
Abstract
We have amplified and sequenced 679 nucleotides of the mitochondrial DNA control-region in 45 Siberian (Capreolus pygargus) and European (C. capreolus) roe deer from two localities in Russia and seven in Italy. Average interspecific sequence divergence was 4.9%. Six different haplotypes were found in Siberian roe deer, and 14 haplotypes in Alpine European roe deer. A population of the endemic Italian subspecies C. c. italicus was monomorphic bearing a single haplotype with one unique nucleotide deletion and a fixed transversion. Phylogenetic relationships among haplotypes indicated that the two species were separated with 100% bootstrap support, and there were two distinct population clusters within each species. These clusters correspond to different geographical locations of the samples: Siberian roe deer were subdivided into west Siberia (Kurgan region) and east Siberia (Amur region), and European roe deer were subdivided into an eastern and a western Alpine group. Average sequence divergence among conspecific populations was 1.2%. Calibrations of evolutionary rates of the different domains of the control-region suggest that Siberian and European roe deer speciated about 2–3 million years ago, and haplotype diversity within species was generated during the last 150 000–370 000 years. Geographical structuring of sequence variability in roe deer allows us to identify historical and recent intraspecific population differences, including the effects of human disturbance. The genetic peculiarities of the endemic Italian subspecies C. c. italicus call for careful conservation of its surviving populations.
Similar content being viewed by others
Introduction
Roe deer (Capreolus Gray, 1821) are distributed in the Palaearctic and in continental Asia (Danilkin, 1996). Two polytypic species are recognized: the larger Siberian (C. pygargus Pallas, 1777) and the smaller European roe deer (C. capreolus Linnaeus, 1758) (Groves & Grubb, 1987; Grubb, 1993). They have different body sizes, morphometric traits and karyotypes, and most hybrid males obtained in captivity were sterile (Sokolov & Gromov, 1990; Danilkin, 1996). Although their genetic divergence has not been investigated, their allopatric distributions, with possible narrow areas of parapatric contact around the banks of the rivers Volga and lower Don in Russia (Heptner et al., 1989), and their fossil record of late Pleistocene age (Heptner et al., 1989) suggest that the species of roe deer are recent and closely related.
Roe deer populations show wide morphological, ethological and ecological variability (Sokolov & Gromov, 1990; Danilkin, 1996). Noteworthy, for example, is the presence of sympatric sedentary and migrating populations in Siberia (Danilkin, 1996). Roe deer populations in the south-west of Europe have been relatively disturbed as a consequence of habitat fragmentation and restocking for hunting purposes. Roe deer populations of the western Italian Alps have been extirpated by overhunting during the last 30 years, and then restored using stocks from the eastern Italian Alps, central Europe and the Balkans. In contrast, roe deer of the eastern Italian Alps have been preserved and still represent the autochthonous Alpine populations. Isolated roe deer populations in central–southern Italy have been recognized as an endemic subspecies, named C. c. italicus by Festa (1925). The level of genetic diversity and the phylogeographical relationships among natural roe deer populations are poorly known (Hartl & Reimoser, 1988). Therefore, the eventual genetic consequences of human disturbances are still unknown. For these reasons, roe deer provide a case study to evaluate the genetic effects of fragmentation and human disturbance on managed populations, within the background of the recent evolutionary divergence of the two species.
The high mutation rates of mitochondrial DNA (mtDNA) can produce intraspecific polymorphism and deep interspecific divergence in relatively short evolutionary times (Avise et al., 1987). Maternal transmission and the absence of recombination can result in highly detectable geographical subdivision of populations, particularly in species with female phylopatry. The control-region (CR), the unique long noncoding nucleotide sequence, is the most variable portion of mtDNA (Moritz et al., 1987). The rapid molecular evolution of CR results from point mutations, insertion–deletions of one or more nucleotides, or different numbers of tandem repeats, which can result in individual heteroplasmy and length differences among species (Hoelzel et al., 1994). Different domains of CR evolve at different rates, and, in particular, peripheral sequences evolve faster than the remarkably conserved central blocks. The peripheral domains of CR can be used for detailed analyses of population structure (e.g. Arctander et al., 1996), whereas the conserved central blocks are informative for reconstructing phylogenetic relationships also among distantly related taxa (e.g. Arnason et al., 1993).
In this paper we study nucleotide sequences of the mtDNA CR of Siberian and European roe deer sampled from different populations in Russia and Italy. We aim: (i) to estimate nucleotide sequence divergence and eventual reciprocal haplotype monophyly between Siberian and European roe deer; (ii) to add information to the relatively scarce data set on mtDNA sequence variability of large mammalian species; (iii) to study the genetic structure in undisturbed vs. disturbed roe deer populations; and (iv) to define the genetic traits of the Italian endemic subspecies of roe deer.
Materials and methods
Sample collection
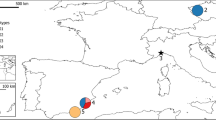
Tissue and blood samples of nine Siberian roe deer were collected from two localities in Russia (five samples from the Kurgan region, west Siberia; four samples from the Amur region), and 36 European roe deer from seven localities in the Italian Alps and central Italy (Fig. 1). The 13 samples collected in central Italy belong to one of the last three surviving populations of the Italian endemic subspecies C. c. italicus, as described by Festa (1925)

Distribution of European roe deer populations in Italy. Cross-hatched areas, autochthonous populations; lined areas, reintroduced populations; dotted areas in central–southern Italy, populations of Capreolus capreolus italicus. Sampling localities are numbered as follows (n, sample size): 1, EUR1 (Trieste; n=2); 2, EUR2 (Asiago; n=4); 3, EUR3 (Monte Bondone; n=5); 4, EUR4 (Sondrio; n=1); 5, EUR5 (Piemonte, Val Chisone; n=4); 6, EUR6 (Savona; n=5); 7, EUR7 (C. c. italicus; Castel Porziano Reserve; n=13).
DNA isolation, amplification and sequencing
Total DNA was extracted through proteinase-k digestion and phenol/chloroform extractions. The entire mtDNA CR was amplified by PCR using primers L-Pro (5§-CGT CAG TCT CAC CAT CAA CCC CCA AAG C-3§), and H-Phe (5§-GGG AGA CTC ATC TAG GCA TTT TCA GTG-3§) (Jäger et al., 1992), which bind to nucleotides nos 15 740 and 420 of the bovine tRNAPro and tRNAPhe genes, respectively (Anderson et al., 1982). Amplifications were performed using a 9600 Perkin Elmer thermocycler, with the following protocol: 94°C for 2 min; 94°C for 15 s, 55°C for 15 s, 72°C for 1 min (30 cycles); 72°C for 10 min; and with 3 mM MgCl2 in the PCR buffer. Purified PCR products were sequenced using ΔTaq Sequenase (Amersham). The entire CR was sequenced in one Siberian roe deer (SIB2.1; GeneBank accession number Z70317) and one European roe deer (EUR2.1; Z70318), using primers L-Pro, H-Phe and two internal primers: L-362 (5§-AAT CAC CAT GCC GCG TGA AAC C-3§), and H-493 (5§-TGA GAT GGC CCT GAA GAA AGA ACC-3§). These primers were used to obtain partial sequences of 679 bp in all the 45 samples of roe deer.
Data analysis
Nucleotide diversity within and between species was computed with the program SEND.FOR (by L. Jin, based on the paper by Nei & Jin, 1989). Haplotypic diversity was computed by Hs=(1−Σ x2i) n/(n−1), where xi is the frequency of haplotype i and n is the sample size (Wenink et al., 1996). The distribution of molecular variance between species and populations was computed using AMOVA (Excoffier et al., 1992), with the observed number of pairwise substitutions among haplotypes as the input matrix of genetic distances (L. Excoffier, pers. comm.). The hierarchical analysis of molecular variance was performed by subdividing the observed 21 different haplotypes into two groups (the two species) and five populations: SIB1=Kurgan region, west Siberia; SIB2=Amur region, east Siberia; eastern Italian Alps (haplotypes EUR1 to EUR4); western Italian Alps (EUR5, EUR6); and C. c. italicus (EUR7). AMOVA estimates the values of haplotypic diversity at the different hierarchical levels (between species, among populations within species, and within populations). The population structure at each level of the hierarchy is estimated by φ-statistics, measures of haplotypic correlations which are equivalent to the hierarchical F-statistics of Cockerham (1973). The significance of the different variance and φ components are obtained by a random permutation procedure (Excoffier et al., 1992).
Sequences were aligned with CLUSTAL W (Thompson et al., 1994). The subfamily Cervinae is the sister group of the Odocoileinae (Groves & Grubb, 1987). Therefore, a new sequence of the entire CR of the fallow deer Dama dama (Cervinae) was used as an outgroup for defining the phylogenetic relationships among CR haplotypes of Capreolus (Odocoileinae). Significance of the phylogenetic signal generated by the aligned sequences was evaluated using the g1 statistic (Hillis & Huelsenbeck, 1992), computed by estimating the skewness of a distribution of 10 000 random trees (RANDOM TREE option in PAUP 3.1.1; Swofford, 1993). Pairwise genetic distances were computed with MEGA as percentage sequence divergence and using Tamura & Nei's γ distance (1993), with α=0.5, as suggested by Kumar et al. (1993). Phylogenetic trees were reconstructed with neighbour joining (NJ) and maximum parsimony (MP; Swofford, 1993) procedures using MEGA and PAUP. Parsimony analyses, excluding uninformative substitutions and indels, were performed through heuristic searches with TBR and MULPARS options in use, and with 10 random additions of sample sequences. Support of the clusters was evaluated by bootstrap, as percentage recurrence of clusters based on 1000 bootstrapped replications, both with MEGA and PAUP.
Results
The aligned 679 nucleotide sequences of the control-region in 45 Siberian (Capreolus pygargus) and European (C. capreolus) roe deer showed 51 substitutions, which defined 21 different haplotypes (Fig. 2 ). Alignments were obtained by assuming two indels, the first at position no. 8 (a single nucleotide gap in European roe) and the second at position no. 517 (a single nucleotide gap in Siberian roe). The 13 samples of the endemic Italian subspecies C. c. italicus were monomorphic and different from all the others, showing a haplotype with one unique nucleotide deletion at position no. 58 and a fixed transversion (position no. 60). The peripheral parts of CR (domains I and III) evolved four and three times faster than central domain II, respectively. In fact, counting each gap as a single mutation, domain I had 28 substitutions over 175 nucleotides (16.0%), domain II had 17/441 substitutions (3.8%), and domain III had 8/63 (12.7%) substitutions (Fig. 2). Among the 53 variable sites there were 45 transitions (84.9%), four transversions (7.5%) and two gaps (3.8%). Four sites have been hit at least twice: position nos 60, 157 and 161 (one transition and one transversion), and position no. 58 (one transition and one gap). Three of the six observed transversions were fixed differences between species (position nos 24, 174 and 662), and one was a fixed substitution of C. c. italicus (position no. 60). Two transversions occurred at hypervariable position nos 157 and 161, which apparently mutated repeatedly both within and between species (Fig. 2). Six different haplotypes were found among Siberian roe deer, and 15 haplotypes among European roe deer. Haplotypic diversity was 0.93 in both species.

Aligned sequences of the variable sites of the mtDNA CR (L-strand) of Siberian (Capreolus pygargus) and European (C. capreolus) roe deer (GenBank accession nos Z70317, Z70318). Variable sites are indicated by numbers. The central domain, part II, was defined by its high sequence similarity to the conserved blocks described in mammals (Anderson et al., 1982), and spans from conserved block I to the beginning of putative CSB1 or 7S DNA region (Anderson et al., 1982). SIB, haplotypes found in Siberian roe deer; EUR, haplotypes found in European roe deer. The first number of each haplotype indicates the population where the haplotype was found; the second number indicates different haplotypes found in each population.
The observed proportions of nucleotide substitutions were 0.3–2.2% (average 1.2%) within species, and 4.1–5.2% (average 4.9%) between species (Table 1). Nucleotide diversity (Nei & Jin, 1989), estimated using both UPGMA and NJ methods, was d=1.2 (SE 0.3) and 1.1 (SE 0.2) within populations of Siberian and European roe deer, respectively. Nucleotide diversity was d=4.9 (SE 0.7) between the two species.
The aligned sequences expressed significant phylogenetic signal (g1=−0.98; P<0.01). Using the fallow deer CR sequence as an outgroup, the two species of roe deer were separated at an observed 4.5% nucleotide divergence with 100% bootstrap support (Fig. 3). CR haplotypes clustered into two groups within each species. Bootstrap support was very high for the two population clusters in Siberian roe deer, and lower for the two clusters in European roe deer (Fig. 3). These clusters correspond to different geographical populations. Siberian roe deer sampled in the Kurgan region (cluster A) clustered apart from roe deer sampled in the Amur region (cluster B). The two groups of European roe deer closely reflect their geographical distributions and recent population histories. In fact, cluster C includes western populations, whereas cluster D includes all populations distributed in the eastern Alps and C. c. italicus (Fig. 1). Eastern populations represent those original Italian populations which have not been restocked, whereas the western ones were almost completely reintroduced. There were two exceptions: (i) haplotype EUR3.3 which was collected in eastern population 3, but groups with western haplotypes in cluster C; and (ii) haplotype EUR5.1, collected in western population 5, but grouping with eastern haplotypes in cluster D. The haplotype of C. c. italicus was included within cluster A, the presumed original Italian populations. We obtained 42 equivalent MP trees of length L=75, consistency index CI=0.64, homoplasy index HI=0.36 and retention index RI=0.88 (Swofford, 1993). The 50% majority-rule consensus of these 42 trees was identical to the NJ tree in Fig. 3, and the 21 CR haplotypes joined in the same four clusters (not shown).

Neighbour-joining tree describing the phylogenetic relationships among 21 mtDNA CR haplotypes of roe deer. Branch lengths are expressed as observed percentage nucleotide substitutions. Bootstrapped values based on 1000 replications are reported within boxes at internodes. Cluster A, western Siberia (Kurgan region); cluster B, eastern Siberia (Amur region); cluster C, western Italian Alps; cluster D, eastern Italian Alps and Capreolus capreolus italicus (haplotype EUR7.1).
None of the CR haplotypes was shared between species and among geographical populations. Exceptions were the already mentioned haplotype EUR3.3, an eastern sample which clusters with the western haplotypes, and EUR5.1, a western sample which clusters with the eastern haplotypes. These haplotypes are marked with asterisks in Fig. 3. The distribution of CR haplotypes among geographical regions was significantly different when analysed using AMOVA. A nested analysis showed that the smaller portion of the total molecular variance was distributed within populations (8.6%), whereas 24.7% and 66.7% were distributed among populations within species and between species, respectively (Table 2). The correlation of haplotypes among individuals within populations (0.91) was higher than the correlation among populations (0.74) and among species (0.67).
Discussion
Sequence diversity at the mtDNA CR was high within populations of Siberian and European roe deer. We have found six haplotypes among nine Siberian roe deer and 15 haplotypes among 21 European roe deer, including C. c. italicus. Haplotypic diversity was 0.93 in both species. Nucleotide diversity was 1.2% and 1.1% within populations of Siberian and European roe deer, respectively. Higher values of Nei's nucleotide diversity, ranging from 1.9% to 6.2%, were reported for 371 nucleotides within the CR left domain in five populations of Kenyan Grant's gazelle (Arctander et al., 1996). In contrast, 13 samples of C. c. italicus were monomorphic. These samples were collected from the roe deer population living in an area of pristine Mediterranean coastal forest in central Italy (Fig. 1). This population has a history of long-lasting isolation from other populations of roe deer, and survived at low numbers for generations. Population fragmentation, bottlenecks and isolation are possible causes of the erosion of gene diversity in C. c. italicus. Small populations of this subspecies live in two other areas in southern Italy (Fig. 1). They probably represent the survivors of roe deer populations isolated in southern refuge areas during the last Pleistocene glaciations, when the Alps were covered by ice and most of the central European plains were tundra and steppes (Roberts, 1989).
Nucleotide variability of CR sequences showed taxonomic and geographical structuring in roe deer. Genetic diversity and distances between the two species were four times higher than within species. CR haplotypes were not shared between species, suggesting a historical interruption of gene flow for a number of generations long enough for species to reach the stage of reciprocal monophyly (Avise et al., 1987). These findings can not exclude local introgression among parapatric populations of roe deer, which might be directly controlled by studying samples collected in areas of putative sympatry (the rivers Volga and lower Don in Russia). An enlarged sampling of geographical populations could estimate with more precision the extent of variability in both species. CR haplotypes clustered into two different groups within each species. Siberian roe sampled in west Siberia and the Amur region had different CR haplotypes and clustered into two different groups. European roe deer clustered according to their geographical locations: cluster D includes C. c. italicus and populations sampled in the eastern Alps, whereas cluster C includes only western Alpine and Apennine populations (Figs. 1 and Fig. 3). Eastern Alpine populations are natural, whereas western populations were reintroduced using stocks obtained from Slovenia, Croatia and the eastern Alps. Eastern roe deer haplotypes could therefore represent original Alpine haplotypes, whereas western haplotypes have various geographical origins. Haplotypes EUR3.3 and EUR5.1, which grouped into the ‘wrong’ clusters (Fig. 3), could represent mtDNA polymorphisms shared among populations, or individuals which were recently translocated for restocking purposes.
The fossil record and molecular calibrations suggest that the divergence between Odocoileinae and Cervinae can be placed at T=8–11 Myr ago (. Miyamoto et al., 1990)These dates can be used to estimate the rate of divergence of the CR conserved central blocks of Cervidae. Total sequence divergence, corrected for multiple hits using Tamura and Nei's γ distance with α=0.5, between Dama and Capreolus is d=0.131, corresponding to 0.008–0.006 substitutions/Myr accumulated during 8–11 Myr of independent evolution. Mean divergence rate r can be computed using d, and the formula: r= d/2 T (Ishida et al., 1995). Therefore, the distance d=0.05 between Siberian and European roe could have been generated in a time T= d/2 r=3.1–4.2 Myr. This date may be overestimated because of saturation of transitions during Dama/ Capreolus divergence. The mtDNA cytochrome b divergence between Siberian and European roe deer is about 0.04 (Randi et al. unpubl. data). Cytochrome b sequences diverged at a rate of 0.02 nucleotide substitutions/Myr in ungulates (Randi et al., 1996), at least for the first few million years before entering a zone of transitional saturation. This calibration leads to a divergence time of 2 Myr. These estimates concordantly suggest that roe deer speciated at the beginning of the Pleistocene, probably at the times of extinction of the ancestral Villafranchian species of Capreolus (about 2–3 Myr ago). In fact, fossil remains attributed to different species of Capreolus (C. crusafonti and C. suessenbornensis) have been found since the late Villafranchian, about 2 Myr ago, in central Europe (A. Azzaroli, pers. comm.).
A range of estimates of divergence rates has been published for the CR of different mammalian species. The rates of interspecific divergence ranged from 0.005 to 0.010/Myr for the entire CR of whales (Hoelzel et al., 1991), and were very similar to the rates we have obtained in this work using the central domains of cervids. Divergence rates were about four to seven times higher for the peripheral domains in whales (Hoelzel et al., 1991) and Equidae (Ishida et al., 1995), as well as among human sequences (values ranging from 0.042 to 0.087/Myr; Vigilant et al., 1991; data corrected by Stewart & Baker, 1994), and among shrew sequences (0.083–0.143/Myr; Stewart & Baker, 1994). Estimated substitution differences for domain I were 0.030 and 0.024 in Siberian and European roe deer, respectively. Assuming a rate of 0.04–0.08 substitutions/Myr in the peripheral domains, the divergence among CR haplotypes might have been generated in 0.37–0.19 Myr for Siberian, and in 0.30–0.15 Myr for European roe deer.
The peripheral domains of CR of Capreolus evolve at substantial rates and provide high levels of intraspecific genetic divergence, which can be used to assess population differences and geographical structuring. Sudden and highly significant differences of haplotype sequence divergence and distribution among the two studied Siberian roe deer populations suggest that this species is fragmented into local demes, which could evolve with some degree of isolation. Further investigations on phylogeographical structure and rates of gene flow among undisturbed roe deer populations will produce the information necessary to preserve their biological diversity within the background of their recent history and evolution. Divergence and distribution of CR haplotypes were significantly different also on the smaller geographical scale occupied by the Alpine roe deer populations in Italy. Autochthonous and translocated populations have distinct haplotypes which cluster separately. Sequence divergence among the two groups of haplotypes should make it possible to identify the origins and recent history of the translocated populations. The genetic uniqueness of the southern C. c. italicus calls for careful conservation of its last three surviving populations.
References
Anderson, S., Debruijn, M. H. L., Coulson, A. R., Eperon, I. C., Sanger, F. and Young, Y. G. (1982). Complete sequence of bovine mitochondrial DNA: conserved features of the mammalian mitochondrial genome. J Mol Biol, 156: 683–717.
Arctander, P., Kat, P. W., Aman, R. A. and Siegismund, H. R. (1996). Extreme genetic differences among populations of Gazella granti Grant's gazelle, in Kenya. Heredity, 76: 465–475.
Arnason, U., Gullberg, A. and Widegren, B. (1993). Cetacean mitochondrial DNA control region: sequences of all extant baleen whales and two sperm whale species. Mol Biol Evol, 10: 960–970.
Avise, J. C., Arnold, J., Ball, R. M. J. R., Bermingham, E., Lamb, T. and Neigel, J. E. (1987). et al. Intraspecific phylogeography: the mitochondrial DNA bridge between population genetics and systematics. Ann Rev Ecol Syst, 18: 489–522.
Cockerham, C. C. (1973). Analyses of gene frequencies. Genetics, 74: 679–700.
Danilkin, A. (1996). Behavioural Ecology of Siberian and European Roe Deer. Chapman and Hall, London.
Excoffier, L., Smouse, P. E. and Quattro, J. M. (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: applications to human mitochondrial DNA restriction data. Genetics, 131: 479–491.
Festa, A. (1925). Il capriolo dell'Italia centrale. Boll Mus Zool Anat Comp Torino, 40: 1–2.
Groves, C. P. and Grubb, P. (1987). Relationships of living deer. In: Wemmer, C. M. (ed.) Biology and Management of the Cervidae, pp. 21–59. Smithsonian Institution Press, Washington.
Grubb, P. (1993). Order Artiodactyla. In: Wilson, D. E. and Reeder, D. M. (eds). Mammal Species of the World. A Taxonomic and Geographic Reference, pp. 377–414. Smithsonian Institution Press, Washington.
Hartl, G. B. and Reimoser, F. (1988). Biochemical variation in roe deer (Capreolus capreolus L.): are r-strategists among deer genetically less variable than K-strategists? Heredity, 60: 221–227.
Heptner, V. G., Nasimovich, A. A. and Bannikov, A. G. (1989). Mammals of the Soviet Union Ungulates. vol. I. E. J. Brill, Leiden.
Hillis, D. M. and Huelsenbeck, J. P. (1992). Signal, noise, and reliability in molecular phylogenetic analyses. J Hered, 83: 89–195.
Hoelzel, R. A., Hancock, J. M. and Dover, G. A. (1991). Evolution of the cetacean mitochondrial d-loop region. Mol Biol Evol, 8: 475–493.
Hoelzel, R. A., Lopez, J. V., Dover, G. and O'brien, S. J. (1994). Rapid evolution of a heteroplasmic repetitive sequence in the mitochondrial DNA control region of carnivores. J Mol Evol, 39: 191–199.
Ishida, N., Oyunsuren, T., Mashima, S., Mukoyama, H. and Andsaitou, N. (1995). Mitochondrial DNA sequences of various species of the genus Equus with special reference to the phylogenetic relationship between Przewalskii's wild horse and domestic horse. J Mol Evol, 41: 180–188.
Jäger, F., Hecht, W. and Herzog, A. (1992). Untersuchungen an mitochondrialer DNS (mtDNS) von hessischem Rehwild (C. capreolus). Z Jagdwiss, 38: 26–33.
Kumar, S., Tamura, K. and Nei, M. (1993). MEGA. Molecular evolutionary genetic analysis. Version 1.0.Pennsylvania State University, University Park, PA.
Miyamoto, M. M., Kraus, F. and Ryder, O. A. (1990). Phylogeny and evolution of antlered deer determined from mitochondrial DNA sequences. Proc Natl Acad Sci USA, 87: 6127–6131.
Moritz, C., Dowling, T. E. and Brown, W. M. (1987). Evolution of animal mitochondrial DNA: relevance for population biology and systematics. Ann Rev Ecol Syst, 18: 269–292.
Nei, M. and Jin, L. (1989). Variances of the average numbers of nucleotide substitutions within and between populations. Mol Biol Evol, 6: 290–300.
Randi, E., Lucchini, V. and Diong, C. H. (1996). Evolutionary genetics of the Suiformes as reconstructed using mtDNA sequencing. J Mamm Evol, 3: 163–194.
Roberts, N. (1989). The Holocene: An Environmental History. Basil Blackwell, Oxford.
Sokolov, V. E. and Gromov, V. S. (1990). The contemporary ideas on roe deer (Capreolus Gray, 1821) systematization: morphological, ethological and hybridological analysis. Mammalia, 54: 431–444.
Stewart, D. T. and Baker, A. J. (1994). Patterns of sequence variation in the mitochondrial D-loop region of shrews. Mol Biol Evol, 11: 9–21.
Swofford, D. L. (1993). PAUP. Phylogenetic analysis using parsimony Version 3.1. Illinois Natural History Survey, Champaign, IL.
Tamura, K. and Nei, M. (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol, 10: 512–526.
Thompson, J. D., Higgins, D. G. and Gibson, T. J. (1994). CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalities and weight matrix choice. Nucl Acid Res, 22: 4673–4680.
Vigilant, L., Stoneking, M., Harpending, H., Hawkes, K. and Andwilson, A. C. (1991). African populations and the evolution of human mitochondrial DNA. Science, 253: 1503–1507.
Wenink, P. W., Baker, A., Rosner, H. U. and Tilanus, M. G. J. (1996). Global mitochondrial DNA phylogeography of Holarctic breeding dunlins (Calidris alpina). Evolution, 50: 318–330.
Acknowledgements
We to thank F. Perco (Trieste), A. P. Rizzoli and I. Cattadori (Trento), M. Paganin (Asiago), P. Durio and G. Meneguz (Torino), S. Spanó (Genova), A. Tinelli (Roma) and J. Darman (Amur district) for collecting roe deer samples. A. Azzaroli updated our information on fossil Odocoileini and Capreolus. V. Lucchini, N. Mucci and E. Maiolini provided assistance during laboratory work. Computer programs were kindly supplied by D. G. Higgins (CLUSTAL W), L. Jin (SEND.FOR) and L. Excoffier (AMOVA).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Randi, E., Pierpaoli, M. & Danilkin, A. Mitochondrial DNA polymorphism in populations of Siberian and European roe deer (Capreolus pygargus and C. capreolus). Heredity 80, 429–437 (1998). https://doi.org/10.1046/j.1365-2540.1998.00318.x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1046/j.1365-2540.1998.00318.x
Keywords
This article is cited by
-
Phylogenetic divergence associated with climate oscillations and topology illustrates the dispersal history of Formosan sambar deer (Rusa unicolor swinhoii) in Taiwan
Mammal Research (2023)
-
Understanding the diversification pattern of three subspecies of swamp deer (Rucervus duvaucelii) during the Pleistocene–Holocene based on mitochondrial and Y chromosome markers
Mammalian Biology (2021)
-
Livestock grazing affects movements and activity pattern of Italian roe deer in Southern Italy
European Journal of Wildlife Research (2021)
-
Strong population structure in a species manipulated by humans since the Neolithic: the European fallow deer (Dama dama dama)
Heredity (2017)
-
Linking annual variations of roe deer bag records to large-scale winter conditions: spatio-temporal development in Europe between 1961 and 2013
European Journal of Wildlife Research (2017)
