
Abstract
The genetic population structure of the Cranberry Fritillary Boloria aquilonaris was studied using both RAPDs (random amplified polymorphic DNA) and allozymes. In Belgium, B. aquilonaris has a naturally fragmented distribution that has been accentuated due to human activity during the last century. The genetic population structure of this butterfly was analysed at the regional (several Ardenne uplands) and at the landscape level (several populations within an Ardenne upland). Both population genetic markers confirmed results from a previous CMR study at the landscape scale. At the regional scale however, important incongruences were observed between RAPDs and allozymes. The average gene diversity for the RAPD data was twice that of the allozyme data. The degree of population subdivision was also much greater for RAPDs than for allozymes. The UPGMA clusters produced by each of these markers differed significantly. We believe that, given the higher rate of mutation of RAPDs and the greater number of loci assayed by this method, RAPDs reveal a more accurate and recent population genetic structure than allozymes.
Similar content being viewed by others
Introduction
Loss of natural habitat due to human activities is widely recognised as the most important factor causing species decline world-wide (eg, Sih et al, 2000). At the landscape scale, natural habitats are more and more fragmented. During the fragmentation process, distance between populations increases, progressively hampering the arrival of immigrants from surrounding populations until complete isolation is reached. Extinctions of small, isolated local populations living in such fragmented landscapes occur frequently because of stochastic demographic processes or disturbances (Fahrig and Merriam, 1994). Conservation of threatened species living in fragmented landscapes therefore requires the establishment of networks of suitable habitat patches. Migration of individuals between patches within such networks is a key factor in species persistence, increasing the size of local populations (rescue effect: Brown and Kodric-Brown, 1977), allowing the re-colonization of empty patches after local extinctions, or insuring the maintenance of genetic diversity between local populations. However, migration events are generally uncommon and therefore difficult to quantify by direct observations in the field. The studies of genetic distances between populations helps detect effective network structure via evidence of gene flow or isolation (ie, by revealing the presence or absence of immigrants). In this study, we use this approach to investigate the distribution of a butterfly species, the Cranberry Fritillary Boloria aquilonaris.
Butterflies have turned out to be excellent model organisms for the studies of small populations: in European fragmented landscapes, many species are located in small, discrete patches of suitable habitat connected by individual movements across the landscape matrix (Petit et al, 2001 and references therein). We focus here on a butterfly with a fragmented distribution at two hierarchical levels. Western European B. aquilonaris populations are located in wet acid peat bogs where their principal larval host plant, Vaccinium oxycoccos, is abundant. Because of the high water requirements of bogs, large areas of suitable habitats are restricted to uplands where climatic conditions are cold and wet. Besides this first level of fragmentation at the regional scale, it is worth noting that, because of the particular edaphic conditions required for the installation of bogs, their distribution on each upland is naturally fragmented. At this landscape level, fragmentation has been accentuated by human activity in Belgium. Over the past century, drainage and afforestation of peat bogs by Norway Spruce Picea abies has not only resulted in the substantial decline of the species (104 recorded populations before 1970, only 25 in 1999), but has also increased the isolation of the remnant populations.
In 1995–1996, we carried out a capture-release-recapture (CMR) study in four sites on one of the four uplands where populations of this butterfly are still present in Belgium. We demonstrated that, at a landscape scale, B. aquilonaris populations form a single metapopulation (Mousson et al, 1999). But such CMR studies are very time consuming and limited to small geographic scales. For this reason, we investigate here the population structure of this butterfly at the regional scale, ie, the four Ardennes uplands, using genetic markers. In this paper, we assess gene diversity, genetic structuring, population differentiation and the association between geographical and genetic distance using both allozymes and random amplified polymorphic DNA (RAPD) markers (Williams et al, 1990). Factors responsible for the incongruence between allozymes and RAPD data are also discussed.
Materials and methods
Genetic analyses
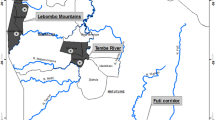
Allozyme and DNA samples were collected from 110 individuals from eight populations in the Belgian Ardennes in 1996 (Figure 1). Samples were collected at two different scales. At the landscape scale, four populations were sampled on the Plateau des Tailles (separated by 2–8 km). At the regional scale, populations were sampled on different Ardenne uplands; ie, Plateau des Tailles, Plateau des Hautes Fagnes, Plateau de St. Hubert and Plateau de Recogne (separated by 11–86 km).

Sampling sites of Boloria aquilonaris with the respective upland on which they are found; PT = Plateau des Tailles, PR = Plateau de Recogne, PSH = Plateau de St. Hubert, PHF = Plateau des Hautes Fagnes. Sampling size is mentioned between parentheses. Shaded areas represent uplands.
Allozymes:
Allozyme analysis was carried out as in Vandewoestijne et al (1999). Only four out of 17 enzymes systems proved to be polymorphic. The four loci studied were: (i) glucose-phosphate isomerase (GPI, EC 5.3.1.9.); (ii) 6-phosphogluconate dehydrogenase (6PGD, EC1 1.1.44); (iii) adenlyte kinase (AK, EC 2.7.4.3); and (iv) malate dehydrogenase (MDH, EC 1.1.1.37).
RAPD:
Genomic DNA was extracted by phenol-chloroform-isoamyl alcohol. Three to four legs of each individual were homogenised in 300 μL of extraction buffer (50mM Tris HCl pH 8.0, 25 mM NaCl, 25 mM EDTA pH 8.0, 0.1% SDS) and one unit of proteinase K. After at least 3 h of incubation at 60°C, 300 μL of phenol/ chloroform/isoamyl alcohol were added and mixed vigorously. Samples were then spun at 2178 g for 5 min. Once the supernatant decanted, 20 μL of 5 mM NaCl and 700 μL of ice cold 100% ethanol were added. Samples were mixed gently by inverting the tubes several times and placed in a freezer for at least 20 min. After a 5-min spin at 2178 g, ethanol was poured off and 200 μL of room temperature 70% ethanol was added. Samples were spun again for 5 min and once the ethanol poured off, dried and resuspended in 50 μL 1*TE (10 mM Tris- HCl pH 8.0, 1 mM EDTA disodium salt.).
The RAPD technique is sensitive to (a) shape of the temperature profile, (b) type of polymerase, and (c) Mg2+, Taq and DNA concentration (Hoelzel and Green 1998). Extracted DNA of all samples was quantified using a fluorometer (TKO100 Dedicated Mini Fluorometer, Hoefer Scientific Instruments), and diluted to obtain identical DNA concentrations. Reactions were standardized and all PCR reactions were run on the same thermal cycler (Perkin-Elmer 9600). For every 25 μL reaction, 20 ng of DNA, 0.4 μM of primer, 3 mM of MgCl2, 200 μM of dNTP and 0.5 U of AmpliTaq polymerase (Perkin-Elmer) were included. The following parameters were used for the amplifications: 4 min denaturation at 94°C, followed by 35 cycles of 1 min at 94°C, annealing at 36°C for 1 min, and elongation at 72°C for 2 min. An additional 5 min period for elongation at 72°C followed this cycle. Negative controls were used to verify for contamination. PCR products were separated on 1.6% agarose gels that were run for 225 min at 100V in 0.5 M TBE buffer. Several DNA size standards were run on every gel to aid in identifying the target bands. Ethidium bromide staining was used to visualize band patterns that were recorded on disc using GelDoc (BIO-RAD).
Four RAPD primers (OPB08, 5′AGGGAACGAG 3′; OPB17, 5′GTCCACACGT 3′; A10, 5′GTGATCGCAG 3′ and A02, 5′TGCCGAGCTG 3′) out of 60 screened were selected based on the polymorphism and reproducibility of the bands they generated. Amplifying the same samples on different days tested reproducibility.
Statistical analyses
Allozyme markers:
Compliance to Hardy-Weinberg equilibrium and linkage equilibrium were tested using GENEPOP (Raymond and Rousset, 1995). Gene diversity and genetic structuring (GST) were calculated with the assistance of GEN-SURVEY (Vekemans and Lefèbvre, 1997). Using TFPGA 1.3 (Miller, 1997), an exact test of population differentiation was carried out with a Markov Chain Monte Carlo approach for an approximation of the exact probability of the observed differences in allele frequencies. Additionally, Fisher’s combined probability test (Fisher, 1954; Sokal and Rohlf, 1995) was employed as a global test over loci to determine the overall significance. A Mantel test was used to assess the association between Nei’s unbiased (1978) genetic distance matrix and the geographical distance matrix using GEN-SURVEY. We produced an UPGMA (ie, unweighted pair-group method using an arithmetic average) tree, based on Nei’s unbiased (1978) genetic distance corrected for small sample size, to further examine the relationship between geographic and genetic distances (TFPGA 1.3).
RAPD markers:
Allele frequencies were calculated from null homozygote frequencies assuming panmixia and corrected for dominance according to Lynch and Milligan (1994) using TFPGA 1.3. Using these allele frequencies, gene diversity, expected heterozygosity (calculated from the null allele frequency and based on Hardy-Weinberg equilibrium), genetic structuring (FST) and a Mantel test were calculated using the same program. We also used genotype data directly to obtain the average gene diversity over all loci (the mean number of differences between all pairs of genotypes divided by the number of loci) and derive FST from the variance components (AMOVA) using ARLEQUIN (Schneider et al, 2000). Pairwise comparisons between populations were carried out with the same program as for the allozyme data. The correlation between Nei’s unbiased (1978) genetic distance and the geographic distance separating populations was assessed with a Mantel test using TFPGA 1.3. An UPGMA cluster analysis was performed using unbiased allele frequencies with TFPGA 1.3.
Results
Genetic analyses
Allozymes:
All sampled populations, with the exception of Logbiermé, were in Hardy-Weinberg equilibrium. No linkage disequilibrium was detected for each locus pair across all populations.
On average, 50% (±13%) of all loci were polymorphic using the 95% criterion. The mean number of alleles per polymorphic locus was 2.31 ± 0.46. The mean expected heterozygosity, or gene diversity, was 0.188 ± 0.029.
The average proportion of the genetic variation found between populations was 10.49% (GST = 0.105; 95% CI = 0.023, 0.210). Significant genetic differentiation was observed for 16 out of the 28 pairs of populations, both within and between different uplands (Table 1).
Genetic distance was positively but not significantly associated with geographic distance (r = 0.414, P = 0.092). The UPGMA dendrogram (Figure 2) grouped the Plateau des Tailles, the Plateau de Recogne and the Plateau St Hubert together. External to this cluster was the population of Eichenbusch, sampled on the Plateau des Hautes Fagnes.

UPGMA dendrograms based on Nei's (1978) genetic distance (a) allozyme-based, (b) RAPD-based.
RAPD:
Primers OPB08, OPB17, A10, A02 produced 10, 4, 7, and 7 reproducible bands respectively. Out of these, 18 were polymorphic. For pooled data, all of these loci exhibited overall frequencies of band presence of less than 1 − 3/N (where N is the number of individuals sampled), reducing potential bias in the allele frequency estimates stemming from low counts of band absence phenotype (Lynch and Milligan, 1994). The results of analyses based on biased and unbiased allele frequencies (Lynch and Milligan, 1994) were very similar, therefore, we only report results based on the latter.
Every sample was characterised by a different RAPD genotype. The average gene diversity over all RAPD loci was 0.402 ± 0.214, indicating that about 40% of the loci differ between all pairs of RAPD genotypes. The expected heterozygosity was 0.405, more than twice that observed for the allozyme data.
FST values of 0.163 and 0.179, based on unbiased allele frequency data and on genotype data respectively, were very similar. The degree of population subdivision indicated by these values, is much greater than that based on the allozyme data. Pairwise comparisons revealed significant genetic differentiation for 15 out of 28 pairs (Table 1). These differences were observed for both populations pairs within and between uplands but not for the same pairs of populations as those based on allozyme data.
The Mantel test showed that the matrices representing genetic and geographic distances are significantly correlated (r = 0.666, P < 0.01). The UPGMA clustering grouped together the Tailles and Hautes Fagnes uplands (Figure 2). Populations sampled on the Plateau de St Hubert and Plateau de Recogne were the most differentiated. The uplands clustered identically, even when populations with small sample sizes were eliminated from the analyses or when different distance measures (Nei, 1972; 1978; Reynolds et al, 1983) were used. The RAPD based cluster differs significantly from the UPGMA cluster based on allozymes.
Discussion
Using the same samples, important differences were observed in the population structure of B. aquilonaris between the allozyme and the RAPD data. With the RAPD markers, populations of B. aquilonaris were characterized by greater genetic diversity than with allozymes. This is consistent with the majority of previous studies comparing these two population genetic markers (Table 2), though there are exceptions to this trend (Liu and Furnier, 1993, LeCorre et al, 1997; Ross et al, 1999). Mostly, this difference is explained by the inherently higher rate of detectable mutations and weaker degree of selective constraint at RAPD compared to allozyme loci since RAPDs can detect variation in both coding and non-coding regions (Szmidt et al, 1996; Aagaard et al, 1998; Waycott, 1998; Sun et al, 1999; Wu et al, 1999; Oiki et al, 2001).
The sampled populations were more subdivided when taking RAPD data (FST = 0.163) into consideration rather than allozyme data (GST = 0.105). Isolation by distance is a more important and significant factor in explaining the genetic differentiation between populations for the RAPD data than it is for the allozyme data (r = 0.666 vs r = 0.414). The greater amount of subdivision and a higher coefficient of correlation between the geographic and genetic distances observed for RAPDs compared to allozymes might be explained by the higher mutation rate of RAPDs. Mutation rates for RAPDs have been shown to lie somewhere in between allozymes (10−6 to 10−9 per locus per generation) and microsatellites (10−3 to 10−4 per locus per generation, Lougheed et al, 2000). A higher mutation rate, generating greater genetic diversity, might make RAPDs more sensitive than allozymes to diminished gene flow and greater genetic drift. Both of these processes have gained importance due to fragmentation and loss of favourable habitat of B. aquilonaris during the last century. Studies by Mamuris et al (1999) and Wu et al (1999), comparing allozyme and RAPD markers have also noted a higher degree of population differentiation and a more pronounced effect of isolation by distance although many other studies do not follow this trend (Table 2). The ability of the RAPD method to screen a larger part of the genome (Mamuris et al, 1999), differences in selection regimes (Oiki et al, 2001) and that allozymes often fail to detect many kinds of nucleotide differences (Wu et al, 1999) are given as possible explanations that could account for the differences observed between these two markers.
UPGMA clustering produced completely different results depending on the molecular marker used. The RAPD-based UPGMA tree seems to reflect more truly the present spatial configuration of favourable habitat patches for this species than does the allozyme based UPGMA tree. In fact, several small favourable habitat patches still exist in valleys between the Plateau des Tailles and the Plateau des Hautes Fagnes, permitting ‘stepping stone’ movements between these two uplands. For about a century, habitat patches are no longer available between the Plateau des Tailles and the Plateau de Recogne and the Plateau de St Hubert, isolating the latter two uplands from the Plateau des Tailles. This distribution of favourable habitat could explain the cluster including the Plateau des Tailles and the Plateau des Hautes Fagnes, from which the Plateau de Recogne and the Plateau de St Hubert are isolated (Figure 2). On the other hand, the allozyme based UPGMA tree may reflect a historical situation. At the end of the 19th century, a large number of potential habitat patches for B. aquilonaris were available between the Plateau des Tailles and the uplands of Recogne and St Hubert. The availability of potential habitat between the Plateau des Tailles and the Plateau des Hautes Fagnes, on the contrary, has not changed substantially since the end of the 19th century. This could explain the grouping of the Plateau des Tailles, the Plateau de Recogne and the Plateau de St Hubert, while the population sampled at the Plateau des Hautes Fagnes is isolated from this cluster (Figure 2). Under the reasonable assumption that RAPDs evolve faster than allozymes, we propose that our allozyme data reflects the population structure of B. aquilonaris that was present more than a century ago, while the RAPD data depicts a more recent population structure of this butterfly. Nève et al (2000) also found that their neighbour-joining dendrogram (based on allozyme data) did not fit the population structure observed in the field (based on MRR data). The authors concluded that since the connectivity of the network of suitable habitats had only significantly weakened since the 1950s, subregional differentiation revealed by allozymes had not yet occurred.
Other studies comparing allozyme and RAPD markers (Table 2) obtained varying results. LeCorre et al (1997, Mamuris et al (1999, Sun et al (1999) and Cole and Kuchenreuther (2001) found that clusters obtained with these markers differed. The larger number of loci resolved by RAPDs compared to allozymes, the selective neutrality of RAPDs and their ability to screen a larger part of the genome were cited as possible factors for the incongruences observed. Ayres and Ryan (1999) and Sun et al (1999) found that the dendrograms generated by combining the different markers reflected the geographical defined populations better than dendrograms based on only one type of marker. Many other studies demonstrated congruence between allozyme and RAPD based clusters (see Table 2).
Differences in gene diversity, population differentiation and population clusters obtained for allozymes and RAPDs have been explained by the following factors: (a) the dominance of RAPDs results in incomplete genotypic information enhancing the sampling variance and inducing bias in parameter estimation (Lynch and Milligan, 1994, b) different part of the genome are sampled by the different markers, (c) the markers are subject to different selection regimes, (d) the inability of allozymes to detect many kinds of genetic variation because only nucleotide changes that lead to electrophoretically detectable changes are observed, and (e) rates of mutation are higher at RAPD loci than at allozyme loci.
In our study, care was taken to obtain the least biased population structure estimators for the RAPD data by including only those markers that were reproducible and clearly polymorphic, by using an asymptotically unbiased estimator to obtain gene and genotype frequencies and by restricting analyses to markers that were not too common (Lynch and Milligan, 1994). Furthermore, FST values calculated directly from RAPD phenotypes and indirectly from RAPD allele frequencies were almost identical.
Since allozymes are different allelic forms of nuclear-encoded enzymes while RAPDs are random amplified fragments of both coding and non-coding region of the genome, these markers could be subject to different selection regimes and different part of the genome are sampled by each marker. Although, in this study populations were in Hardy-Weinberg equilibrium, selection, except at a very strong intensity, cannot be ruled out as a factor responsible for the incongruencies observed between allozyme and RAPD data.
We believe that, in this study, the inability of allozymes to reveal all genetic variation and the ability of RAPDs to generate a greater number of loci are partially responsible for the important differences observed between these two markers. Both of these factors will result in a higher genetic diversity for RAPDs than for allozymes. Genetic drift, a process that has been accentuated in the last century for populations of B. aquilonaris in Belgium, will be more easily detected in the more variable marker, ie RAPDs. This could then explain the greater degree of genetic differentiation and isolation by distance observed between populations with the RAPD method. We also believe that, given the higher rate of mutation in RAPDs, this marker represents a more recent and accurate ‘picture’ of the population structure than allozymes.
Conclusions
The genetic population structure of B. aquilonaris was studied at two different scales using both RAPDs and allozymes. At the landscape level, both markers confirm the previous CMR study, ie populations within an upland form a metapopulation. In constrast, at the regional scale, these markers produced very different population structures. We do not believe that the incongruence between RAPDs and allozymes can be explained by factors such as the dominant nature of RAPDs, different selection regimes acting on the different population genetic marker, or the sampling of different parts of the genome with different markers due to the general congruence found in the majority of studies comparing allozymes and RAPDs. We rather think that, in this study, allozymes and RAPDs represent the population genetic structure of this butterfly at different time scales. The inherently higher mutation rate and the greater amount of genetic diversity displayed by RAPDs make this a marker that reveals a more ‘recent’ population genetic structure than do allozymes. The greater and more significant correlation between genetic and geographic distances obtained with RAPDs compared to allozymes confirms this hypothesis, knowing that the already naturally fragmented populations of B. aquilonaris have been subject to increased isolation during the last century. Another observation that may confirm this hypothesis is the genetic population structure represented in the clusters based on Nei’s genetic distance. The RAPDs based clusters correspond to the availability of favourable habitat observed now, while the allozyme-based clusters correspond to habitat availability at the end of the 19th century.
References
Aagard, JE, Krutovskii, KV, Strauss, SN (1998). RAPDs and allozymes exhibit similar levels of diversity and differentiation among populations and races of Douglas fir. Heredity, 81: 69–78.
Apostol, BL, Black, WC, Reiter, P, Miller, BR (1996). Population genetics with RAPD-PCR markers: the breeding structure of Aedes aegypti in Puerto Rico. Heredity, 76: 325–334.
Ayres, DR, Ryan, FJ (1999). Genetic diversity and structure of the narrow endemic Wyethia reticulata and its congener W. bolanderi (Asteraceae) using RAPD and allozyme techniwues. Am J Botany, 86: 344–353.
Bartish, IV, Garkava, LP, Rumpunen, K, Nybom, H (2000). Phylogenetic relationships and differentiation among and within populations of Chaenomeles Lindl. (Rosaceae) estimated with RAPDs and isozymes. Theoret Applied Genet, 101: 554–563.
Baruffi, L, Damiani, G, Guglielmino, CR, Bandi, C, Malacrida, AR, Gasperi, G (1994). Polymorphism within and between populations of Ceratitis capitata: comparison between RAPD and multilocus enzyme electrophoresis data. Heredity, 74: 425–437.
Brown, JA, Kodric-Brown, A (1977). Turnover rates in insular biogeography: effect of immigration on extinction. Ecology, 58: 445–449.
Cole, CT, Kuchenreuther, MA (2001). Molecular markers reveal little genetic differentiation among Aconitum noveboracense and A. columbionum (Ranunculaceae) populations. Am J Botany, 88: 337–347.
De Wolf, H, Backeljau, T, Verhagen, R (1998). Congruence between allozyme and RAPD data in assessing macrogeographical genetic variation in the periwinkle Littorina striata (Mollusca, Gastropoda). Heredity, 81: 486–492.
Fahrig, E, Merriam, G (1994). Conservation of fragmented populations. Conserv Biol, 8: 50–59.
Fisher, RA (1954). Statistical Methods for Research Workers, 12th edn. Oliver and Boyd: Edinburgh.
Haig, SM, Rhymer, JM, Heckel, DG (1994). Population differentiation in randomly amplified polymorphic DNA of red-cockaded woodpeckers Picoides borealis. Mol Ecol, 3: 581–595.
Hoelzel, AR, Green, A (1998). PCR protocols and population analysis by direct DNA sequencing and PCR-based DNA fingerprinting. In: Hoelzel AR (ed) Molecular Genetic Analysis of Populations. Oxford University Press: Oxford. Pp 230–231.
Isabel, N, Beaulieu, J, Bousquet, J (1995). Complete congruence between gene diversity estimates derived from genotypic data at enzyme and random amplified polymorphic DNA loci in black spruce. Proc Nat Acad Sci USA, 92: 6369–6373.
Jenczewski, E, Prosperi, JM, Ronfort, J (1999). Differentiation between natural and cultivated populations of Medicago sativa (Leguminosae) from Spain: analysis with random amplified polymorphic DNA (RAPD) markers and comparison to allozymes. Mol Ecol, 8: 1317–1330.
Lecorre, V, Dumolinlapegue, S, Kremer, A (1997). Genetic variation at allozyme and RAPD loci in sessile oak Quercus petraea (Matt) Liebl: The role of history and geography. Mol Ecol, 6: 519–529.
Liu, Z, Furnier, GR (1993). Comparison of allozyme, RFLP, and RAPD markers for revealing genetic variation within and between trembling aspen and bigtooth aspen. Theoret Applied Genet, 87: 97–105.
Lougheed, SC, Gibbs, HL, Prior, KA, Weatherhead, PJ (2000). A comparison of RAPD versus microsatellite DNA markers in population studies of the Massasauga rattlesnake. J Hered, 91: 458–463.
Lynch, M, Milligan, BG (1994). Analysis of population genetic structure with RAPD markers. Mol Ecol, 3: 91–99.
Mamuris, Z, Stamatis, C, Triantaphyllidis, C (1999). Intraspecific genetic variation of striped red mullet (Mullus surmuletus L.) in the Mediterranean Sea assessed by allozyme and random amplified polymorphic DNA (RAPD) analysis. Heredity, 83: 30–38.
Miller, MP (1997). Tools for Population Genetic Analysis. Version 1.3. Department of Biological Sciences, Northern Arizona University, Flagstaff.
Mousson, L, Nève, G, Baguette, M (1999). Metapopulation structure and conservation of the cranberry fritillary Boloria aquilonaris (lepidoptera, nymphalidae) in Belgium. Biol Conserv, 87: 285–293.
Nei, M (1972). Genetic distance between populations. Am Nat, 106: 283–293.
Nei, M (1978). Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics, 89: 583–590.
Nève, G, Barascud, B, Descimon, H, Baguette, M (2000). Genetic structure of Proclossiana eunomia populations at the regional scale (Lepidoptera, Nymphalidae). Heredity, 84: 657–666.
Oiki, S, Kawahara, T, Inoue, K, Ohara, M, Maki, M (2001). Random amplified polymorphic DNA (RAPD) variation among populations of the insular endemic plant Campanula microdonta (Campanulaceae). Annals of Botany, 87: 661–667.
Peakall, R, Smouse, PE, Huff, DR (1995). Evolutionary implications of allozyme and RAPD variation in diploid populations of dioecious buffalograss Buchloe sactyloides. Mol Ecol, 4: 135–147.
Petit, S, Moilanen, A, Hanski, I, Baguette, M (2001). Metapopulation dynamics of the bog fritillary butterfly: movements between habitat patches. Oikos, 92: 491–500.
Puterka, GJ, Black, WC, Steiner, WM, Burton, RL (1993). Genetic variation and phylogenetic relationships among world-wide collections of the Russian wheat aphid, Diuraphis noxia (Mordvilko), inferred from allozyme and RAPD-PCR markers. Heredity, 70: 604–618.
Raymond, M, Rousset, F (1995). GENEPOP (Version 1.2), A population genetics software for exact tests and eucumenicism. J Hered, 86: 45–249.
Reynolds, J, Weir, BS, Cockerham, CC (1983). Estimation of the coancestry coefficient: basis for a short-term genetic distance. Genetics, 105: 767–779.
Ross, KG, Shoemaker, DD, Krieger, MJB, Deheer, CJ, Keller, L (1999). Assessing genetic structure with multiple classes of molecular markers: a case study involving the introduced fire ant Solenopsis invicta. Mol Biol Evol, 16: 525–543.
Schneider, S, Roessli, D, Excoffier, L (2000). Arlequin ver. 2.000: A software for population genetics data analysis. Genetics and Biometry Laboratory, University of Geneva, Switzerland.
Sih, A, Jonsson, BG, Luikart, G (2000). Habitat loss: ecological, evolutionary and genetic consequences. Trends Ecol Evol, 15: 132–134.
Sokal, R, Rohlf, FJ (1995). Biometry. W.H. Freeman: New York.
Sun, G -L, Dpaz, O, Salomon, B, von Bothmer, R (1999). Genetic diversity in Elymus caninus as revealed by isozyme, RAPD, and microsatellite markers. Genome, 42: 420–431.
Szmidt, AE, Wang, X-E, Meng-Zhu, L (1996). Empirical assessment of allozyme and RAPD variation in Pinus sylvestris (L.) using haploid tissue analysis. Heredity, 76: 412–420.
Vandewoestijne, S, Nève, G, Baguette, M (1999). Spatial and temporal population genetic structure of the butterfly Aglais urticae L. (Lepidoptera, Nymphalidae). Mol Ecol, 8: 1539–1543.
Vekemans, X, Lefèbvre, C (1997). On the evolution of heavy metal tolerant populations in Armeria maritima: evidence from allozyme variation and reproductive barriers. J Evol Biol, 10: 175–191.
Waycott (1998). Genetic variation, its assessment and implications to the conservation of seagrasses. Mol Ecol, 7: 793–800.
Williams, JGK, Kubelik, AR, Livak, KJ, Rafalski, JA, Tingey, SV (1990). DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucl Acids Res, 18: 6531–6535.
Wu, J, Krutovskii, KV, Strauss, SH (1999). Nuclear diversity, population differentiation, and phylogenetic relationships on RAPD and allozyme markers. Genome, 42: 893–908.
Acknowledgements
We would like to thank L Mousson whose contributions to this study are invaluable, JPh Colson and L Dhondt for help with the laboratory work and M Evrard for graphics. We would also like to thank two anonymous reviewers and J Brookfield for helpful comments. This work was funded by the Université catholique de Louvain through a ‘teaching assistant’ (mandat d’assistant) grant to SV, by a grant from the European Commission ‘Training and Mobility of Researchers’ programme on ‘survival and evolution of species in fragmented landscapes’ (TMR-FRAGLAND) to MB, by a grant from the Ministère de la Région Wallonne to MB and by a grant from the Office of Scientific, Technical and Cultural Affairs (Belgian Federal Government) to MB (contract OSTC-SPSDII EV10/16A). Special capture licenses for B. aquilonaris and site access were provided by the Ministère de la Région Wallonne. This is contribution number BRC008 of the Biodiversity Research Centre (UCL).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Vandewoestijne, S., Baguette, M. The genetic structure of endangered populations in the Cranberry Fritillary, Boloria aquilonaris (Lepidoptera, Nymphalidae): RAPDs vs allozymes. Heredity 89, 439–445 (2002). https://doi.org/10.1038/sj.hdy.6800161
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6800161
Keywords
This article is cited by
-
Conservation genetics of a threatened butterfly: comparison of allozymes, RAPDs and microsatellites
BMC Genetics (2014)
-
Selection on the wing in Heliconius butterflies
BMC Genetics (2011)
-
The Genetic Structure of Anophelines (Culicidae: Diptera): RAPD Versus Allozymes
Proceedings of the Zoological Society (2011)
-
Positive correlation between genetic diversity and fitness in a large, well-connected metapopulation
BMC Biology (2008)
-
Molecular and morphometric variation in chromosomally differentiated populations of the grasshopper Sinipta dalmani (Orthopthera: Acrididae)
Genetica (2008)
