (P<0.05). Apart from affecting size, adipocyte number was also modulated in 2K1C rats, showing a reduction, when compared with the control animals (Figure 4b; P<0.05). The number of adipocytes increased in rats treated with the PPARδ agonist, and treatment with SnMP decreased this adipocyte number (P<0.05). Though the body weight of rats did not change among the different groups (Table 1), visceral fat content was significantly higher in 2K1C rats when compared with the control animals (Figure 4c; P<0.05). 2K1C rats treated with the PPARδ agonist showed a significant reduction in visceral fat content and the concurrent administration of SnMP produced an increase in visceral fat mass (Figure 4c).

(a) Hematoxylin–eosin staining of visceral adipose tissue and quantitative analysis of adipocyte size, expressed in μm2, in 2K1C animals—treated with and without GW 501516 or GW 501516+SnMP. Results are means±s.e.; *P<0.05 vs control, #P<0.05 vs 2K1C,+P<0.05 vs 2K1C+GW. (b) The graph shows number of adipocytes in 2K1C animals—treated with and without GW 501516 or GW 501516+SnMP. * P<0.05 vs control, #P<0.05 vs 2K1C,+P<0.05 vs 2K1C+GW. (c) Effects of the PPARδ-agonist on visceral fat in 2K1C animals—treated with and without GW 501516 or GW 501516+SnMP. Results are means±s.e., n=6/group; *P<0.05 vs control, #P<0.05 vs 2K1C,+P<0.05 vs 2K1C+GW.
We also examined the hepatic lipid-oxidation pathways to ascertain their role in bringing about adipose tissue changes in the 2K1C rats as compared with the sham-operated rats. Rats with a clipped kidney had levels of carnitine palmitoyltransferase 1A (CPT1a) and mitochondrial uncoupling protein 2 (UCP2) mRNA not dissimilar from the controls (CPT1a: sham—1.41±0.07 vs 2K1C—1.18±0.15; UCP2: sham—1.38±0.08 vs 2K1C—1.13±0.10). It is noteworthy, however that treatment with the PPARδ agonist increased (P<0.05) gene expression of both CPT1a and UCP2 in the 2K1C rats (CPT1a: 2K1C+GW—2.45±0.06; UCP2: 2K1C—2.15±0.08; P<0.05 vs 2K1C). SnMP blocked this stimulatory effect of the PPARδ agonist (CPT1a: 2K1C+GW+SnMP—1.70±0.10; UCP2: 2K1C+GW+SnMP—1.67±0.11; P<0.05 vs 2K1C+GW).
Effect of PPARδ agonist on adipogenic markers and adiponectin expression in visceral adipose tissue
Our next set of experiments examined possible mechanisms involved in RAS-dependent stimulation of visceral adiposity. We first studied the levels of Wnt10b, β-catenin and adiponectin to see whether they could account for the decrease in fat content in adipose tissues of 2KIC rats treated with the PPARδ agonist. Wnt10b and β-catenin levels decreased in 2K1C rats as compared with control animals (Figures 5a and b respectively; P<0.01). Administration of the PPARδ agonist increased the Wnt10b and β-catenin expression; SnMP blocked this effect (P<0.05). Activation of the Wnt-signalling cascade promotes a lower differentiation state of pre-adipocytes,22 and this was tested in our study via pre-adipocyte factor 1 (pref1) expression analysis (Figure 5c). Pref1 is an adipocyte marker, expressed abundantly in pre-adipocytes. Animals with a clipped kidney expressed lower levels of pref1 (P<0.05). Pref1 expression was rescued by the PPARδ agonist (P<0.05), an effect blocked by SnMP (P<0.05). Additionally, western blot analysis of adiponectin levels showed a significant reduction (P<0.05) in 2K1C rats as compared with control animals (Figure 5d); P<0.05. The concurrent administration of the PPARδ agonist increased (P<0.05) adiponectin expression and SnMP decreased this effect. These data suggest that an increase in PPARδ-HO-1 axis improves adipocyte function and reduces adiposity through upregulation of adipocyte-protective adiponectin and activation of Wnt10b and β-catenin pathway.

Effects of the PPARδ-agonist on adipogenic markers and adiponectin expression in adipose tissue in 2K1C animals—treated with and without GW 501516 or GW 501516+SnMP. (a, b) Western blot and densitometry analysis of Wnt10b and β-catenin expression respectively; *P<0.05 vs control, #P<0.05 vs 2K1C,+P<0.05 vs 2K1C+GW. Results are means±s.e., n=6/group; data are shown as mean band density normalized to β-actin. (c) Western blot and densitometry analysis of pref1 expression; *P<0.05 vs control, #P<0.05 vs 2K1C,+P<0.05 vs 2K1C+GW. Results are means±s.e., n=6/group; data are shown as mean band density normalized to β-actin. (d) Western blot and densitometry analysis of adiponectin expression; *P<0.05 vs control, #P<0.05 vs 2K1C,+P<0.05 vs 2K1C+GW. Results are means±s.e., n=6/group; data are shown as mean band density normalized to β-actin.
Effect of PPARδ agonist on adipogenic markers in visceral adipose tissue
Experiments studying positive regulators of lipid accumulation and adipogenesis were examined next. Our results showed that positive regulators of adipogenesis mest, C/EBPα and Wnt5b levels were all increased significantly in 2K1C rats as compared with control animals (Figures 6a–c, respectively; P<0.05). The concurrent administration of the PPARδ agonist decreased mest, C/EBPα and Wnt5b expression, and this effect was increased by SnMP treatment in 2K1C animals (P<0.05). Additionally, fatty acid synthase expression (Figure 6d; P<0.05) was elevated in rats with a clipped kidney. PPARδ agonist treatment blocked this increase (P<0.05), which was restored when animals were co-treated with SnMP (P<0.05).

Effects of the PPARδ-agonist on adipogenic marker expression in adipose tissue in 2K1C animals—treated with and without GW 501516 or GW 501516+SnMP. (a–d) Western blot and densitometry analysis of mest, C/EBPα, Wnt5b and fatty acid synthase (FAS) levels, respectively; *P<0.05 vs control, #P<0.05 vs 2K1C,+P<0.05 vs 2K1C+GW. Results are means±s.e., n=6/group; data are shown as mean band density normalized to β-actin.
Effect of PPARδ agonist on HO-1 levels in visceral adipose tissue
We evaluated the proposed interplay between PPARδ and HO systems in the 2K1C animal model. Western blot analysis showed a significant increase in HO-1 expression (P<0.05) in the visceral adipose tissue of 2K1C rats treated with the PPARδ agonist, as compared with the 2K1C rats without treatment (Figure 7a). Furthermore, our results showed that the concurrent administration of SnMP also increased HO-1 expression. These findings confirm our previous results with SnMP, which induced a significant increase in HO-1 expression but is a potent inhibitor of HO activity.9 This effect of SnMP prevents heme degradation and inhibits formation of HO products, that is, CO and biliverdin.

(a) Effect of PPARδ-agonist on HO-1 expression in adipose tissue in 2K1C animals—treated with and without GW 501516 or GW 501516+SnMP. Western blot and densitometry analysis of HO-1 levels; *P<0.05 vs control, #P<0.05 vs 2K1C. Results are means±s.e., n=6/group; Data are shown as mean band density normalized to β-actin. (b) HO-1 promoter activity, as determined in Cos 7 cells by the luciferase assay. ***P<0.05 vs without PPARδ; **P<0.05 vs with PPARδ but without GW.
To study the potential mechanistic link between PPARδ and HO-1, we transiently transfected the HO-1 promoter luciferase construct into Cos 7 cells. Overexpression of PPARδ cDNA and treatment with its agonist GW501516 significantly increased HO-1 promoter activity (Figure 7b; P<0.05). In addition, our results show that there was no significant increase in HO-1 promoter activity in the absence of PPARδ cDNA overexpression or with GW501516, suggesting that HO-1 is a PPARδ-target gene.
Discussion
Angiotensin II induces oxidative stress and contributes to various pathological conditions, including insulin resistance and the metabolic syndrome.8, 23 In this regard, recent reports have indicated that inhibitors of the RAS attenuate oxidative stress and improve the metabolic profile in various animal models.24 However, evidence linking Ang II with adipocyte size and function is scarce, thereby prompting the present study. Two complimentary lines of evidence highlight the first key finding of this study,that is, a stimulatory effect of Ang II on adipocyte size and lipid accumulation. First sets of experiments demonstrate a dose-dependent increase in lipogenesis: in rodent preadipocytes with exogenous Ang II. Complementary in vivo studies reveal that elevation of endogenous Ang II levels also lead to adipocyte hypertrophy and visceral adiposity. Redox imbalance has been shown to increase adipogenesis5 that is characterized by adipocyte hypertrophy and dysfunction.5 Goldblatt’s 2K1clip animals are an established model of reno-vascular hypertension where upregulated RAS precipitates redox imbalance and oxidative stress.9 These animals exhibit high blood pressure with increased plasma levels of renin and Ang II, including in adipose tissues. Increased visceral adiposity was evident in these animals in spite of less than significant change in their body and liver weight, blood glucose, serum triglycerides and low-density lipoprotein levels. This suggests that Ang II is directly responsible for the adipocyte hypertrophy that appears to be defined by its ability to modulate redox balance. Relevant to these findings, Ang II-induced increase in adiposity was characterized by depleted proliferation and enhanced lipid accumulation by visceral adipocytes: morphological characteristics frequently accompanied by adipocyte dysfunction.
Increased circulating levels of inflammatory cytokines, and reduced adiponectin levels, both in the plasma and in the adipose tissues, substantiate Ang II-induced dysfunctional adipogenesis in the 2K1clip rats. Adiponectin is a cytoprotective adipokine used as a marker of adipocyte function.3 Attenuation of adiponectin levels is frequently associated with enlarged, hypoxic adipocytes, which also demonstrate increased inflammatory infiltration.25 These morpho-physiological alterations of the adipocytes are the hallmarks of obesity and metabolic syndrome.22, 26 Association of these functional perturbations of adipocytes with elevated levels of Ang II implicates its role in bringing about these dysfunctions.
Redox-dependent modulation of Wnt10b/β-catenin (the canonical pathway) is an effect that underlies increased lipid accumulation and hypertrophic adipogenesis; this is commonly observed in clinico-pathological scenarios associated with oxidative stress.22, 25 Wnt10b is an endogenous regulator of adipogenesis that functions as a ‘switch’ that retards adipocyte differentiation and maturation into lipid-laden cells. Activation of Wnt10b contributes to the inactivation/phosphorylation of glycogen synthase kinase-3β and, consequently, elevated level of β-catenin (Figure 8); it is the molecular node of the canonical Wnt signaling pathway.27 Conversely, Wnt5b, another component of the Wnt family, stimulates adipogenesis by inhibiting the β-catenin and the Wnt-signaling cascade.28 Consistent with these findings, we found that the Wnt10b and β-catenin protein expression were downregulated and that the Wnt5b protein levels were increased in 2K1clip rats. Wnt10b maintains adipocytes in a reduced state of differentiation22 as evident in our study by the expression of pref1. Pref1 is employed as a marker for adipocyte differentiation; abundant in pre-adipocytes, while the expression declines progressively as they mature and accumulate lipid droplets. Rats with overexpression of the RAS demonstrated attenuation of the Wnt-signalling cascade along with lower levels of pref1 protein. These observations, in conjunction with the morphological characteristics of the visceral adipose tissues, implicate RAS-mediated promotion of adipocyte growth and differentiation in these animals. Supportive evidence in this direction shows that RAS-induced dysfunctional adipogenesis is accompanied by increased expression of adipogenic regulators—mest and CCAAT/enhancer binding protein (C/EBPα). An increase in Wnt/β-catenin signaling inhibits C/EBPα and mest levels, which are expressed during adipogenesis and have a crucial role in the transcriptional regulation of adipocyte genes.25

A schematic summary of our hypotheses and obtained results. Angiotensin II, via redox-dependent mechanisms, promotes adipocyte growth and lipid accumulation. This involves the suppression of the Wnt-canonical pathway and leads to dysfunctional hypertrophic adipogenesis—characterized by increased visceral adiposity, increased inflammation and decreased adiponectin levels. A synthetic PPARδ-agonist binds to and activates the HO-1 promoter, with increased HO-1 expression as a consequence. Activation of this pathway stimulates the Wnt-canonical signaling cascade in adipose tissues, subjected to higher levels of Ang II and oxidative stress. Consequentially, preadipocyte maturation and lipid accumulation is retarded, leading to an increased number of smaller adipocytes with improved adipocytokine profile. Thus, PPARδ-agonist-mediated HO-1 activation prevents oxidative stress and associated dysfunctional adipogenesis in animals with an overactive renin–angiotensin system.
The redox-dependent increase in adipogenesis and adipocyte hypertrophy is attenuated by exogenous antioxidants.22 In this regard, PPARδ—a ligand-activated transcription factor—regulates inflammation, energy homeostasis and reduces oxidative stress.29 The second key finding presented in this study shows that a synthetic PPARδ agonist, GW501516, attenuates Ang II-mediated increase in adipogenesis. Although the biological roles of PPARα/PPARγ in modulating metabolic homeostasis are well established,30 contributions of PPARδ to this process remain unclear. Our results show that the pro-adipogenic effects of AngII, both in vitro and in vivo, are prevented by treatment with a PPARδ agonist, manifest by an improvement in adipocyte function and an abatement of the inflammatory response. A concomitant attenuation in adipose tissue oxidative stress and adiponectin rescue by GW501506 underscores the ability of PPARδ activation to restore redox balance. This could, in turn, contribute towards the restoration of adipose tissue abnormalities observed in a setting of elevated levels of AngII. The patho-physiological recovery, in animals treated with a PPARδ agonist, was accompanied by the rescue of the Wnt-canonical pathway that, in turn, retards adipocyte maturation into large lipid-laden cells. PPARδ agonist-induced effects on adipocyte differentiation are embodied by the increased expression of pre-adipocyte marker—pref1. In addition, the observed suppression of pro-adipogenic regulators, mest1 and C/EBPα, support the hypothesis that activation of PPARδ via an exogenous synthetic agonist leads to the attenuation of lipid accumulation and adipocyte hypertrophy. These results are bolstered with reports characterizing increased lipolysis and reduced adiposity in transgenic animals treated with the PPARδ agonist.12
Although 2K1C animals have similar hepatic lipid oxidation as their sham-operated counterparts, PPARδ agonist did induce these biochemical processes in the treated animals. This may be one of the contributing factors towards the agonist-induced attenuation of adiposity, in the in vivo setting. These results are unsurprising, as the PPARs have been shown to modulate lipid-oxidation pathways in models of diet-induced obesity.12, 31 What was unexpected was the failure of this agonist to affect the metabolic profile of the animals in our study. Despite reducing visceral adiposity and inflammation, the PPARδ agonist had no effect on the metabolic profile of the treated animals. These observations, while unexplained by the study, lend themselves to a few hypotheses: firstly, enhanced adiposity in these animals was not initiated by dietary alterations, rather by induction of tissue oxidative stress; it is also possible that a more chronic study with or without additional dietary modifications may allow a better understanding of the patho-physiological mechanisms involved in the redox-initiated adipogenic processes.
The heme-HO system has therapeutic potential in clinico-pathological conditions frequently associated with chronic oxidative stress.17, 19 That AngII-dependent cardio-vascular pathologies in the 2k1clip model of hypertension are rescued in animals with increased levels of HO1 is evidence of the ability of this antioxidant system to reverse the ROS-dependent effects of AngII.9, 17, 32 In light of this evidence, we examined the role of transcriptional stimulation of HO1 by the PPARδ agonist, and its role in restoring adipocyte structure and function in models with elevated AngII. The third key finding of this study shows that the PPARδ agonist-mediated rescue of effects of AngII on adipose tissues is reliant upon activation of the heme-HO system. Multiple lines of evidence support this conclusion: firstly, attenuation of visceral adiposity and improvement in adipocyte function, observed in animals with clipped kidney and treated with GW501516, was paralleled by upregulation of HO1. Secondly, morpho-physiological rescue of the adipose tissues by the PPARδ agonist, both in vitro and in vivo, was prevented in a setting where HO activity is blocked. And finally, stimulation of HO1 promoter by the PPARδ-agonist implicates PPARδ binding to the HO promoter. Further supporting evidence to the involvement of the heme-HO system is provided by recovery of the Wnt-canonical pathway in animals treated with the PPARδ agonist. Previous reports have shown the stimulatory effects of heme-HO on Wnt10b signaling,22 and we have evidence linking this effect of HO-1 to its anti-adipogenic properties (unpublished work).
To conclude as represented in the schematic (Figure 8), we show here that AngII has a stimulatory effect on adipogenesis and adipocyte lipid accumulation, with a concomitant decline in adipocyte function, as evidenced by the downregulation of the protective adipokine—adiponectin. These effects of AngII contribute towards enhanced visceral adiposity in an animal model displaying elevation of the RAS. Suppression of the Wnt-canonical-signaling pathways with a concomitant increase in the pro-adipogenic regulators, such as mest, C/EVBPα and the fatty acid synthase, appear to underlie the effects of Ang II. An exogenous PPARδ agonist suppresses the AngII-induced dysfunctional adipogenesis and lipid accumulation, while stimulating the Wnt-canonical pathway. Also contributing to this effect is the upregulation of the hepatic lipid-oxidation processes by the PPARδ agonist. These actions of the PPARδ agonist are, in turn, reliant upon the transcriptional activation of the heme-HO system. Antiredox properties of the HO system underlie, at least partly, the therapeutic benefits obtained from GW501506.
References
Ueno T, Fujimori K . Novel suppression mechanism operating in early phase of adipogenesis by positive feedback loop for enhancement of cyclooxygenase-2 expression through prostaglandin F2alpha receptor mediated activation of MEK/ERK-CREB cascade. FEBS J 2011; 278: 2901–2912.
Matsushita K, Wu Y, Okamoto Y, Pratt RE, Dzau VJ . Local renin angiotensin expression regulates human mesenchymal stem cell differentiation to adipocytes. Hypertension 2006; 48: 1095–1102.
Sodhi K, Puri N, Inoue K, Falck JR, Schwartzman ML, Abraham NG . EET agonist prevents adiposity and vascular dysfunction in rats fed a high fat diet via a decrease in Bach 1 and an increase in HO-1 levels. Prostaglandins Other Lipid Mediat 2012; 98: 133–142.
Kim DH, Burgess AP, Li M, Tsenovoy PL, Addabbo F, McClung JA et al. Heme oxygenase-mediated increases in adiponectin decrease fat content and inflammatory cytokines, tumor necrosis factor-alpha and interleukin-6 in Zucker rats and reduce adipogenesis in human mesenchymal stem cells. J Pharmacol Exp Ther 2008; 325: 833–840.
Puri N, Sodhi K, Haarstad M, Kim DH, Bohinc S, Foglio E et al. Heme induced oxidative stress attenuates sirtuin1 and enhances adipogenesis in mesenchymal stem cells and mouse pre-adipocytes. J Cell Biochem 2012; 113: 1926–1935.
Iyer A, Fairlie DP, Prins JB, Hammock BD, Brown L . Inflammatory lipid mediators in adipocyte function and obesity. Nat Rev Endocrinol 2010; 6: 71–82.
Hung WW, Hsieh TJ, Lin T, Chou PC, Hsiao PJ, Lin KD et al. Blockade of the renin-angiotensin system ameliorates apelin production in 3T3-L1 adipocytes. Cardiovasc Drugs Ther 2011; 25: 3–12.
Cassis LA, Police SB, Yiannikouris F, Thatcher SE . Local adipose tissue renin-angiotensin system. Curr Hypertens Rep 2008; 10: 93–98.
Botros FT, Schwartzman ML, Stier CT Jr, Goodman AI, Abraham NG . Increase in heme oxygenase-1 levels ameliorates renovascular hypertension. Kidney Int 2005; 68: 2745–2755.
Mogi M, Iwai M, Horiuchi M . Emerging concept of adipogenesis regulation by the renin-angiotensin system. Hypertension 2006; 48: 1020–1022.
Ehrenborg E, Krook A . Regulation of skeletal muscle physiology and metabolism by peroxisome proliferator-activated receptor delta. Pharmacol Rev 2009; 61: 373–393.
Wang YX, Lee CH, Tiep S, Yu RT, Ham J, Kang H et al. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell 2003; 113: 159–170.
Kim HJ, Ham SA, Paek KS, Hwang JS, Jung SY, Kim MY et al. Transcriptional up-regulation of antioxidant genes by PPARdelta inhibits angiotensin II-induced premature senescence in vascular smooth muscle cells. Biochem Biophys Res Commun 2011; 406: 564–569.
Abraham NG, Kappas A . Pharmacological and clinical aspects of heme oxygenase. Pharmacol Rev 2008; 60: 79–127.
Abraham NG, Cao J, Sacerdoti D, Li X, Drummond G . Heme oxygenase: the key to renal function regulation. Am J Physiol Renal Physiol 2009; 297: F1137–F1152.
Abraham NG, Kappas A . Heme oxygenase and the cardiovascular-renal system. Free Radic Biol Med 2005; 39: 1–25.
Cao J, Sodhi K, Inoue K, Quilley J, Rezzani R, Rodella L et al. Lentiviral-human heme oxygenase targeting endothelium improved vascular function in angiotensin II animal model of hypertension. Hum Gene Ther 2011; 22: 271–282.
Peterson SJ, Kim DH, Li M, Positano V, Vanella L, Rodella LF et al. The L-4F mimetic peptide prevents insulin resistance through increased levels of HO-1, pAMPK, and pAKT in obese mice. J Lipid Res 2009; 50: 1293–1304.
Li M, Kim DH, Tsenovoy PL, Peterson SJ, Rezzani R, Rodella LF et al. Treatment of obese diabetic mice with a heme oxygenase inducer reduces visceral and subcutaneous adiposity, increases adiponectin levels, and improves insulin sensitivity and glucose tolerance. Diabetes 2008; 57: 1526–1535.
Peterson SJ, Drummond G, Kim DH, Li M, Kruger AL, Ikehara S et al. L-4F treatment reduces adiposity, increases adiponectin levels and improves insulin sensitivity in obese mice. J Lipid Res 2008; 49: 1658–1669.
Sodhi K, Inoue K, Gotlinger K, Canestraro M, Vanella L, Kim DH et al. Epoxyeicosatrienoic acid agonist rescues the metabolic syndrome phenotype of HO-2-null mice. J Pharmacol Exp Ther 2009; 331: 906–916.
Vanella L, Li M, Kim D, Malfa G, Bellner L, Kawakami T et al. ApoA1: mimetic peptide reverses adipocyte dysfunction in vivo and in vitro via an increase in heme oxygenase (HO-1) and Wnt10b. Cell Cycle 2012; 11: 706–714.
Ferder L, Inserra F, Martinez-Maldonado M . Inflammation and the metabolic syndrome: role of angiotensin II and oxidative stress. Curr Hypertens Rep 2006; 8: 191–198.
Dandona P, Dhindsa S, Ghanim H, Chaudhuri A . Angiotensin II and inflammation: the effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockade. J Hum Hypertens 2007; 21: 20–27.
Cao J, Peterson SJ, Sodhi K, Vanella L, Barbagallo I, Rodella LF et al. Heme oxygenase gene targeting to adipocytes attenuates adiposity and vascular dysfunction in mice fed a high-fat diet. Hypertension 2012; 60: 467–475.
Immenschuh S, Hinke V, Katz N, Kietzmann T . Transcriptional induction of heme oxygenase-1 gene expression by okadaic acid in primary rat hepatocyte cultures. Mol Pharmacol 2000; 57: 610–618.
Bennett CN, Ross SE, Longo KA, Bajnok L, Hemati N, Johnson KW et al. Regulation of Wnt signaling during adipogenesis. J Biol Chem 2002; 277: 30998–31004.
Zalesin KC, Franklin BA, Miller WM, Peterson ED, McCullough PA . Impact of obesity on cardiovascular disease. Med Clin North Am 2011; 95: 919–937.
Schnegg CI, Robbins ME . Neuroprotective mechanisms of PPARdelta: modulation of oxidative stress and inflammatory processes. PPAR Res 2011; 2011: 373560.
Wang YX . PPARs: diverse regulators in energy metabolism and metabolic diseases. Cell Res 2010; 20: 124–137.
Olson EJ, Pearce GL, Jones NP, Sprecher DL . Lipid effects of peroxisome proliferator-activated receptor-delta agonist GW501516 in subjects with low high-density lipoprotein cholesterol: characteristics of metabolic syndrome. Arterioscler Thromb Vasc Biol 2012; 32: 2289–2294.
Hu CM, Chen YH, Chiang MT, Chau LY . Heme oxygenase-1 inhibits angiotensin II-induced cardiac hypertrophy in vitro and in vivo. Circulation 2004; 110: 309–316.
Acknowledgements
We had full access to the data and take responsibility for its integrity. We have read and agreed with the manuscript as written. This work was supported by National Institutes of Health grants DK56601, HL-34300 and Brickstreet Foundation Inc. We thank Jennifer Brown for her outstanding editorial assistance in the preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Sodhi, K., Puri, N., Kim, D. et al. PPARδ binding to heme oxygenase 1 promoter prevents angiotensin II-induced adipocyte dysfunction in Goldblatt hypertensive rats. Int J Obes 38, 456–465 (2014). https://doi.org/10.1038/ijo.2013.116
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ijo.2013.116
Keywords
This article is cited by
-
Six Decades of History of Hypertension Research at the University of Toledo: Highlighting Pioneering Contributions in Biochemistry, Genetics, and Host-Microbiota Interactions
Current Hypertension Reports (2022)
-
Self-regulation of the inflammatory response by peroxisome proliferator-activated receptors
Inflammation Research (2019)
-
PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD
Nature Reviews Endocrinology (2017)
