
Abstract
Sympathetic hyperactivity has an important role in cardiovascular mortality in patients with type 2 diabetes (T2D). Thyrotropin-releasing hormone (TRH)-containing fibers innervate autonomic motor and premotor nuclei of the brainstem and spinal cord that regulate cardiovascular functions. We compared cardiovascular responses to application of TRH-analog in the brainstem of Wistar and T2D Goto–Kakizaki (GK) rats. GK rats exhibited basal systolic hypertension (152±2 mm Hg) and had a significantly potentiated, dose-related hypertensive response to intracisternal (i.c.) injection of the TRH-analog RX77368 (10–60 ng). In GK rats only, i.c. RX77368 (30–60 ng) markedly increased heart rate (HR; +88 b.p.m.) and induced acute cardiac mortality (100%), concurrent with extreme hyperglycemia (>26 mmol l−1), increased plasma H2O2 and 8-isoprostane, and enhanced heart expression of NADPH oxidase 4 and vascular cell adhesion molecule-1 mRNAs. GK rats also had elevated basal plasma epinephrine, higher adrenal gene expression of tyrosine hydroxylase and dopamine β-hydroxylase (DβH), and greater plasma catecholamine and adrenal DβH responses to i.c. TRH-analog, compared with Wistar rats. In GK rats, hexamethonium blocked i.c. RX77368-induced hypertensive and tachycardic responses, and reduced mortality by 86%, whereas phentolamine abolished the hypertensive response but enhanced tachycardia (+160 b.p.m.), and reduced mortality by 50%. The angiotensin II type 1 receptor antagonist irbesartan prevented i.c. RX77368-induced increases in blood pressure, HR and mortality. In conclusion, sympathetic overactivation triggered by brainstem TRH contributes to the mechanism of cardiovascular morbidity and mortality in T2D, which involves heightened cardiac inflammation and peripheral oxidative stress responses to sympathetic drive, and a mediating role of the renin–angiotensin system.
Similar content being viewed by others

Introduction
Cardiovascular disease is the leading cause of mortality in patients with type 2 diabetes (T2D). T2D patients have a high incidence of hypertension and nonischemic heart failure, and worse outcomes in acute cardiovascular events compared with nondiabetic controls.1, 2 A key mechanism underlying cardiovascular disorders is an increase in sympathetic nerve activity.3, 4 In addition to pathological cardiovascular changes due to inflammation and overactivity of the renin–angiotensin system,2, 5, 6, 7 T2D is associated with autonomic dysregulation, including enhanced sympathetic neural drive and vagal impairment. This autonomic dysregulation contributes to increased blood pressure (BP), cardiac arrhythmias and atrial fibrillation, and the resulting progression to heart failure.8, 9, 10, 11, 12, 13 Although autonomic imbalance in T2D has been extensively documented, the neural mechanisms underlying the sympathetic hyperactivity involved in high cardiac morbidity and mortality in T2D patients remains elusive.
Thyrotropin-releasing hormone (TRH) is a three amino-acid neuropeptide originally discovered in the hypothalamic paraventricular nucleus, where it regulates pituitary thyrotropin release. Other major loci of central TRH synthesis include the brainstem raphe nuclei, which project TRH-containing fibers to sympathetic and vagal motor neurons located respectively in the spinal intermediolateral cell column and the dorsal vagal complex consisting the nucleus tractus solitarius and the dorsal motor nucleus of the vagus (DMV). These areas are densely supplied with TRH-immunoreactive nerve terminals and TRH receptor 1.14, 15, 16 TRH-containing fibers also innervate autonomic premotor loci, particularly the rostral ventrolateral medulla (RVLM), a brainstem sympathetic center that has a pivotal role in the autonomic control of cardiovascular function.17, 18, 19 These TRH-innervated brainstem and spinal nuclei are important central components of the baroflex pathways.3 Injection of TRH or its stable analog RX77368 into the brain ventricles increases both sympathetic and vagal motor output, inducing sympathetically driven hyperglycemia, hypertension and tachycardia, and vagal-mediated stimulation of gastric secretion/contractility and pancreatic insulin secretion.17, 20, 21, 22, 23, 24, 25 Brainstem microinjection and intrathecal injection studies revealed that the pontine locus coeruleus, medullary RVLM and spinal intermediolateral cell column are key sites at which TRH acts to stimulate sympathetic output,17, 20, 26, 27 whereas the dorsal motor nucleus of the vagus and nucleus ambiguus are among those responsible for the resulting stimulation of vagal efferent outflow.28, 29 This TRH pathways appear to be important in responding to metabolic challenges, as brainstem TRH gene expression is upregulated by energy deficiency or increased energy demand, such as occurs in starvation, hypothermia and hypothyroidism.30, 31, 32 The dual role of TRH-containing fibers in regulating both sympathetic and vagal outflow indicates that brainstem TRH has an important physiological role in balancing sympathovagal control of multiorgan visceral function to achieve metabolic homeostasis.
Dysfunction of brainstem TRH contributes to autonomic abnormalities in stress responses and diseases. For example, brainstem TRH is involved in the strong sympathovagal activation during acute cold exposure, resulting in gastric ulceration and in the autonomic disorders presenting in hypo- and hyperthyroidism.31, 32, 33, 34 We recently established a role for disrupted autonomic responses to brainstem TRH receptor activation in diabetic pathophysiology, by demonstrating that an unbalanced ‘sympathetic-over-vagal’ excitation by brainstem TRH is responsible for the impaired insulin response to glucose in the Goto–Kakizaki (GK) rat,17, 25 an inbred polygenetic model of spontaneous non-obese T2D with a wide range of abnormalities directly relevant to components of the cardiometabolic syndrome and diabetes complications.35, 36, 37
The objective of the present study was to test the hypothesis that in T2D, brainstem TRH evokes exaggerated sympathetic activation that is responsible for the increased cardiovascular morbidity. Heart rate (HR), BP and indices of sympathetic–adrenal system activity, heart/plasma inflammation and oxidative stress were compared between nondiabetic Wistar rats and T2D GK rats under basal conditions and after intracisternal (i.c.) injection of the stable TRH-analog RX77368. Mechanisms responsible for the altered cardiovascular responses in GK rats were studied using pharmacological and molecular biological approaches.
Methods
Animals
Male nondiabetic Wistar rats (250–280 g) were purchased from Harlan Laboratories (San Diego, CA, USA) and maintained in the animal facilities of the Department of Veterans Affairs (VA) Greater Los Angeles Healthcare System (GLAHS) for 1 week before experiments. Sex- and weight-matched T2D GK rats were bred in the VA GLAHS animal facilities with a breeding protocol approved by the Institutional Animal Care and Use Committee (IACUC) of VA GLAHS, and were used at the age of 3 months when their body weights were 250–280 g. Rats were housed under controlled conditions (21–23 °C, light on from 0600 to 1800) with free access to standard rat chow (Prolab Lab Diet, PMI Nutrition International, Brentwood, MO, USA) and tap water. The animal study protocol was approved by the IACUC of VA GLAHS. All animal studies were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Chemicals
RX77368 (p-Glu-His-(3,3′-dimethyl)-Pro-NH2, Reckitt and Colman, Kingston Upon Hull, UK) is a metabolically stable TRH-analog with potent and relatively long-lasting central effects compared with TRH in rats and mice. RX77368 was freshly diluted with normal saline, from aliquots (30 μg/50 μl in 0.1%BSA/saline) stored at −80 °C. Hexamethonium chloride (Sigma Chemical Co., St Louis, MO, USA), a nicotinic acetylcholine receptor antagonist, and phentolamine (Sigma), a nonselective α-adrenergic antagonist, were freshly dissolved in normal saline. Irbesartan (Bristol-Meyers Squibb US Pharmaceuticals, Princeton, NJ, USA), an angiotensin II type 1 receptor blocker, was dissolved in water before administration by gavage.
Experimental procedure
Prior to the experiment, animals were acclimated to restraint in a rodent restrainer used for BP measurement three episodes per day and 10–15 min per episode for 3 days, in order to reduce the influence of restraint stress per se on HR and BP. On the day of experiment, non-fasted conscious rats were warmed (27–29 °C ambient) before the measurement of basal systolic BP and HR by tail cuff plethysmography (IITC Inc. Woodland Hills, CA, USA). Then, vehicle (normal saline, 10 μl) or RX77368 [10–60 ng (25–150 pmol) per 10 μl] were i.c. injected in rats under brief (2–3 min) isoflurane anesthesia. Animals awakened within 1 min, and BP and HR were measured at 30 min intervals thereafter for 120 min. In some groups, one of the following pretreatments was applied after basal BP/HR measurement and before i.c. injection at t=0 min: hexamethonium chloride (10 mg kg−1) injected s.c. at −30 min; phentolamine (2 mg kg−1) injected i.p. at −30 min; irbesartan (50 mg kg−1 per 10 ml) administered by gavage at −60 min. At the end of the experiment, rats were deeply anesthetized with i.p. pentobarbital (75 mg kg−1) and 3 ml of blood was drawn from the superior mesenteric vein for measurement of circulating compounds. Blood glucose levels were measured using the One Touch Ultra Blood Glucose Monitoring System (Lifescan, Milpitas, CA, USA). Tissue from the left heart ventricle (50–100 mg) was collected for gene expression tests. For GK rats that survived <120 min after the i.c. injection, the blood and tissue collection were performed at the time of the death. Samples from GK rats receiving i.c. 60 ng RX77368 were not processed because of the difference in sample-collecting times due to early death.
In another three groups each of Wistar and GK rats, blood was collected by needle-punch from the capillary bed of the tail tip under basal conditions and 60 min after saline or RX77368 (25 or 50 ng) i.c. injection, for measuring plasma epinephrine and norepinephrine levels. The rats were then euthanized by pentobarbital overdose (100 mg kg−1), and adrenal glands were harvested for measuring gene expression of catecholamine-synthesizing enzymes.
Intracisternal injection
Isoflurane-anesthetized rats were individually positioned on a stereotaxic instrument (David Kopf Instruments, Tujunga, CA, USA, model 900). The needle of a Hamilton syringe was inserted through the skin, posterior atlanto-occipital membrane, dura matter and the underlying arachnoid membrane into the cisterna magna. Successful insertion of the needle into the cisterna magma was verified by aspirating clear cerebrospinal fluid into the syringe before and after the injection of the tested substance.
Quantitative determination of mRNA expression in the heart and the adrenal glands
Total RNAs were extracted from the harvested left heart ventricle tissue and bilateral adrenal glands using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA). Heart tissue complementary DNA was synthesized using the Omniscript RT system (Qiagen Inc., Valencia, CA, USA). Real-time PCR in heart samples was performed using TaqMan reagents (Applied Biosystems (ABI), Foster City, CA, USA) in an ABI 9300 Real-Time PCR System (ABI). TaqMan gene expression probes for NADPH oxidase 4 (NOX4), vascular cell adhesion molecule-1 (VCAM-1) and glyceraldehydes-3-phosphate dehydrogenase (GAPDH) were obtained from ABI. Standard curves for each gene of interest were constructed, and relative concentrations of each gene of interest were quantified and standardized to the internal GAPDH expression for that sample. Total RNA isolated from adrenal glands was reverse-transcribed using a QuantiTect Reverse Transcription Kit (Qiagen Inc.). The genes of interests were detected by PCR with 30 cycles of amplification. Primers used were: forward (F): 5′-GCTACCGAGAGGACAGCATC-3′, reverse (R): 5′-GCACCATAAGCCTTCAGCTC-3′ for adrenal tyrosine hydroxylase (TH); F: 5′-TGAAGCCTGAGGTCTCCACT-3′, R: 5′-ACCGGCTTCTTCTGGGTAGT-3′ for dopamine β-hydroxylase (DβH); F: 5′-AGCCTTCGACTGGAGTGTGT-3′, R: 5′-GGTCTCGGACCTCATAACCA-3′ for phenylethanolamine N-methyltransferase; and F: 5′-CAAGTTCAACGGCACAG-3′, R: 5′-GCCAGTAGACTCCACGACAT-3′ for GAPDH. PCR products were analyzed by electrophoresis on a 1.5% agarose gel. The mRNA expression level was scanned and quantified using NIH imaging software (nih-image163, http://rsbweb.nih.gov/nih-image/download.html).
Blood chemistry
Blood samples were collected into EDTA-containing tubes and separated plasma were stored at −80 °C. ELISA was used to measure levels of plasma epinephrine and norepinephrine (Rocky Mountain Diagnostics Inc., Colorado Springs, CO, USA), 8-isoprostane (Cayman, Ann Arbor, MI, USA) and monocyte chemotactic protein-1 (MCP-1, Assay Designs, Madison, WI, USA). Hydrogen peroxide (H2O2) in plasma samples was determined using an Amplex Red assay (Molecular Probes, Eugene, OR, USA). Total plasma cholesterol was measured using the enzymatic colorimetric method (Wako Pure chemical Industries, Ltd, Richmond, VA, USA).
Statistical analysis
Data are expressed as mean±s.e.m. for each experimental group. Statistical comparisons between group-mean values for Wistar and GK rats receiving the same treatment or between RX77368-treated and vehicle-treated rats of the same strain were performed using unpaired Student's t-test. Comparisons between values obtained before and after i.c. injection in the same group were performed using a paired Student's t-test. Comparisons of dose-related response to i.c. RX77368 between Wistar and GK rats were performed using two-way analysis of variance. All statistical tests were performed using SigmaStat 2.0 software (Aspire Software International, Ashburn, VA, USA). P-values <0.05 were considered statistically significant.
Results
Dose-related BP, HR, cardiac mortality and blood glucose responses to i.c. TRH-analog RX77368 in T2D GK rats compared with nondiabetic Wistar rats
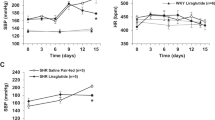
Basal systolic BP in T2D GK rats was 152±2 mm Hg (n=29) as compared with 124±3 mm Hg (n=29) in Wistar rats (P<0.05). There was no difference in basal HR between the two strains (Figure 1). The TRH-analog RX77368 given i.c. at the dose of 10 or 20 ng did not significantly change systolic BP in either strain. However, there was a decrease in HR in Wistar rats (10 ng at 60 min) and an increase in GK rats (10 and 20 ng at 30 min), respectively (Figure 1). After 20 ng RX77368 i.c., two of eight (25%) GK rats died, at 60 and 90 min, respectively, from acute congestive heart failure, characterized by pulmonary edema found in autopsy (Figure 1). Injection of 30 ng RX77368 i.c. induced significant BP increases in both strains and, in GK rats only, a potent and long-lasting tachycardia (Figure 1). BP in GK rats remained elevated persistently until death in all rats. In contrast, in Wistar rats, BP did not exceed 170 mm Hg and declined from its peak at 120 min post-injection, without causing any cardiac mortality (Figure 1). The hypertensive response to 60 ng RX77368 i.c. was stronger than that to 30 ng in GK rats, but was similar in Wistar rats. At the 60 ng dose, four of five GK rats died within 60 min and the remaining rat by 90 min post-injection, while all the Wistar rats survived this treatment (Figure 1). Clear dose-related responses to i.c. RX77368 were observed in the systolic BP at 60 min and HR at 30 min in both Wistar and GK rats. Due to the inability of the tail cuff technique to continuously monitor BP, the possibility of missing severe spikes in BP during these experiments cannot be excluded.

Time course of systolic blood pressure, heart rate and mortality in response to intracisternal (i.c.) injection of the stable thyrotropin-releasing hormone-analog RX77368 (10–60 ng) in nondiabetic Wistar and type 2 diabetic Goto–Kakizaki (GK) rats. Each point represents mean±s.e.m. of the number of rats indicated beside each line in the left panel (in the case of no mortality) or the number of surviving rats in the group at the time point. The numerator in a fraction in the graphs of the left panel indicates the number of rats expired in the previous interval and the denominator indicates the total number of rats in this group. *P<0.05 compared with the value for Wistar rats at the same time point. #P<0.05 compared with the basal levels of the same group.
RX77368 i.c. (30 or 60 ng) did not alter blood glucose levels in Wistar rats at 120 min post-injection compared with saline controls (in mmol l−1, saline: 7.4±0.3, RX77368 30 ng: 7.1±0.1, RX77368 60 ng: 6.7±0.1), whereas it induced potent hyperglycemia in GK rats at the time of death (saline: 17.4±1.0, RX77368 30 ng: 33.2±1.0, RX77368 60 ng: 26.1±3.2).
Cardiac NOX4 and VCAM-1 mRNA responses to i.c. TRH-analog RX77368 in Wistar and GK rats
In GK rats, basal cardiac mRNA levels of NOX4, an important source of reactive oxygen species production, were 30% higher than in Wistar rats (NOX4/GAPDH: 2.093±0.076 vs. 1.608±0.148, P<0.05), and VCAM-1, an indicator of vascular inflammation, was fourfold higher (VCAM-1/GAPDH: 1.445±0.160 vs. 0.354±0.034, P<0.05). Expression of these cardiac genes was not altered in Wistar rats following i.c. injection of RX77368 (10–60 ng) but was significantly elevated by three- to fourfold in GK rats, both compared with basal levels in GK rats and to Wistar controls post-injection (two-way analysis of variance, Figure 2).

Cardiac mRNA expression of NAPDH oxidase 4 (NOX4) and vascular cell adhesion molecule-1 (VCAM-1) in response to intracisternal (i.c.) thyrotropin-releasing hormone-analog RX77368 in Wistar and Goto–Kakizaki (GK) rats. Each column represents mean±s.e.m. of the number of rats indicated at the bottom of the column at 120 min after i.c. injection for surviving rats or at the time of death for GK rats. Two-way analysis of variance showed significant differences between the responses in GK rats and Wistar rats (P=0.001 for NOX4 and P=0.007 for VCAM-1). *P<0.05 compared with Wistar rats receiving same treatment. #P<0.05 compared with the basal levels of the group. GAPDH, glyceraldehydes-3-phosphate dehydrogenase.
Plasma H2O2, 8-isoprostane, cholesterol and MCP-1 responses to i.c. TRH-analog RX77368 in Wistar and GK rats
Basal plasma levels of H2O2 and 8-isoprostane, indicators of oxidative stress, MCP-1, an indicator of vascular inflammation, and cholesterol were not significantly different between Wistar and T2D GK rats. H2O2 and 8-isoprostane were significantly elevated in GK rats at 120 min after RX77368 (10–30 ng) i.c., compared with the unchanged responses in Wistar rats (two-way analysis of variance, Figures 3a,b). Plasma MCP-1 did not change in either Wistar or GK rats (Figure 3c). RX77368 i.c. at 20 and 30 ng significantly reduced cholesterol levels by 68% and 75%, respectively, in Wistar rats but not in GK rats (Figure 3d).

Plasma hydrogen peroxide (H2O2, a), 8-isoprostane (b), monocyte chemotactic protein-1 (MCP-1, c) and cholesterol (d) levels in response to intracisternal (i.c.) thyrotropin-releasing hormone-analog RX77368 in Wistar and Goto–Kakizaki (GK) rats. Each column represents mean±s.e.m. of the number of rats indicated at the bottom of the column at 120 min after the injection for surviving rats or at the time of death for GK rats. Two-way analysis of variance showed significant difference in the responses of H2O2 (P=0.004), 8-isoprostane (P=0.005) and cholesterol (P=0.001) between GK rats and Wistar rats. *P<0.05 compared with Wistar rats receiving same treatment. #P<0.05 compared with the basal levels of the group.
Sympathetic–adrenal system responses to i.c. TRH-analog RX77368 in Wistar and GK rats
GK rats had significantly higher basal levels of plasma epinephrine compared with Wistar rats (0.52±0.19 vs. 0.04±0.01 ng ml−1). Basal norepinephrine was similar (1.18±0.41 vs. 0.89±0.11 ng ml−1) in Wistar rats and GK rats. At 60 min after 25 ng RX77368 i.c. injection, epinephrine and norepinephrine levels were significantly increased in both strains, but the effect was much more pronounced in GK rats, showing about threefold higher (epinephrine:2.06±0.29 ng ml−1; norepinephrine: 8.18±0.95 ng ml−1) than in Wistar rats (epinephrine: 0.65±0.18 ng ml−1; norepinephrine: 2.74±0.24 ng ml−1), coincident with the changes in blood glucose levels.
Adrenal gland mRNA expression of TH, DβH and phenylethanolamine N-methyltransferase, which catalyze the first and rate-limiting step of catecholamine synthesis, dopamine conversion to norepinephrine, and norepinephrine conversion to epinephrine, respectively, were measured in rats 60 min after i.c. saline or 50 ng RX77368. In i.c. saline groups, adrenal TH mRNA levels in GK rats were more than double that in Wistar rats. RX77368 i.c. injection significantly enhanced adrenal TH mRNA expression in both strains. Although the relative increase was smaller in GK rats compared with that seen in Wistar rats due to the much higher basal levels in the former compared with the latter, the absolute levels of adrenal TH mRNA in GK rats post-injection was still 48% higher than that in Wistar controls (Figure 4). DβH mRNA levels were 37% higher after i.c. RX77368 in GK rats than they were following i.c. saline, resulting in an absolute level significantly higher, by 2.3-fold, than that seen following i.c. RX77368 in Wistar rats, whose DβH mRNA levels were unchanged by the treatment (Figure 4). Insignificant increases in adrenal phenylethanolamine N-methyltransferase mRNA levels were observed in both strains after i.c. RX77368 (Figure 4).

Adrenal gland mRNA expression of tyrosine hydroxylase (TH), dopamine β-hydroxylase (DβH) and phenylethanolamine N-methyltransferase (PNMT) in response to intracisternal (i.c.) injected saline or RX77368 (50 ng) at 60 min after the injection in Wistar and Goto–Kakizaki (GK) rats. Each column represents mean±s.e.m. of the number of rats indicated at the bottom of the column. *P<0.05 compared with the value of i.c. saline. #P<0.05 compared with Wistar rats receiving same treatment.
Effects of pretreatment with hexamethonium or phentolamine on systolic BP, HR, blood glucose and mortality in response to i.c. TRH-analog RX77368 in GK rats
Pretreatment of GK rats with the nicotinic receptor antagonist hexamethonium (10 mg kg−1, s.c., −30 min) followed by i.c. saline injection resulted in a significant decrease (∼40 mm Hg) in systolic BP that lasted for 90 min (Figure 5a), whereas HR remained unchanged (Figure 5b). RX77368 i.c. at 30 ng did not change systolic BP in hexamethonium-pretreated GK rats, in contrast to the decrease seen following i.c. saline, while slightly increased HR that reached statistical significance only at 120 min (Figure 5b). Following hexamethonium pretreatment, mortality was reduced to one in seven GK rats (14%), compared with five of five GK rats (100%) in the non-pretreatment group (Figure 5a). The hyperglycemic response of GK rats to 30 ng RX77368 i.c. was also significantly reduced by the hexamethonium pretreatment (Figure 5).

Effects of pretreatment of hexamethonium (10 mg kg−1, s.c., −30 min, a, b) or phentolamine (2 mg kg−1, i.p., −30 min, c, d) on intracisternal (i.c.) thyrotropin-releasing hormone-analog RX77368 (30 ng)-induced hypertension and tachycardia in type 2 diabetes Goto–Kakizaki (GK) rats. The values of control groups, shown in open circles in each pane, are the same data shown in Figure 1 representing the effects of i.c. RX77368 (30 ng) on blood pressure or heart rate of GK rats without pretreatment. Each point represents mean±s.e.m. of the number of rats indicated within the parentheses. The numerator in each fraction in the graphs of the left panel indicates the number of rats that died in the preceding interval, and the denominator indicates the total number of rats in the group. *P<0.05 compared with the value of i.c. RX77368 without pretreatment. #P<0.05 compared with the basal levels of the same group.
Pretreatment with the nonselective α-adrenergic antagonist phentolamine (2 mg kg−1, i.p., −30 min) in GK rats followed by i.c. saline resulted in a nonsignificant reduction in systolic BP at 30 min (Figure 5c). HR increased slightly but was not significantly different from basal levels (Figure 5d). Phentolamine pretreatment completely prevented the hypertensive effect of i.c. 30 ng RX77368 in GK rats, and significantly potentiated the tachycardic effect at 60 min (Figures 5c, d). The cardiac mortality induced by i.c. 30 ng RX7738 in GK rats dropped from 100% in non-pretreated animals to 50% in phentolamine pretreated animals, and the hyperglycemic response was completely abolished (Figure 5).
Effects of pretreatment with irbesartan on systolic BP, HR, blood glucose and mortality in response to i.c. TRH-analog RX77368 in GK rats
Pretreatment with irbesartan (50 mg kg−1 by gavage, −60 min), an angiotensin II type 1 receptor antagonist, prevented the hypertensive and tachycardic responses to 30 ng RX77368 i.c. in GK rats (Figure 6). Compared with the 100% mortality in non-pretreatment group, all irbesartan-pretreated rats survived at the end of the experiment. The hyperglycemic response to i.c. injected RX77368 was reduced (23.7±2.6 vs. 33.2±1.3 mmol l−1, P=0.05) compared with non-pretreated GK rats.

Effect of pretreatment with the angiotensin II type 1 receptor blocker irbesartan (50 mg kg−1 by gavage, −60 min) on intracisternal (i.c.) thyrotropin-releasing hormone-analog RX77368 (30 ng)-induced hypertension and tachycardia in type 2 diabetes Goto–Kakizaki (GK) rats (close circles). The effects of i.c. RX77368 (30 ng) alone without pretreatment on blood pressure or heart rate that have been shown in Figure 1 are presented as the line with open circles in each panel, to serve as controls. Each point represents mean±s.e.m. of the number of rats indicated within the parentheses in the left panel (in the case of no mortality) or the number of surviving rats in the group at the time point. The numerator in each fraction in the left panel indicates the number of rats that died in the preceding interval, and the denominator indicates the total number of rats in this group. *P<0.05 compared with the value of i.c. RX77368 alone without pretreatment. #P<0.05 compared with the basal levels of the same group.
Discussion
Cardiovascular function is tightly regulated by the brain via exquisitely precise reflexes, which ensure prompt adjustments in sympathetic and parasympathetic outflow to the heart and blood vessels. Previous findings show that TRH acts in specific pontine, medullary and spinal cord nuclei comprising baroreflex pathways to regulate sympathetic and vagal motor output.20, 22, 28, 29, 38, 39 The moderate cardiovascular changes evoked by i.c. TRH-analog in nondiabetic Wistar rats reflect normal autonomic reflex control with balanced sympathovagal efferent responses, which maintain cardiovascular functions within relatively normal ranges. By contrast, the extreme cardiovascular responses to i.c. injected TRH-analog in T2D GK rats clearly indicates an abnormally profound sympathetic overactivation. As indicated by the ameliorative effect of adrenergic antagonist pretreatment, the acute heart failure occurring in GK rats results from the combined effect of increased peripheral resistance, because of α-adrenergic-mediated vascular contraction, and increased cardiac activity, because of β-adrenergic-mediated excitation. The persistent increases in systolic BP and HR after i.c. TRH-analog in GK rats indicate that in addition to the sympathetic overactivation there was an impaired vagal counter regulation.
Sympathetic outflow to the cardiovascular system under strong baroreceptor control is regulated through a key site in the brainstem, the RVLM, including the C1 area, which contains a group of catecholamine-synthesizing neurons.40 Recently, we found that the RVLM is richly innervated with prepro-TRH-immunoreactive nerve terminals that form putative axodendritic and axosomatic interactions with TH-positive neurons.17 In addition, TRH-analog RX77368 microinjected into the C1 area of the RVLM induced remarkably potentiated hyperglycemic responses in GK rats, compared with Wistar rats.17 This latter observation suggests that overactivation of the catecholamine-synthesizing neurons in the RVLM may be involved in the mechanism of sympathetic hyperactivity in GK rats after i.c. TRH-analog injection, and possibly under basal conditions as well. Extensive studies have firmly established the physiological and pathophysiological roles of brainstem TRH in autonomic regulation.17, 20, 21, 22, 23, 24, 25, 28, 29 The present data indicate that these brainstem pathways are likely critically involved in the cardiovascular autonomic dysregulation in T2D. However, due to the potential spread of TRH-analog through the ventricular system following i.c. injection, it is possible that in addition to exerting brainstem pathways, other effects of the injection may have resulted via TRH action at more rostral sties, including hypothalamic sites.41 Further studies will be needed to define the role of particular nuclei in each aspect of the sympathetic hyperactivity that has been shown here to be involved in the cardiovascular mortality evoked by i.c. TRH-analog in GK rats, and to assess the role of endogenous brainstem TRH signaling in the persistent sympathetic hyperactivities and impaired vagal activation present in diabetes.
A curious observation of the present study was that GK rats in the BP measurement studies appeared more susceptible to developing heart failure in response to i.c. RX77368 (rats received 20–30 ng RX77368 i.c.) than were GK rats in the studies in which blood and tissues were harvested (rats received i.c. 25 or 50 ng RX77368). This is likely due to enhanced sympathetic reactivity to the higher ambient temperatures and the restraint required for cuff plethysmography in the BP measurement study.
Increased sympathetic activity is associated with multiple organs-mediated metabolic abnormalities in T2D.8, 9, 10 The role of brainstem TRH in disturbed autonomic balance has been previously evidenced in the impaired insulin response to hyperglycemia in GK rats.25 Peripheral inflammation, oxidative stress, dyslipidemia and altered renin–angiotensin system function have important roles in the pathology of obesity, T2D and hypertension, and are all associated with overactivation of adrenal-sympathetic functions.3, 42, 43, 44 These factors may also contribute to the fatal cardiovascular responses to i.c. injection of TRH-analog observed here in GK rats. Basal heart mRNA levels of NOX4 and VCAM-1, indicators of vascular inflammation, were significantly higher in GK rats than in Wistar rats. TRH-analog i.c. injection in GK rats further significantly elevated cardiac mRNA levels of NOX4 and VCAM-1, and in addition, increased plasma levels of H2O2 and 8-isoprostane. Plasma cholesterol was decreased by i.c. TRH-analog in Wistar rats but remained high in GK rats. These findings suggest that i.c. TRH-analog-induced lethal cardiovascular responses in T2D GK rats is associated with an acutely elevated sympathetic-adrenal activation superimposed over a prevailing state of peripheral inflammation, oxidative stress and dyslipidemia, pathological changes related to chronic sympathetic overactivation. Because GAPDH is involved in glycolysis and its gene expression in certain tissues may change in diabetic conditions,45, 46 further studies using other housekeeping genes, such as β-actin, will be needed to further validate the present data. It is well established that abnormal angiotensin II function is involved in cardiovascular dysregulation, including angiotensin II actions in the forebrain and brainstem to activate sympathetic premotor neurons.3 Pretreatment with the angiotensin type 1 receptor blocker irbesartan prevented the cardiac mortality in GK rats, suggesting an important mediating role of angiotensin system in the cardiovascular effects of i.c. TRH-analog. Taken together, the present data suggest that sympathetic overactivation by brainstem TRH, resulting in sympathovagal imbalance in T2D, may have profound chronic effects on systemic oxidative stress and inflammation, which contribute to the cardiac morbidity in T2D. Acute effects of brainstem TRH-triggered sympathetic hyperactivation may interact with existing cardiovascular pathology in T2D, leading to nonischemic heart failure and cardiac mortality.
T2D is the world's fastest growing human disease with high morbidity and mortality rates, predominantly as a result of heart failure. Animal models provide the possibility to investigate the dysfunctional brainstem circuits involved in the mechanisms contributing to diabetic heart failure. GK rats have been reported to have hypertension and accelerated progression of heart failure following myocardial infarction.47, 48 Left ventricular hypertrophy with enhanced activity of the myocardial Na/H exchanger, increased myofilament sensitivity to calcium concentration, and impaired energy metabolism resulting from cardiac mitochondrial alterations, in conjunction with insulin resistance, endothelial dysfunction, and perivascular inflammation, have all been found to contribute to the cardiovascular morbidity in GK rats.48, 49, 50, 51 The absence of age-dependent reductions in HR variability suggested an autonomic dysfunction in GK rats.52 The present study investigated brainstem mechanism of cardiovascular mortality in T2D and focuses on TRH, an critical signaling molecule involved in the control of sympathovagal coordination and the focus of our research for many years.17, 23, 25 The initial dose of TRH-analog we investigated was set at 60 ng, which is substantially lower than the safe and non-life-threatening dose of 100 ng used in nondiabetic rats in previous studies.29, 53 The 60 ng dose did not cause mortality or even significant change in HR and blood glucose in Wistar rats in the present study. Thereafter, the dose was decreased repeatedly when we encountered the lethal effects in GK rats, until we reached a dose (10 ng) that did not induce heart failure. Our findings provide evidence that dysfunction of neurocircuitry responsive to a brainstem neuropeptide can cause fatal cardiovascular outcome and establish a reliable animal model for investigating central mechanisms of cardiovascular dysfunction in T2D, especially those responsible for acute heart failure and sudden death. That high incidence of cardiovascular mortality in T2D GK rats following i.c. TRH-analog suggests a cardinal role of brainstem TRH in chronic and acute autonomic imbalance that is involved in the high cardiovascular mortality in T2D, albeit this is still to be confirmed in more diabetic and/or obese animal models. Correction of sympathetic hyperaction and avoiding stressful acute sympathetic activation, such as hypoglycemia and hyperthermia, should be an important strategy for the prevention of cardiovascular death in T2D.
References
Kannel WB, McGee DL . Diabetes and cardiovascular disease. The Framingham study. JAMA 1979; 241: 2035–2038.
Lockhart CJ, Hamilton PK, McVeigh KA, McVeigh GE . A cardiologist view of vascular disease in diabetes. Diabetes Obes Metab 2008; 10: 279–292.
Malpas SC . Sympathetic nervous system overactivity and its role in the development of cardiovascular disease. Physiol Rev 2010; 90: 513–557.
Palatini P, Julius S . The role of cardiac autonomic function in hypertension and cardiovascular disease. Curr Hypertens Rep 2009; 11: 199–205.
Taegtmeyer H, Golfman L, Sharma S, Razeghi P, van AM . Linking gene expression to function: metabolic flexibility in the normal and diseased heart. Ann NY Acad Sci 2004; 1015: 202–213.
Boudina S, Abel ED . Diabetic cardiomyopathy revisited. Circulation 2007; 115: 3213–3223.
Guha A, Harmancey R, Taegtmeyer H . Nonischemic heart failure in diabetes mellitus. Curr Opin Cardiol 2008; 23: 241–248.
Urbancic-Rovan V, Meglic B, Stefanovska A, Bernjak A, Azman-Juvan K, Kocijancic A . Incipient cardiovascular autonomic imbalance revealed by wavelet analysis of heart rate variability in type 2 diabetic patients. Diabet Med 2007; 24: 18–26.
Aso Y, Wakabayashi S, Nakano T, Yamamoto R, Takebayashi K, Inukai T . High serum high-sensitivity C-reactive protein concentrations are associated with relative cardiac sympathetic overactivity during the early morning period in type 2 diabetic patients with metabolic syndrome. Metabolism 2006; 55: 1014–1021.
Perin PC, Maule S, Quadri R . Sympathetic nervous system, diabetes, and hypertension. Clin Exp Hypertens 2001; 23: 45–55.
Dublin S, Glazer NL, Smith NL, Psaty BM, Lumley T, Wiggins KL, Page RL, Heckbert SR . Diabetes mellitus, glycemic control, and risk of atrial fibrillation. J Gen Intern Med 2010; 25: 853–858.
Esler M . Sympathetic nervous activation in essential hypertension: commonly neglected as a therapeutic target, usually ignored as a drug side effect. Hypertension 2010; 55: 1090–1091.
Voulgari C, Papadogiannis D, Tentolouris N . Diabetic cardiomyopathy: from the pathophysiology of the cardiac myocytes to current diagnosis and management strategies. Vasc Health Risk Manag 2010; 6: 883–903.
Lynn RB, Kreider MS, Miselis RR . Thyrotropin-releasing hormone-immunoreactive projections to the dorsal motor nucleus and the nucleus of the solitary tract of the rat. J Comp Neurol 1991; 311: 271–288.
Sasek CA, Wessendorf MW, Helke CJ . Evidence for co-existence of thyrotropin-releasing hormone, substance P and serotonin in ventral medullary neurons that project to the intermediolateral cell column in the rat. Neuroscience 1990; 35: 105–119.
Rinaman L, Miselis RR, Kreider MS . Ultrastructural localization of thyrotropin-releasing hormone immunoreactivity in the dorsal vagal complex in rat. Neurosci Lett 1989; 104: 7–12.
Ao Y, Ko M, Chen A, Marvizon JC, Adelson D, Song MK, Go VL, Liu YY, Yang H . Potent hyperglycemic and hyperinsulinemic effects of thyrotropin-releasing hormone microinjected into the rostroventrolateral medulla and abnormal responses in type 2 diabetic rats. Neuroscience 2010; 169: 706–719.
Underwood MD, Iadecola C, Reis DJ . Lesions of the rostral ventrolateral medulla reduce the cerebrovascular response to hypoxia. Brain Res 1994; 635: 217–223.
Ross CA, Ruggiero DA, Park DH, Joh TH, Sved AF, Fernandez-Pardal J, Saavedra JM, Reis DJ . Tonic vasomotor control by the rostral ventrolateral medulla: effect of electrical or chemical stimulation of the area containing C1 adrenaline neurons on arterial pressure, heart rate, and plasma catecholamines and vasopressin. J Neurosci 1984; 4: 474–494.
Paakkari I, Siren AL, Nurminen ML, Svartstrom-Fraser M . Injection of thyrotropin releasing hormone into the locus coeruleus increases blood pressure. Eur Heart J 1987; 8 (Suppl B): 147–151.
Koskinen LO . Effects of TRH on cerebral and peripheral blood flows; role of submesencephalic brain stem centres. Acta Physiol Scand 1986; 128: 277–288.
Yan J, Chen H, Liao W, Lu S . Cardiovascular effects of thyrotropin-releasing hormone in normotensive and hypotensive rats: role of rostral ventrolateral medulla. Circ Shock 1992; 37: 253–261.
Taché Y, Yang H . Role of medullary TRH in the vagal regulation of gastric function. In: Wingate DL, Butkd TF (eds) Innervation of the Gut: Pathophysiological Implications. CRC: Boca Raton, 1994, pp 67–80.
Yang H, Tache Y, Ohning G, Go VL . Activation of raphe pallidus neurons increases insulin through medullary thyrotropin-releasing hormone (TRH)-vagal pathways. Pancreas 2002; 25: 301–307.
Ao Y, Toy N, Song MK, Go VL, Yang H . Altered glucose and insulin responses to brain medullary thyrotropin-releasing hormone (TRH)-induced autonomic activation in type 2 diabetic Goto-Kakizaki rats. Endocrinology 2005; 146: 5425–5432.
Assadian H, Ishikawa Y, Shimatsu A, Tanoh T, Imura H . Serotoninergic denervation suppresses the sympathetic outflow induced by thyrotropin-releasing hormone in conscious rats. J Auton Nerv Syst 1991; 35: 193–198.
Ishikawa Y, Shimatsu A, Kato Y, Murakami Y, Imura H . Effects of intrathecal administration of thyrotropin releasing hormone and its analogue, DN1417, on plasma glucose and catecholamine levels in conscious rats. Brain Res 1990; 514: 1–4.
Ishikawa T, Yang H, Taché Y . Medullary sites of action of the TRH analogue, RX 77 368, for stimulation of gastric acid secretion in the rat. Gastroenterology 1988; 95: 1470–1476.
Stephens RL, Ishikawa T, Weiner H, Novin D, Taché Y . TRH analogue, RX 77 368, injected into dorsal vagal complex stimulates gastric secretion in rats. Am J Physiol 1988; 254: G639–G643.
Ao Y, Go VL, Toy N, Li T, Wang Y, Song MK, Reeve JRJ, Liu Y, Yang H . Brainstem thyrotropin-releasing hormone regulates food intake through vagal-dependent cholinergic stimulation of ghrelin secretion. Endocrinology 2006; 147: 6004–6010.
Yang H, Wu SV, Ishikawa T, Taché Y . Cold exposure elevates thyrotropin-releasing hormone gene expression in medullary raphe nuclei: relationship with vagally mediated gastric erosions. Neuroscience 1994; 61: 655–663.
Yang H, Yuan P, Wu V, Tache Y . Feedback regulation of thyrotropin-releasing hormone gene expression by thyroid hormone in the caudal raphe nuclei in rats. Endocrinology 1999; 140: 43–49.
Niida H, Takeuchi K, Okabe S . Role of thyrotropin-releasing hormone in acid secretory response induced by lowering of body temperature in the rat. Eur J Pharmacol 1991; 198: 137–142.
Yuan PQ, Yang H . Hypothyroidism increases Fos immunoreactivity in cholinergic neurons of brain medullary dorsal vagal complex in rats. Am J Physiol Endocrinol Metab 2005; 289: E892–E899.
Goto Y, Suzuki K, Sasaki M, Ono TS . GK rat as a model of nonobese, noninsulin-dependent diabetes. Selective breeding over 35 generations. In: Renold AE, Renold (eds) Frontiers in Diabetes Research. Lessons from Animal Diabetes II. John Libbey: London, UK, 1988, pp 301–303.
Porte DJ, Kahn SE . beta-cell dysfunction and failure in type 2 diabetes: potential mechanisms. Diabetes 2001; 50 (Suppl 1): S160–S163.
Wallis RH, Collins SC, Kaisaki PJ, Argoud K, Wilder SP, Wallace KJ, Ria M, Ktorza A, Rorsman P, Bihoreau MT, Gauguier D . Pathophysiological, genetic and gene expression features of a novel rodent model of the cardio-metabolic syndrome. PLoS One 2008; 3: e2962.
Riedel W, Burke SL . Selective autonomic nervous control of thyroid hormone and calcitonin secretion during metabolic and cardiorespiratory activation by intracisternal thyrotropin-releasing hormone (TRH). J Auton Nerv Syst 1988; 24: 157–173.
Carter DA, Lightman SL . Cardio-respiratory actions of substance P, TRH and 5-HT in the nucleus tractus solitarius of rats: evidence for functional interactions of neuropeptides and amine neurotransmitters. Neuropeptides 1985; 6: 425–436.
Brown DL, Guyenet PG . Electrophysiological study of cardiovascular neurons in the rostral ventrolateral medulla in rats. Circ Res 1985; 56: 359–369.
Badoer E . Role of the hypothalamic PVN in the regulation of renal sympathetic nerve activity and blood flow during hyperthermia and in heart failure. Am J Physiol Renal Physiol 2010; 298: F839–F846.
Johnson JD, Campisi J, Sharkey CM, Kennedy SL, Nickerson M, Greenwood BN, Fleshner M . Catecholamines mediate stress-induced increases in peripheral and central inflammatory cytokines. Neuroscience 1910; 135: 1295–1307.
Pauletto P, Scannapieco G, Pessina AC . Sympathetic drive and vascular damage in hypertension and atherosclerosis. Hypertension 1991; 17: III75–III81.
Nash DT . Alpha-adrenergic blockers: mechanism of action, blood pressure control, and effects of lipoprotein metabolism. Clin Cardiol 1990; 13: 764–772.
Nishikawa T, Araki E . Impact of mitochondrial ROS production in the pathogenesis of diabetes mellitus and its complications. Antioxid Redox Signal 2007; 9: 343–353.
Seo E, Park EJ, Park MK, Kim DK, Lee HJ, Hong SH . Differential expression of metabolism-related genes in liver of diabetic obese rats. Korean J Physiol Pharmacol 2010; 14: 99–103.
Chandler MP, Morgan EE, McElfresh TA, Kung TA, Rennison JH, Hoit BD, Young ME . Heart failure progression is accelerated following myocardial infarction in type 2 diabetic rats. Am J Physiol Heart Circ Physiol 2007; 293: H1609–H1616.
Cheng ZJ, Vaskonen T, Tikkanen I, Nurminen K, Ruskoaho H, Vapaatalo H, Muller D, Park JK, Luft FC, Mervaala EM . Endothelial dysfunction and salt-sensitive hypertension in spontaneously diabetic Goto-Kakizaki rats. Hypertension 2001; 37: 433–439.
Darmellah A, Baetz D, Prunier F, Tamareille S, Rucker-Martin C, Feuvray D . Enhanced activity of the myocardial Na+/H+ exchanger contributes to left ventricular hypertrophy in the Goto-Kakizaki rat model of type 2 diabetes: critical role of Akt. Diabetologia 2007; 50: 1335–1344.
Desrois M, Clarke K, Lan C, Dalmasso C, Cole M, Portha B, Cozzone PJ, Bernard M . Upregulation of eNOS and unchanged energy metabolism in increased susceptibility of the aging type 2 diabetic GK rat heart to ischemic injury. Am J Physiol Heart Circ Physiol 2010; 299: H1679–H1686.
Howarth FC, Qureshi MA . Myofilament sensitivity to Ca2+ in ventricular myocytes from the Goto-Kakizaki diabetic rat. Mol Cell Biochem 2008; 315: 69–74.
Howarth FC, Jacobson M, Shafiullah M, Adeghate E . Long-term effects of type 2 diabetes mellitus on heart rhythm in the Goto-Kakizaki rat. Exp Physiol 2008; 93: 362–369.
Stephens RL, Tache Y . Intracisternal injection of a TRH analogue stimulates gastric luminal serotonin release in rats. Am J Physiol 1989; 256: G377–G383.
Acknowledgements
This work was supported by Department of Veterans Affairs Merit Award (H Yang), National Institute of Diabetes and Digestive and Kidney Diseases Grant DK-41301 (Center for Ulcer Research and Education Center grant Animal Core) and American Heart Association (M Tuck). We thank Dr Ke-Wei Zhao for his contribution on the manuscript preparation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Yang, H., Nyby, M., Ao, Y. et al. Role of brainstem thyrotropin-releasing hormone-triggered sympathetic overactivation in cardiovascular mortality in type 2 diabetic Goto–Kakizaki rats. Hypertens Res 35, 157–165 (2012). https://doi.org/10.1038/hr.2011.154
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hr.2011.154
Keywords
This article is cited by
-
Voltage dependence of the Ca2+ transient in endocardial and epicardial myocytes from the left ventricle of Goto–Kakizaki type 2 diabetic rats
Molecular and Cellular Biochemistry (2018)
