
Abstract
‘Living fossil’ taxa, by definition, have no close relatives, and therefore no outgroup to provide a root to phylogenetic trees. We identify and use a molecular outgroup in the sole extant lineage of sphenodontid reptiles, which separated from other reptiles 230 million years ago. We isolated and sequenced a partial nuclear copy of the mitochondrial cytochrome b gene. We confirm the copy is indeed not mitochondrial, is older than all extant mitochondrial copies in Sphenodon (tuatara), and is therefore useful as a molecular outgroup. Under phylogenetic analysis, the nuclear copy places the root of the tuatara mitochondrial gene tree between the northern and the southern (Cook Strait) groups of islands of New Zealand that are the last refugia for Sphenodon. This analysis supports a previous mid-point rooted mitochondrial gene tree. The mitochondrial DNA tree conflicts with allozyme analyses which place a Cook Strait population equidistant to all northern and other Cook Strait populations. This population on North Brother Island is the only natural population of extant S. guntheri; thus, we suggest that the current species designations of tuatara require further investigation.
Similar content being viewed by others


Introduction
The outgroup is a requisite part of molecular phylogenetic studies, as it provides a root for phylogenetic trees to determine the polarity of evolution among the taxa of interest. However, this can present a problem for phylogenetically isolated taxa which have no close relatives that can be used as outgroups. Studies of such taxa result in unrooted trees. This is especially a problem for the so-called ‘living fossil’ taxa that are virtually monotypic. In these cases, a solution may be to use a molecular outgroup rather than an organismal outgroup. Nuclear copies of mitochondrial genes (numt, Lopez et al, 1994) can act as outgroups if they are shown to have been transposed to the nucleus before the origin of the common ancestor of all extant mitochondrial copies. Conversely, the mitochondrial copies can act as outgroups to the nuclear copies if it can be established that there was a single transposition event and subsequent evolution occurred independently within the nucleus. Hence, one data set may yield two independent gene trees, one mitochondrial and one nuclear.
Many numts have been reported in the last 20 years (eg, reviews Zhang and Hewitt, 1996; Sorenson and Quinn, 1998; Bensasson et al, 2001 and references therein; Vartanian and Wain-Hobson, 2002; Hazkani-Covo et al, 2003), but usually as problems to beware of in studies of mitochondrial genes. We are aware of only three papers in which they have been used as phylogenetic outgroups, two for geese (Quinn, 1992; Ruokonen et al, 2000), and one for humans (Zischler et al, 1995). We are not aware of the previous use of numts as outgroups for species for which there is no possible organismal outgroup because of their remote phylogenetic status.
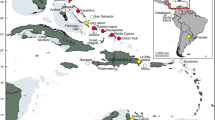
Sphenodon (tuatara) is an extreme case of phylogenetic isolation. Sphenodontian reptiles separated from the Order Squamata (lizards and snakes) in the early-middle Triassic, and were globally distributed in the Jurassic and Cretaceous (Benton, 1993; 2000). Other than Sphenodon, the most recent sphenodontid fossil is ∼115 million years old (Benton, 1993). A remnant of this once widespread Order Sphenodontia survived on the New Zealand landmass that separated from Gondwana 82 million years ago. Late Pleistocene tuatara fossil remains are found throughout the two main islands of current day New Zealand (Worthy and Holdaway, 2002, pp 459–461), but human-introduced predators and habitat modification reduced the extant populations to ∼30 offshore islands in 12 island groups (Figure 1). Currently, two species, Sphenodon punctatus and Sphenodon guntheri, are recognised (Daugherty et al, 1990). The latter is known naturally only from North Brother Island. Apparently conflicting data sets on the phylogenetic position of S. guntheri (Figure 2) require rooted trees to determine if the differences in the genetic phylogenies are real or merely branch length differences to North Brother Island tuatara (Hay et al, 2003). Allozymes clearly differentiate S. guntheri from all other populations (Daugherty et al, 1990), whereas mitochondrial DNA (mtDNA) sequence groups S. guntheri among other nearby islands in Cook Strait (Hay et al, 2003). Both data sets recognise a northern–southern (Cook Strait) islands split (Figure 2).

Distribution of Sphenodon in New Zealand. ○ are past and subfossil records (from Crook, 1975). Solid symbols are natural extant populations: • are northern S. punctatus, ▾ are western Cook Strait S. punctatus, ▪ is S. guntheri. 1 is the Poor Knights Islands (Tawhiti Rahi, Aorangi, Aorangaia, Stack B), 2 is the Hen and Chickens Islands (Hen, Lady Alice, Whatupuke, Coppermine), 3 is Little Barrier Island, 4 is Cuvier Island, 5 is the Mercury Islands (Stanley, Red, Middle and Green), 6 is the Aldermen Islands, (Ruamahua-iti, Ruamahua-nui, Hongiora, Hernia), 7–9 are the Bay of Plenty Islands, 7 is Karewa Island, 8 is Motunau Island, 9 is Moutoki Island, 10 is Stephens Island, 11 is the Trios Islands (Middle, North, South), 12 is North Brother Island.

Unrooted phylograms of tuatara gene trees based on (a) allozyme data, and (b) mtDNA sequence data, from Hay et al (2003).
In the course of sequencing mtDNA for their phylogenetic study, Hay et al (2003) were unable to obtain clean sequence for cytochrome b (CYTB). In the present study, we demonstrate that there are two copies of CYTB, and that one copy has been inserted into the nuclear genome. We use that nuclear copy of the mitochondrial CYTB (nCYTB) as an outgroup to provide a root for the mtDNA CYTB (mtCYTB) tree. We had intended to do the converse, to use the mtCYTB genes as an outgroup to the nCYTB gene tree, but this was not possible due to lack of variation in the nuclear copy. As far as we can find, this is the first published record of a nuclear mitochondrial pseudogene (numt) in a diapsid reptile, although one has been recorded in an anapsid reptile, a turtle (Stuart and Parham, 2004).
Materials and methods
Blood sampling of tuatara from all 12 island groups was described in Hay et al (2003). Samples are listed in Appendix A. Genomic DNA (whole purified DNA containing nuclear and mitochondrial genomes) was extracted from erythrocytes stored at −80°C according to the protocol of Millar et al (1992).
The entire mitochondrial genome of a Stephens Island Sphenodon punctatus was amplified with the TaKaRa LA PCR kit (Takara Shuzo Co., Ltd). The kit protocol was followed with the 2.5 mM MgCl2 buffer, adding an extra annealing step for the tuatara-specific control region primers PETL and CSB3H (Hay et al, 2003) under the following cycling conditions: 94°C for 5 min; (98°C for 20 s, 64°C for 30 s, 68°C for 15 min) × 15 cycles; (98°C for 20 s, 64°C for 30 s, 68°C for 15 min+15 s per cycle) × 15 cycles on a Perkin-Elmer 9600 thermalcycler.
Subsequent work revealed two control regions in tuatara mtDNA (Rest et al, 2003). In case the primers in the above amplicon were located in two different control regions, the entire tuatara mitochondrial genome was reamplified, this time in two overlapping segments using new primers (Table 1) designed to the tuatara sequence (then unpublished sequence courtesy of P Waddell, J Ast and D Mindell, see Rest et al, 2003). These long-range PCRs of ∼11 (ND4L-ND2H) and ∼8 (ND1L-ND6H) kilobases, respectively, were accomplished using Roche Expand Long Template PCR System according to manufacturer's instructions with buffer system I, and the following cycling regime: 93°C for 2 min; (93°C for 30 s, 55°C for 30 s, 68°C for 9 min) × 10 cycles; (93°C for 30 s, 55°C for 30 s, 68°C for 9 min+20 s per cycle) × 25 cycles; 68°C for 7 min on a Bio-Rad iCycler thermalcycler.
The long-range mitochondrial amplicons were used as templates for the amplification of 307 base pairs (bp) of cytochrome b sequence using standard CYTB primers (Kocher et al, 1989; Hedges et al, 1992). This clean sequence was compared to CYTB sequence previously obtained from genomic DNA from the same animal, and which included double bands at some positions. The nonmatching base pairs in the positions containing double bands were inferred to be the nonmitochondrial DNA sequence. From these two sequences, primers specific to the mitochondrial and nonmitochondrial copies, respectively, were designed (Table 1, TcbL/H mt/nc). The mtCYTB primers yielded 169 bp of clean sequence from both genomic DNA and from the ND4-ND2 and the entire mitochondrial amplicons; the sequence matched the mtCYTB sequence obtained above. The mtCYTB primers did not amplify from the ND1-ND6 mtDNA amplicon. The nCYTB primers amplified 169 bp clean sequence from genomic DNA only, and not from any of the mitochondrial genome amplicons; the sequence matched the inferred nCYTB sequence obtained above. These sets of amplifications confirm that the nCYTB copy is not in the mitochondrial genome.
To enable robust phylogenetic analyses, we increased the size of the nCYTB fragment by using bubble PCR (Munroe et al, 1994) to amplify some of the unknown flanking region of the nuclear sequence, according to the protocol and linker sequences of Vázquez et al (1999). Freshly extracted tuatara DNA was digested with NdeII. The complementary bubble linkers were annealed to each other, and then ligated to the digested DNA. For the bubble PCR, we used a standard touchdown PCR protocol from 65 to 60°C annealing temperature decreasing 0.5°C per cycle, with 1.5 mM MgCl2, 1 mg/ml BSA and 1 M betaine (N, N, N-trimethylglycine), using one primer matching one bubble linker (M13Rev) and the other primer matching the known nCYTB sequence. This bubble PCR reaction used primers TcbLnc and M13Rev; the PCR product was reamplified in a semi-nested PCR with primers TinvLnc and M13Rev (Table 1). This gave a PCR fragment of 260 bp, within which H primers specific to tuatara mtCYTB and nCYTB were designed (Table 1, TcbH2). In conjunction with the TcbL primers, these TcbH2 primers yield 445 bp of sequence. There are sufficient informatively variable sites in this fragment to provide a conclusive root for the mtCYTB gene tree. DNA of one to two tuatara from each of the 12 island groups (see Appendix A and Figure 1) was sequenced for both mtCYTB and nCYTB on both L and H strands, on Applied BioSystems ABI 377 or ABI 3730 automated sequencers (Genbank Accession Numbers AY426627-AY426670).
Sequences were aligned with Sequencher ver. 4.1(Gene Codes Corporation) with base calling and alignment accuracy checked by eye. Mitochondrial sequences are read on the L strand. Where sequences were identical (eg, both individuals from an island), only one was used for analysis. Sequences of representative squamates were aligned with tuatara CYTB and analysed to ensure that the nonmitochondrial sequence was indeed tuatara and not contamination (Eumeces obsolete GenBank Accession # AB016606, Iguana iguana # NC002793, Lacerta vivipara # U69834 and Anguis fragilis # AY099996). MEGA2 (Kumar et al, 2001) was used to construct minimum evolution (ME) trees using Kimura-2 distances. The inferred translated amino-acid data set was also analysed (using p-distances), but gave less resolution than nucleotide analyses, and results are not shown. ME tree topologies were tested with 2000 replications for both bootstrap confidence levels (BCL; Felsenstein, 1985), and confidence probability tests of interior branches (CP; Rzhetsky and Nei, 1992; Nei and Kumar, 2000). PAUP*4.0b10 (Swofford, 2002) was used to construct maximum parsimony (MP) and maximum likelihood (ML) trees. The parsimony topology was found using a full heuristic search and tested with 1000 bootstrap replications. For the ML analyses, a fast-heuristic search was conducted on a reduced data set containing only two nCYTB sequences and tested with × 100 bootstrap replications. An HKY85+G+I with empirically estimated values of the gamma parameter, invariable sites and base frequencies was used. MP and ML analyses yielded the same essential topologies as the ME trees, and so are not shown separately. However, their BCL values above 50% are put on the ME tree in Figure 3 where relevant.

Minimum evolution (ME) tree of 445 bp tuatara mtCYTB and nCYTB, and mtCYTB of representative squamates, nucleotide Kimura-2 distances with MEGA complete deletion option. Only values above 50% shown on nodes: first number is the ME CP value (× 2000 replications), where relevant the second number is the ME BCL value (× 2000), third is the maximum parsimony BCL value (× 1000), fourth is the maximum likelihood BCL (× 100). N=northern islands, CS=Cook Strait islands, including Brothers tuatara, mt=mitochondrial, nc=nuclear, *=only two samples for ncCYTB for ML tree BCL value, × 2 on population names indicates identical sequences from two individuals.
Results and Discussion
Tuatara cytochrome b numt
Alignment of the mitochondrial and nonmitochondrial copies of CYTB in tuatara is unambiguous with only one indel in one population (see Poor Knights nCYTB below). Contamination is rejected as an explanation for the nonmitochondrial cytochrome b sequence for a number of reasons. First, the nCYTB sequence is not close to any likely contaminant based on (a) a BLAST search of the National Center for Biotechnology Information databases, and (b) the phylogenetic analyses of tuatara nCYTB and mtCYTB and representative squamate CYTB sequences (Figure 3), which grouped the mtCYTB and nCYTB tuatara sequences together at 99% confidence probability and 96–100% bootstrap confidence levels. Second, genetic distances between tuatara nCYTB and mtCYTB (0.21–0.22) were much higher than those among tuatara mtCYTB (0–0.03), but lower than those between any tuatara CYTB and all snakes and lizards tested (0.27–0.45). Third, the original ‘double sequence’ obtained using universal CYTB primers was found in all populations, using samples collected at different times by different people under different field conditions, and in independent DNA extraction, PCR and sequencing procedures. In contrast to CYTB, clean sequences of mitochondrial control region and ND1, and nuclear aldolase intron were obtained from the same DNA extractions (Hay et al, 2003).
We confirmed that the second copy of tuatara CYTB is nuclear and not mitochondrial gene duplication or heteroplasmy, as all nuclear-specific primers amplified genomic DNA only and did not amplify any of the three separate long amplifications of mtDNA. Rest et al (2003) have confirmed that there is a single copy only of cytochrome b in the tuatara mitochondrial genome. In addition, one would expect a higher percentage of guanine (G) in nuclear than in L-strand mitochondrial sequences (Saccone et al, 1999). We see this in the putative tuatara numt, which has an average of 17.5% G compared to 14.7% in tuatara mtCYTB. More tellingly, at third codon positions which are under reduced selective constraint because fewer mutations at those sites alter the amino acid, nCYTB has 10.8% G compared to 2.8% in mtCYTB. The nCYTB gives one stop codon when translated with the vertebrate mitochondrial genetic code and four stop codons with the standard nuclear genetic code. All lines of evidence indicate that the nCYTB is a nonfunctional nuclear pseudogene.
There are no transversions among nuclear sequences in this data set. On average, there are 3.6 to 1 transitions to transversions (R) between the mtCYTB and nCYTB tuatara sequences compared with an average R of 6.9 among mtCYTB sequences, matching expectations of a lower R between mitochondrial and nuclear sequences than among mitochondrial DNA sequences (Saccone et al, 1999). Between the mitochondrial and nuclear sequences, there are 0.57 synonymous substitutions/synonymous site and 0.05 nonsynonymous substitutions/nonsynonymous site (based on the vertebrate mitochondrial genetic code), and an average distance (Kimura 2-parameter) of 0.219. Mitochondrial gene transposition to the nuclear genome renders numts nonfunctional pseudogenes (Fukuda et al, 1985) and results in all sites in the pseudogene becoming freed from selection constraints and equally subject to mutation. However, the rate of substitution is much slower in nuclear pseudogenes than in mitochondrial genes due to the superior proofreading activity of the nuclear DNA polymerase (Brown et al, 1979; Kunkel and Loeb, 1981) and the many fewer cycles of replication in chromosomes compared to mitochondrial genomes. Therefore, we suggest that many of the differences between the tuatara numt and mtCYTB occurred under selective constraint in the mitochondrial genome prior to transposition, as calculated by Sunnucks and Hales (1996) in aphids. The numt may be derived from a now extinct mitochondrial lineage, whereby the associated numt persisted in the separate chromosomal DNA lineage (Sunnucks and Hales, 1996).
There is only minor heteromorphic variation in the nCYTB sequence among populations. The Poor Knights tuatara has a single substitution at site 295 and a deletion heteromorphy at sites 137–138 (Figure 4). Similarly, both individuals sequenced from each of the Hen and Chickens, Cuvier and Mercury populations (island groups 2, 4 and 5 in Figure 1) contain the same set of transition double bases at four sites in the numt (sites 20, 22, 172 and 259, Figure 4). When these heterozygous sites are coded as distinct character states, the nCYTB sequences of Mercury, Cuvier and Hen and Chickens tuatara group together at 93% BCL under parsimony analysis (JMH and Tim White, Massey University, unpublished data). We infer that this set of substitutions is a synapomorphy. We cannot tell whether these substitutions were present but lost in the geographically intervening population of Little Barrier (island 3 in Figure 1), or whether that population was independently derived. Nor do we know whether the two numt copies represent allelic heterozygosity or a duplication of the numt in the nuclear genome. Duplications of numts are quite common (eg, Fukuda et al, 1985; Sunnucks and Hales, 1996; Mirol et al, 2000; Bensasson et al, 2001 and references therein), although most authors have not distinguished between gene duplication and allelic variation.

Variable sites in the 445 bp of Cytochrome b in tuatara.
The high sequence conservation of the numt among all tuatara populations suggests that either the nuclear pseudogene is located in a highly conserved region of the nuclear genome, or the extant populations are so recently related that there has been insufficient time for variation to occur in chromosomal DNA. We favour the latter explanation because there was no variation in aldolase intron sequences from all island groups either (Hay et al, 2003).
Phylogenetic analyses
All tuatara cytochrome b sequences group together at 99% CP and 89–100% BCL (Figure 3). The genetic divergence is much higher between tuatara nCYTB and mtCYTB (D=0.21–0.22) than among mtCYTB sequence of all populations (0–0.03), indicating that the branching of the nuclear copy is older than the common ancestor of all extant mitochondrial copies, and so appears to be an ideal outgroup. It has been noted that numts may have significantly longer or shorter branch lengths than their mitochondrial homologues, which may affect phylogenetic reconstruction (Lopez et al, 1997; Zhang and Hewitt, 1997). The internal branch leading to the tuatara numt sequences looks much longer than that leading to the mtCYTB sequences. However, this is a tree-construction artefact, as the branch length differences lessen when Jukes–Cantor distances or the neighbour-joining method is used, and lessen more in MP and ML trees. All these methods give the same overall tree topology. For all the above reasons, nCYTB is an appropriate outgroup with which to root a tuatara mitochondrial gene tree. Other squamates, separated by 230 million years of evolution, are too distant to act as reliable outgroups to the mtCYTB of tuatara populations; the nCYTB sequences break the long branch between the tuatara and squamates (Figure 3). The short-terminal branches in the tuatara mtCYTB are likely indicative of a recent species bottleneck that lost most mtDNA genetic variation in Sphenodon, as discussed in Hay et al (2003).
All the tree-building methods used here place the root of the mitochondrial DNA tree between northern and Cook Strait populations (except the ML tree, which did not resolve tuatara branching patterns), with the Cook Strait populations grouping together at 93% CP and 85–97% BCL, and the northern populations clustering at 82% CP and 74–76% BCL (Figure 3). This northern–Cook Strait islands split is the best supported node in the mitochondrial part of the tree.
The present analysis with a suitable outgroup supports the previous mtDNA estimate with mid-point rooting (Hay et al, 2003): that for the mitochondrial genome, Cook Strait Sphenodon punctatus group more closely with S. guntheri (also Cook Strait) than with northern S. punctatus. This topology is concordant with geographic distribution, but is discordant with allozyme genetic diversity. The discrepancy between the two molecular approaches (Figure 2) requires resolution if the current taxonomic division of tuatara into two species is to be confirmed. For example, the allozyme and mtDNA phylogenies and the current division into S. guntheri (Brothers Island) and S. punctatus (all other populations) could be reconciled satisfactorily if it were demonstrated that there had been Pleistocene or recent introgression of mtDNA into the Brother's tuatara population through admixture of populations during glacial cycles, as was observed in Drosophila simulans (Ballard, 2000). The Cook Strait islands were last joined to the mainland 8–12 000 years ago, but there were up to 50 glacial cycles of joining and separating land masses as sea levels fell and rose again (Worthy and Holdaway, 2002). Alternatively, it may be that an anomaly of historical partitioning of allozyme alleles from other Cook Strait, northern or extinct mainland populations combined with genetic drift and other population processes has resulted in a phylogenetic signal that has led to an incorrect division into two species. Resolving the organismal history will require independent nuclear DNA information, which is being gathered (Aitken et al, 2001; Hay unpublished data). We are also examining genetic variation of mtDNA from subfossil tuatara bones from now extinct mainland populations, to test whether former genetic variation was continuous or disjunct, and to see whether Brothers tuatara are allied more closely with an extinct population than with any of the remnant extant populations. The present study demonstrates the difficulties of elucidating the organismal history of a once widespread taxon from the fragmented remnant populations. This problem is common in New Zealand and other archipelagos where terrestrial species evolved in an environment free of mammalian predators and competitors, and whose post-human colonisation distribution is greatly reduced and fragmented.
References
Aitken N, Hay JM, Sarre SD, Lambert DM, Daugherty CH (2001). Microsatellite markers for tuatara (Sphenodon spp.). Conserv Genet 2: 183–185.
Ballard JWO (2000). When one is not enough: introgression of mitochondrial DNA in Drosophila. Mol Biol Evol 17: 1126–1130.
Bensasson D, Zhang D-X, Hartl DL, Hewitt GM (2001). Mitochondrial pseudogenes: evolution's misplaced witnesses. Trends Ecol Evol 16: 314–321.
Benton MJ (1993). Reptilia. In: Benton MJ (ed) The Fossil Record 2, Chapman & Hall: London. pp 681–715.
Benton MJ (2000). Vertebrate Palaeontology, 2nd edn. Blackwell Science: Oxford.
Brown WM, George Jr M, Wilson AC (1979). Rapid evolution of animal mitochondrial DNA. Proc Natl Acad Sci USA 76: 1967–1971.
Crook IG (1975). The tuatara. In: Kuschel G (ed) Biogeography and Ecology in New Zealand, Junk: The Hague. pp 331–352.
Daugherty C, Cree A, Hay JM, Thompson MB (1990). Neglected taxonomy and continuing extinctions of tuatara (Sphenodon). Nature 347: 177–179.
Felsenstein J (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39: 783–791.
Fukuda M, Wakasugi S, Tsuzuki T, Nomiyama H, Shimada K, Miyata T (1985). Mitochondrial DNA-like sequences in the human nuclear genome: characterization and implications in the evolution of mitochondrial DNA. J Mol Biol 186: 257–266.
Hay JM, Daugherty CH, Cree A, Maxson LR (2003). Low genetic divergence obscures phylogeny among populations of Sphenodon, remnant of an ancient reptile lineage. Mol Phylog Evol 29: 1–19.
Hazkani-Covo E, Sorek R, Graur D (2003). Evolutionary dynamics of large numts in the human genome: rarity of independent insertions and abundance of post-insertion duplications. J Mol Evol 56: 169–174.
Hedges SB, Bogart JP, Maxson LR (1992). Ancestry of unisexual salamanders. Nature 356: 708–710.
Kocher TD, Thomas WK, Meyer A, Edwards SV, Pääbo S, Villablanca FX et al (1989). Dynamics of mitochondrial DNA evolution in animals: amplification and sequencing with conserved primers. Proc Natl Acad Sci USA 86: 6196–6200.
Kumar S, Koichiro T, Jakobsen, Nei M (2001). MEGA2: molecular evolutionary genetics analysis software. Bioinformatics 17: 1244–1245.
Kunkel TA, Loeb LA (1981). Fidelity of mammalian DNA polymerases. Science 213: 765–767.
Lopez JV, Stephens JC, O'Brien SJ (1997). The long and short of nuclear mitochondrial DNA (Numt) lineages. Trends Ecol Evol 12: 114.
Lopez JV, Yuhki N, Masuda R, Modi W, O'Brien SJ (1994). Numt, a recent transfer and tandem amplification of mitochondrial DNA to the nuclear genome of the domestic cat. J Mol Evol 39: 174–190.
Millar CD, Lambert DM, Bellamy AR, Stapleton PM, Young EC (1992). Sex-specific restriction fragments and sex ratios revealed by DNA fingerprinting in the brown skua. J Hered 83: 350–355.
Mirol PM, Mascheretti S, Searle JB (2000). Multiple nuclear pseudogenes of mitochondrial cytochrome b in Ctenomys (Caviomorpha, Rodentia) with either great similarity to or high divergence from the true mitochondrial sequence. Heredity 84: 538–547.
Munroe DM, Haas M, Bric E, Whitton T, Aburatani H, Hunter K et al (1994). IRE-bubble PCR: a rapid method for efficient and reproductive amplification of human genomic DNA sequences and complex sources. Genomics 19: 506–514.
Nei M, Kumar S (2000). Molecular Evolution and Phylogenetics, Oxford University Press: New York.
Quinn TW (1992). The genetic legacy of Mother Goose: phylogeographic patterns of lesser snow goose Chen caerulescens caerulescens maternal lineages. Mol Ecol 1: 105–117.
Rest JS, Ast JC, Austin CC, Waddell PJ, Tibbetts EA, Hay JM et al (2003). Molecular systematics of primary reptilian lineages and the tuatara mitochondrial genome. Mol Phylog Evol 29: 289–297.
Ruokonen M, Kvist L, Lumme J (2000). Close relatedness between mitochondrial DNA from seven Anser goose species. J Evol Biol 13: 532–540.
Rzhetsky A, Nei M (1992). A simple method for estimating and testing minimum-evolution trees. Mol Biol Evol 9: 945–967.
Saccone C, de Giorgi C, Gissa C, Pesole G, Reyes A (1999). Evolutionary genomics in Metazoa: the mitochondrial DNA as a model system. Gene 238: 195–209.
Sorenson MD, Quinn TW (1998). Numts: a challenge for avian systematics and population biology. The Auk 115: 214–221.
Stuart BL, Parham JF (2004). Molecular phylogeny of the critically endangered Indochinese box turtle (Cuora galbinifrons). Mol Phylog Evol 31: 164–177.
Sunnucks P, Hales DF (1996). Numerous transposed sequences of mitochondrial cytochrome oxidase I–II in aphids of the genus Sitobion (Hemiptera: Aphididae). Mol Biol Evol 13: 510–524.
Swofford DL (2002). PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods) Version 4, Sinauer Associates: Sunderland, MA.
Vartanian J-P, Wain-Hobson S (2002). Analysis of a library of macaque nuclear mitochondrial sequences confirms macaque origin of divergent sequences from old oral polio vaccine samples. Proc Natl Acad Sci USA 99: 7566–7569.
Vázquez M, Lorenzi H, Schijman AG, Ben-Dov C, Levin MJ (1999). Analysis of the distribution of SIRE in the nuclear genome of Trypanosoma cruzi. Gene 239: 207–216.
Worthy TH, Holdaway RN (2002). The Lost World of the Moa: Prehistoric Life of New Zealand, Canterbury University Press: Christchurch.
Zhang D-X, Hewitt GM (1996). Nuclear integrations: challenges for mitochondrial DNA markers. Trends Ecol Evol 11: 247–251.
Zhang D-X, Hewitt GM (1997). Reply from D-X Zhang and GM Hewitt. Trends Ecol Evol 12: 114.
Zischler H, Geisert H, von Haeseler A, Pääbo S (1995). A nuclear ‘fossil’ of the mitochondrial D-loop and the origin of modern humans. Nature 378: 489–492.
Acknowledgements
Blood sampling of tuatara was conducted with the permission and assistance of the New Zealand Department of Conservation. We thank Gwilym Haynes for increasing the sample size of sequences, Leon Huynen for introducing JMH to bubble PCR and providing the linkers, and Tim White for devising methods of coding heterozygous sites for phylogenetic analysis. JMH and all laboratory work were funded by a New Zealand Foundation for Research, Science and Technology Postdoctoral Fellowship.
Author information
Authors and Affiliations
Corresponding author
Appendix A
Appendix A
All tuatara samples used in this study are from the National Frozen Tissue Collection (NFTC), collected and held by CHD and are shown in Table A1.
Rights and permissions
About this article
Cite this article
Hay, J., Sarre, S. & Daugherty, C. Nuclear mitochondrial pseudogenes as molecular outgroups for phylogenetically isolated taxa: a case study in Sphenodon. Heredity 93, 468–475 (2004). https://doi.org/10.1038/sj.hdy.6800525
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6800525
Keywords
This article is cited by
-
First Report of a Mitochondrial Pseudogene in Agnathan Vertebrates (Cyclostomata: Petromyzontidae)
Journal of Molecular Evolution (2018)
-
Genetic diversity and taxonomy: a reassessment of species designation in tuatara (Sphenodon: Reptilia)
Conservation Genetics (2010)
-
Primate numts and reticulate evolution of capped and golden leaf monkeys (Primates: Colobinae)
Journal of Biosciences (2008)
