
Abstract
Populations of Proclossiana eunomia (Lepidoptera, Nymphalidae) occur in middle Europe in patchy habitats of hay meadows along valleys or peat bogs. Samples of P. eunomia populations from the Ardennes region (northern France and southern Belgium) were analysed by allozyme electrophoresis. Patches isolated by more than 2 km of mature forests proved genetically distinct from their neighbouring populations. Mantel tests and regression analysis showed that the degree of genetic differentiation between the 26 studied populations is related to the geographical distances between them. Autocorrelation analysis (Moran’s I) showed that allele frequencies are positively correlated for populations up to 13 km apart and that the genetic neighbourhood of individuals is in the range of 0.9 km, which is in accordance with movement studies in this species conducted in the same area. Analysis using Wright’s F-statistics revealed that the highest differentiation occurs between populations of the same subregion, whereas the whole Ardennes region is not genetically partitioned into subregions. This is probably because the connectivity of the network of suitable habitats has significantly weakened only since the 1950s, and thus subregional differentiation has not yet occurred.
Similar content being viewed by others

Introduction
Following Sewall Wright’s research on the theory of population differentiation (e.g. Wright, 1978), numerous studies have dealt with the genetic structure of natural populations (cf. reviews in Endler, 1977, 1986). Wright suggested a population genetic structure in which the main factors are population density and dispersal rates within a large and continuous habitat. In this classical model, the probability of mating with a given conspecific is inversely proportional to the distance between the potential mates, resulting in an isolation-by-distance effect. Further developments of this model introduced the hierarchical structure of populations, in which the probability that two individuals will mate depends on the relationship between the populations they belong to. The genetic consequences of a given spatial population structure depend on the dispersive power of individuals (Slatkin, 1994).
In butterfly species different types of population structure can be found. Some species show an open population structure and high observed dispersal (e.g. Aglais urticae, Vandewoestijne et al., 1999); such species may then comply with Wright’s isolation-by-distance model, as has been shown for Pieris napi (Porter & Geiger, 1995). Other butterfly species occur in discrete populations showing restricted dispersal between neighbouring patches (Ehrlich, 1984). Several species, especially those living in temporary habitats, have been shown to have a balance of colonization/extinction where, on average, the number of populations which go extinct is balanced by the same number of new populations being founded (Thomas & Hanski, 1997).
Mark–release–recapture surveys have shown that there are frequent exchanges between nearby populations of Proclossiana eunomia within the same valley, and the analysis of the colonization process in Morvan (central France) revealed that the (re-)colonization ability of this species is high, with steps of up to 6 km having been observed (Nève et al., 1996a). The combination of ecological and genetical approaches in this species confirmed the results of dispersal ability (Nève et al., 1996b) and validated the genetics approach at a higher scale (Bossart & Prowell, 1998).
The present paper addresses the question of genetic structure and its relationship with the observed pattern of movement and colonization. Are the populations of P. eunomia from a given upland range organized genetically in a hierarchical way, or are exchanges throughout the area sufficient to counteract genetic differentiation? Do geographical distances between populations determine the genetic differentiation of populations?
Methods
The species
Proclossiana eunomia is confined to mountain ranges of southern Europe, and to the uplands of middle Europe; these groups of populations are thus isolated from each other by hundreds of kilometres of unsuitable habitats (Nève et al., 1996a). Other subspecies occur from Scandinavia through Siberia to North America. In middle Europe, P. eunomia is a specialist butterfly species restricted to wet hay meadows and peat bogs where Polygonum bistorta is its only food plant. The species is monovoltine; imagines are on the wing in middle Europe from late May to the beginning of July. Populations of this species are distributed patchily at different scales. Within the Ardennes uplands, in southern Belgium and nearby France, the species is distributed widely (Fig. 1), and suitable habitats are usually spaced in an ‘archipelago’ fashion, in peat bog areas, or along valleys with wet hay meadows.

Historically known range of Proclossiana eunomia in Belgium, based on 10 × 10 km UTM squares. The enlarged map indicates the location of the studied populations; ellipses indicate the grouping into five subregions.
Sampling
Between 1991 and 1996, 31 samples totalling 1236 individuals were collected in the Belgian and French Ardennes (Fig. 1, Table 1); samples of fewer than 20 individuals were merged with nearby samples, when exact tests (see below) showed that the relevant samples did not differ significantly in their allele frequencies (Table 2). In such cases the co-ordinates of the pooled sample were considered as being the weighted mean of the co-ordinates of each of its parts. Population ELS (Elsenborn) was sampled annually from 1992 to 1996 (32, 20, 58, 66 and 53 individuals, respectively), and populations TAI and ROC were sampled twice (1992 and 1994, and 1994 and 1995, respectively) in order to control for any temporal variation.
Genetic analysis
Upon collection the specimens were deep-frozen in liquid nitrogen (−196°C), and later transferred to a −80°C freezer until analysis. When thawed, the wings were kept for morphometric analysis, and the thorax and the anterior part of the abdomen were squashed in 0.5 mL of pH 7.1 buffer [15% (w/v) sucrose, 50 mM Tris/HCl pH 7.1, 0.5% (v/v) Triton X-100, with one drop of Bromophenol Blue as runner marker]. The homogenate was then centrifuged at 2200 g for 3 min; the supernatant was frozen as 10 μL droplets in liquid nitrogen. One droplet per specimen was sufficient for cellulose acetate electrophoresis, using the buffer and staining recipes of Richardson et al. (1986) and Wynne et al. (1992).
Ten presumptive polymorphic loci were tested: adenylate kinase (Ak; EC 2.7.4.3); phosphoglucomutase (Pgm; EC 5.4.2.2.); glucose-phosphate isomerase (Pgi; EC 5.3.1.9); 6-phosphogluconate dehydrogenase (6Pgd; EC 1.1.1.44); glucose-6-phosphate dehydrogenase (EC 1.1.1.49); aspartate aminotransferase, two loci (EC 2.6.1.1); mannose-6-phosphate isomerase (EC 5.3.1.8), β-hydroxybutyrate dehydrogenase (EC 1.1.1.30); glycerol-3-phosphate dehydrogenase (EC 1.1.1.8). Of these loci, only the first four proved to be polymorphic in the studied area.
Genotypic data were studied using different software. The GENEPOP population genetics software (version 3.1; Raymond & Rousset, 1995a): (i) assessed differentiation between pairs of samples (Raymond & Rousset, 1995b); (ii) calculated pairwise FST values between populations, using Weir & Cockerham’s (1984) method; (iii) tested compliance to Hardy–Weinberg expectations in the sampled populations using Fisher’s exact test; and (iv) tested for isolation by distance, using a Mantel permutation procedure according to Rousset (1997). For a hierarchical analysis, populations were grouped as five subregions, all within the Ardennes region, according to the geographical distance between samples (Fig. 1); this analysis was performed using the BIOSYS-1 software (version 1.7; Swofford & Selander, 1981). A dendrogram of the 26 studied populations was produced by bootstrapping among the loci and calculating a consensus of 100 trees obtained using the neighbour-joining algorithm on Nei’s genetic distances; a 30-individual sample collected in June 1993 in Targassonne (French Pyrenees, 42°30′N, 1°58′E) was used as an outgroup. This dendrogram was constructed using the PHYLIP program (Felsenstein, 1993). Autocorrelation analyses of allele frequencies were computed using Moran’s I index, by the ‘R’ package (Legendre & Vaudor, 1991), on the geographical distance data divided into 20 equifrequent classes of distances between all possible pairs of populations. Slatkin (1993) showed that effective migration between populations (Nm) may be estimated by the relationship

Neighbourhood area may then be estimated by the intercept of the function of the pairwise FST = a + b/(4x + 1), where x is the geographical distance between populations (Porter & Geiger, 1995).
Results
Pairwise comparisons of allele frequencies, using Raymond & Rousset’s (1995b) exact test, were performed when a sample amounted to fewer than 20 individuals; such samples were compared to the nearest other sample. In none of these cases was there a significant difference between the allele frequencies, and the paired samples were pooled (Table 2).
In the resulting 26 populations, allozyme frequencies varied greatly (Table 1). Pgi was polymorphic in only three of the 26 populations. 6Pgd showed fixation in one case, population WIT, which occurs in a forest clearing 3.7 km from the nearest other population.
Exact tests showed a significant deviation from Hardy–Weinberg expectation in six of the 26 studied populations (Table 1), always with a deficit of heterozygotes. In the remaining 20 populations, four showed a significant deviation from H–W expectation at one locus, but in each case Fisher’s exact test was nonsignificant for the four loci studied together (Table 1). The allele frequencies of populations ELS and ROC did not show significant temporal differentiation (χ28=12.63, P=0.11 and χ26=4.07, P=0.68, respectively). However, deviation from Hardy–Weinberg expectation occurred in the ELS population only at the 6Pgd locus, and only for two of the five-year samples, each time with a deficit in heterozygotes, and hence an excess of rare homozygotes (1994: 1 observed, 0.026 expected, P=0.0261; 1995: 3 observed, 0.16 expected, P < 0.001).
In population TAI, both samples (1992 and 1994) agreed with HW expectations (χ26=9.2, P=0.16 and χ26=6.7, P=0.34). However, these samples had different allele frequencies for both 6Pgd (P=0.037) and Ak (P=0.047), but not for Pgm (P=0.549), giving an overall significant difference when considering the three loci together (χ26=13.9, P=0.0296). The deviation from H–W expectation for the pooled sample resulted from a Wahlund effect caused by two samples from a population which had undergone a temporal variation in allele frequency between the sampling sessions.
In both LIE and GAU, the merging of samples of slightly different allele frequencies caused the significant departure from H–W expectation. Exact tests on the individual samples indicated that only LIE1 showed a significant deviation from H–W expectation (Table 2).
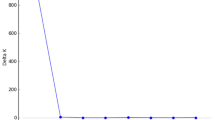
Spatial autocorrelation analyses of the frequencies of the most common alleles of 6Pgd, Pgm and Ak (Fig. 2) showed that: (i) Ak and 6Pgd allele frequencies are positively correlated within the first distance class, i.e. up to 5.6 km (P < 0.05 and P < 0.01, respectively); 6Pgd is also positively correlated in the second distance class, i.e. from 5.6 to 8.0 km; (ii) significant negative correlation is shown in the 17th (73.0–78.2 km, P < 0.001) and 20th classes (92.5–121.1 km, P < 0.01) for Ak and in the 3rd (8.0–12.6 km), 8th (38.2–42.7 km) and 9th (42.7–45.0 km) for 6Pgd; (iii) Ak is also positively correlated at the 5th and 8th distance classes, i.e. from 18.6 to 26.6 and from 38.2 to 42.7 km. In the case of 6Pgd, the sequence of positive and negative values may be interpreted as originating from a general spatial pattern of waves or bumps, with no overall gradient. For Ak, the sequence of significant positive values in the first half of the graph and the significant negative values in the second part of the graph may be interpreted to have originated from an overall gradient (Legendre & Legendre, 1998). The allele frequency variation at the Pgm locus does not show any significant spatial pattern. The autocorrelation analysis was repeated for Ak and 6Pgd with different numbers of distance classes in order to estimate the upper length of the first autocorrelation class for which a significant I-value would still be obtained (Fig. 3). In both cases the value of Moran’s I decreased from a length for the first class at 1.8 km up to 13.3 km, then levelled off at a low value (Moran’s I < 0.25, Fig. 3). For Ak, the 0.05 significance level was not met at the first distance class within 13.3 km (P=0.09).

Spatial autocorrelation analysis of the most common allele at the 6Pgd, Ak and Pgm loci in Proclossiana eunomia. Filled symbols indicate an associated P < 0.05.

Moran’s I for varying upper limit of the first distance class for the most common alleles of 6Pgd and Ak. The open circle at 13.3 km indicates a nonsignificant (P=0.09) I-value; all the other values are significant at the 0.05 level.
The absolute number of migrants, Nm, between populations was estimated from the pairwise FST, and log(Nm) was regressed against the geographical distances between populations, to detect any isolation-by-distance effect (Slatkin, 1993). Discarding meaningless negative values of Nm (25 cases), the linear relationship between log(Nm) and the geographical distance (x, measured in km) was estimated as log(Nm)=1.617–0.0050x (F1,297=4.757, P=0.030), giving a significantly negative slope (95% CI: −0.0005, −0.0096). As the geographical distance data are not independent, a Mantel test was also applied, using GENEPOP 3.1, between the matrix of pairwise FST and the log of the distances; the results also proved significant (2000 iterations; P=0.012).
The relationship between pairwise FST and the geographical distance between populations gave a best fit with FST=0.0841 − 0.394/(4x + 1) (F1,323=11.17, P= 0.0009), which has an intercept at x=0.92 km, thus giving the estimated average local genetic neighbourhood.
A consensus dendrogram constructed by the neighbour-joining method on Nei’s genetic distance shows a limited congruence with the geographical distance between the populations (Fig. 4). For example, populations LIE, TAI, BIE, BIW, SOM and REC make up a geographically sound set, but BOE and BOW ought to have been included in the same branch. Most other groups do not cluster according to their relative geographical proximity.

Neighbour-joining dendrogram: consensus tree built from 100 trees obtained by bootstrapping on Nei’s distance between 26 Proclossiana eunomia populations from the Ardennes region. Population codes follow Table 2 and Fig. 1; PYR indicates the population from Targassonne (French Pyrenees), used as an outgroup. The numbers at the forks indicate the number of times the group consisting of the populations which are to the right of that fork occurred among the 100 trees.
The hierarchical analysis of F-statistics shows that the highest differentiation is between localities within subregions, whereas the whole Ardennes region is not partitioned into subregions (Table 3).
Discussion
In the Ardennes region, populations of P. eunomia situated close together within a river basin or a peat bog area usually show strong genetic inter-relationships (Nève et al., 1996b). Direct observation of emigration, including up to 6 km colonization steps, had suggested such patterns (Nève et al., 1996a). Geographically isolated populations, however, may show more distinct allele frequency patterns from their neighbours because of genetic drift.
We did not find any direct evidence of movements between river basins in any of our mark–release–recapture studies. Colonization patterns in Morvan have shown, however, that movement to adjacent river basins does occur, albeit rarely (Nève et al., 1996a). Hierarchical F analysis suggests that each of the five subregions studied contains most of the genetic diversity present at the regional level. Differentiation between populations occurring on European mountain ranges (Ardennes, Pyrenees, Cantabricous mountains, Balkans, Czechia) within Europe is much higher, with a mean FST for the four loci considered here of 0.177 (Barascud, Descimon & Nève, unpubl. data), which indicates a ‘great’ genetic differentiation (Wright, 1978).
The practical use of the relationship FST=a + b/(4x + 1), with x as the distance between the pairs of populations considered, in inferring the genetic neighbourhood size leads to problems linked with the autocorrelation of geographical and genetic distance data sets. From a sample of n populations, a total of n(n − 1)/2 points arise in the regression test. To avoid this pseudoreplication problem autocorrelation analyses were performed, revealing a positive correlation between populations up to about 13 km apart. This distance does not correspond to the radius of Wright’s (1978) genetic neighbourhood area, i.e. the area from which individual matings occur at random, but rather to the way allele frequencies are correlated between populations according to the geographical distance between them.
Our mark–release–recapture data suggest a genetic neighbourhood in the order of several hundred metres (Nève et al., 1996a), well in accordance with the neighbourhood radius estimated here (0.9 km). The pattern of the autocorrelation for the most common allele of the locus Ak shows significantly positive values at small distances and negative values at the longest recorded distances. This is typical of clinal variation, with long-distance differentiation (Sokal et al., 1989). Endler (1977) showed that movements between adjacent populations are sufficient to induce clines, which involve correlation of populations that do not exchange individuals directly.
Heterozygote deficiency in several populations may result from nonpanmictic pairing having occurred in at least partly divided populations, from recent immigration having not yet reached equilibrium or from natural selection (Endler, 1986). Our data are not sufficient to test these hypotheses.
Despite an isolation-by-distance effect within the whole studied area, the consensus dendrogram constructed from Nei’s distance between populations does not fit the geographical distribution of the samples. This suggests that factors other than geographical distance may be responsible for population differentiation (Slatkin, 1994). These may include recent colonization (Meglécz et al., 1999), especially if dispersal, which is related to sexual behaviour and density in P. eunomia (Baguette et al., 1998), is variable in time. For this species, a colonization process within Morvan (France) over a 25-year period has led to significant differentiation between populations (Barascud et al., 1999). Genetic drift occurring by chance in the same way in different populations, regardless of the relative locations of these populations, may also obscure the differentiation-by-distance effect. This has also been shown to occur in the Western Seep Fritillary (Speyeria nokomis apacheana), a butterfly confined to isolated mesic areas in the American Great Basin region, in an area roughly 300 × 400 km (Britten et al., 1994).
Small populations which have long been isolated are usually subject to genetic drift and founder effect, both of which may obliterate the isolation-by-distance effect (Slatkin, 1993). In Morvan, P. eunomia colonized the region from a small number of females originating from a single population. Within a 25-year period, significant population differentiation occurred, but no isolation-by-distance effect could be detected, suggesting a stepping-stone colonization process (Barascud et al., 1999). In the Great Basin area, with distances ranging from 12 to 500 km, independent genetic drift occurred in long-isolated populations of Euphydryas editha, and a low level of isolation-by-distance could be detected. By contrast, in populations of E. editha which have not experienced such a long isolation, as in the Rocky Mountain Cordillera, the isolation-by-distance effect is significant for a similar range of distances (300–400 km; Britten et al., 1995).
In the context of metapopulation dynamics and genetics (cf. Hedrick & Gilpin, 1997), dynamics of the colonization/extinction of populations at the landscape scale may maintain a high genetic diversity between populations. The lack of subregional differentiation in Ardennes populations of P. eunomia may result from: (i) a comparatively recent colonization or (ii) occasional stepping-stone movements across the whole regional distribution of the species, at least until recently; or (iii) a combination of both factors. Variation of the Diamond-back Moth (Plutella xylostella) follows a similar pattern of differentiation within Oahu and Maui on the Hawaiian archipelago, where it was introduced in the 19th century: low variation between subregions (islands) and a higher level of population differentiation between subpopulations (Caprio & Tabashnik, 1992). These authors conclude that ‘there has probably been sufficient time since its [Plutella xylostella’s] introduction for local populations to have differentiated, but it is less likely that enough time has passed for larger regional population units to have differentiated from each other.’ A similar pattern has been shown for the P. eunomia colonization of Morvan (France) from 1970 to the 1990s (Barascud et al., 1999).
Lack of correlation between geographical distance and genetic distance is a characteristic of species with high dispersal power, such as in the migratory butterfly Aglais urticae (Vandewoestijne et al., 1999). Given the fact that the increase in fragmentation of P. eunomia’s habitats in Belgium is recent (since the 1950s; Goffart et al., 1992), the connections between the populations of the subregions have only weakened recently, and subregional differentiation has not yet occurred. In the closely related Boloria improba, studied in the Rocky Mountain Cordillera by Britten & Brussard (1992), most of the differentiation is also displayed at the population level (Fpopulation–region=0.379) whereas the regional differentiation is weaker (Fregion-total=0.172). The higher within-differentiation of B. improba within the Rocky Mountain Cordillera, compared with the within-Ardennes nil differentiation of P. eunomia, is probably caused by stronger barriers to dispersal and to the much larger area of the Rocky Mountain range of B. improba, from the Yukon Territory to Colorado (over 3000 km), compared with the Ardennes populations of P. eunomia. Gene flow within large areas is generally the case for either generalist species, such as Pieris napi (Porter & Geiger, 1995), or for species linked with a widely distributed host species, such as Aglais urticae feeding on Urtica dioica (Vandewoestijne et al., 1999).
Populations of specialist butterflies have often been described as ‘closed’, mainly from ecological studies recording the smallest patches in which the species have been reported as occurring in a viable population (e.g. Thomas, 1984). However, several studies have demonstrated that low-frequency emigration/immigration events and founders may have a strong impact on the genetic structure of populations. Brussard & Vawter (1975) were the first to show that a seemingly highly sedentary butterfly, Euphydryas phaeton, for which no recaptures over 100 m from release locations were observed, still had a high level of gene flow between its seemingly ecologically separated populations. It seems, therefore, that an ecological description of the long survival of a small population is not sufficient to assess its effective genetic isolation.
The low genetic differentiation of P. eunomia populations within the Ardennes region seems to be a result of emigration/immigration occurring within the whole area up to a recent time. In the perspective of the conservation of P. eunomia, the present work shows that several levels are implied, in particular a spatial one. Maintenance of a habitat network seems to be essential to avoid the loss of gene diversity through drift in overly small, isolated populations. The ways of maintaining significant connectivity between the colonies are subject to debate, however. Managing or restoring habitat corridors allowing the exchange of individuals and, hence, gene flow should be the best way, even if it may look unrealistic under present socio-economic conditions. Translocation of individuals between colonies could be a lower-cost solution, but this raises problems of genetic management that should be carefully studied, and would preserve the genetic diversity only of the target species, without considering other species sharing the same habitat requirements.
References
Baguette, M., Convié, I., Vansteenwegen, C. and Nève, G. (1998). Sex-biased density-dependent dispersal in a metapopulation of the butterfly Proclossiana eunomia. Acta Œcol, 19: 17–24.
Barascud, B., Martin, J. F., Baguette, B. and Descimon, H. (1999). Genetic consequences of an introduction-colonization process in an endangered butterfly species. J Evol Biol, 12: 697–709.
Bossart, J. L. and Prowell, D. P. (1998). Genetic estimates of population structure and gene flow: limitations, lessons and new directions. Trends Ecol Evol, 13: 202–206.
Britten, H. B. and Brussard, P. F. (1992). Genetic divergence and the Pleistocene history of the alpine butterflies Boloria improba (Nymphalidae) and the endangered Boloria acrocnema (Nymphalidae) in western North America. Can J Zool, 70: 539–548.
Britten, H. B., Brussard, P. F., Murphy, D. D. and Austin, G. T. (1994). Colony isolation and isozyme variability of the western seep fritillary, Speyeria nokomis apacheana (Nymphalidae), in the Western Great Basin. Great Basin Naturalist, 54: 97–105.
Britten, H. B., Brussard, P. F., Murphy, D. D. and Ehrlich, P. R. (1995). A test for isolation-by-distance in central Rocky Mountain and Great Basin populations of Edith’s checkerspot butterfly (Euphydryas editha). J Hered, 86: 204–210.
Brussard, P. F. and Vawter, A. T. (1975). Population structure, gene flow and natural selection in populations of Euphydryas phaeton. Heredity, 34: 407–415.
Caprio, M. A. and Tabashnik, B. E. (1992). Allozymes used to estimate gene flow among populations of Diamondback Moth (Lepidoptera, Plutellidae) in Hawaii. Environ Entomol, 21: 808–816.
Ehrlich, P. R. (1984). The structure and dynamics of butterfly populations. In: Vane-Wright, R. I. and Ackery, P. R. (eds) The Biology of Butterflies. Symposium 11 of the Royal Entomological Society, pp. 25–40. Academic Press, London.
Endler, J. A. (1977). Geographic Variation, Speciation, and Clines. Princeton University Press, Princeton, NJ.
Endler, J. A. (1986). Natural Selection in the Wild. Princeton University Press, Princeton, NJ.
Felsenstein, J. (1993). PHYLIP (Phylogeny Inference Package). Version 3.5c. Department of Genetics, University of Washington, Seattle.
Goffart, P. H., Baguette, M. and de Bast, B. (1992). La situation des Lépidoptères Rhopalocères en Wallonie ou Que sont nos papillons devenus?. Bull Ann Soc R Belge Entomol, 128: 355–392.
Hedrick, P. W. and Gilpin, M. E. (1997). Genetic effective size of a metapopulation. In: Hanski, I. and Gilpin, M. E. (eds) Metapopulation Biology: Ecology, Genetics and Evolution, pp. 165–181. Academic Press, San Diego, CA.
Legendre, P. and Legendre, L. (1998). Numerical Ecology. Elsevier, Amsterdam.
Legendre, P. and Vaudor, A. (1991). Le Progiciel R — Analyse Multidimentionnelle, Analyse Spatiale. Département de Sciences Biologiques, Université de Montréal, Montréal.
Meglécz, E., Nève, G., Pecsenye, K. and Varga, Z. (1999). Genetic variations in space and time in Parnassius mnemosyne (Lepidoptera) populations in northeast Hungary. Biol Cons, 89: 251–259.
Nève, G., Barascud, B., Hughes, R., Aubert, J., Descimon, H., Lebrun, P. H. and Baguette, M. (1996a). Dispersal, colonization power and metapopulation structure in the vulnerable butterfly Proclossiana eunomia (Lepidoptera, Nymphalidae). J Appl Ecol, 33: 14–22.
Nève, G., Mousson, L. and Baguette, M. (1996b). Adult dispersal and genetic structure of butterfly populations in a fragmented landscape. Acta Oecol, 17: 621–626.
Porter, A. H. and Geiger, H. (1995). Limitations to the inference of gene flow at regional geographic scales — an example from the Pieris napi group (Lepidoptera: Pieridae) in Europe. Biol J Linn Soc, 54: 329–348.
Raymond, M. and Rousset, F. (1995a). GENEPOP (Version 1.2), a population genetics software for exact tests and ecumenicism. J Hered, 86: 248–249.
Raymond, M. and Rousset, F. (1995b). An exact test for population differentiation. Evolution, 49: 1280–1283.
Richardson, B. J., Baverstock, P. R. and Adams, M. (1986). Allozyme Electrophoresis. Academic Press, Sydney.
Rousset, F. (1997). Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics, 145: 1219–1228.
Slatkin, M. (1993). Isolation by distance in equilibrium and non-equilibrium populations. Evolution, 47: 264–279.
Slatkin, M. (1994). Gene flow and population structure. In: Real, L. A. (ed.) Ecological Genetics, pp. 3–17. Princeton University Press, Princeton, NJ.
Sokal, R. R., Harding, R. M. and Oden, N. L. (1989). Spatial patterns of human gene frequencies in Europe. Am J Phys Anthropol, 80: 267–294.
Swofford, D. L. and Selander, R. B. (1981). BIOSYS-1: a FORTRAN program for the comprehensive analysis of data in population genetics and systematics. J Hered, 72: 281–283.
Thomas, C. D. and Hanski, I. (1997). Butterfly metapopulations. In: Hanski, I. and Gilpin, M. E. (eds) Metapopulation Biology: Ecology, Genetics and Evolution, pp. 359–396. Academic Press, San Diego, CA.
Thomas, J. A. (1984). The conservation of butterflies in temperate countries: past efforts and lessons for the future. In: Vane-Wright, R. I. and Ackery, P. R. (eds) The Biology of Butterflies. Symposium 11 of the Royal Entomological Society, pp. 333–353. Academic Press, London.
Vandewoestijne, S., Nève, G. and Baguette, M. (1999). Spatial and temporal population genetic structure of the butterfly Aglais urticae (Lepidoptera Nymphalidae). Mol Ecol, 8: 1539–1544.
Weir, B. S. and Cockerham, C. C. (1984). Estimating F-statistics for the analysis of population structure. Evolution, 38: 1358–1370.
Wright, S. (1978). Evolution and the Genetics of Populations, vol. 4, Variability Within and Among Natural Populations. University of Chicago Press, Chicago.
Wynne, I. R., Loxdale, H. D. and Brookes, C. P. (1992). Use of a cellulose acetate system for allozyme electrophoresis. In: Berry, R. J., Crawford, T. J. and Hewitt, G. M. (eds) Genes in Ecology, pp. 494–499. Blackwell Scientific Publications, Oxford.
Acknowledgements
Ph. Lebrun gave constant support and interest throughout the study. We would like to thank E. Meglécz, A.H. Porter and the late F.A. Lints for their critical reading and constructive comments on an earlier draft of this manuscript. Ph. Goffart and Th. Martin helped in the field, in the laboratory and in numerous fruitful discussions. Data for the Belgian distribution of P. eunomia were made available from the Insect Distribution Database (Gembloux) and the Lepidoptera Research Group. The mapping was carried out using M. Dufrêne’s “CartoBelgique” programme. This work was funded partly by grants from the ‘Institut pour l’Encouragement de la Recherche Scientifique dans l’Industrie et l’Agriculture’ (Brussels), from the ‘Ministère de la Recherche et de la Technologie’ (Paris) and from the European Commission ‘Training and Mobility of Researchers’ programme on ‘Survival and evolution of species in fragmented landscapes’. Special capture licences were granted from the ‘Ministère de la Région Wallonne’ (Belgium) and the ‘Ministère de l’Environnement’ (France).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nève, G., Barascud, B., Descimon, H. et al. Genetic structure of Proclossiana eunomia populations at the regional scale (Lepidoptera, Nymphalidae). Heredity 84, 657–666 (2000). https://doi.org/10.1046/j.1365-2540.2000.00699.x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1046/j.1365-2540.2000.00699.x
Keywords
This article is cited by
-
Phylogeographic patterns of steppe species in Eastern Central Europe: a review and the implications for conservation
Biodiversity and Conservation (2016)
-
Conservation genetics of a threatened butterfly: comparison of allozymes, RAPDs and microsatellites
BMC Genetics (2014)
-
Matrix quality and habitat configuration interactively determine functional connectivity in a widespread bush cricket at a small spatial scale
Landscape Ecology (2012)
-
High dispersal ability and low genetic differentiation in the widespread butterfly species Melanargia galathea
Journal of Insect Conservation (2010)
-
The genetic consequence of differing ecological demands of a generalist and a specialist butterfly species
Biodiversity and Conservation (2009)
