
Abstract
The genetic relationships between the Pacific and the Atlantic populations of marine coastal biota in Southern South America have been analyzed in few studies, most of them relying on a single mitochondrial locus. We analyzed 10 polymorphic microsatellite loci, isolated from a dinucleotide-enriched Eleginops maclovinus genomic library, in a total of 240 individuals (48 from each of 5 sampled sites: 2 Atlantic, 2 Pacific and 1 in Beagle Channel). The results were contrasted against a previous work on the same species with mitochondrial DNA (mtDNA). Observed heterozygosity within localities ranged from 0.85 to 0.88 with the highest overall number of alleles observed at the northernmost locality on the Pacific side (Concepción), but no clear geographic pattern arose from the data. On the other hand, the number of private alleles was negatively correlated with latitude (Spearman’s rs test, P=0.017). Among-population variance was low but significant (1.35%; P<0.0001, analysis of molecular variance (AMOVA)) and low genetic differentiation between populations was observed (pairwise FST values ranged from 0 to 0.021). A Mantel test revealed a significant correlation between geographic distances and FST (r=0.56, P=0.047). This could be partially accounted by the Atlantic versus Pacific population differentiation detected in three different analyses (STRUCTURE, SAMOVA (Spatial Analysis of MOlecular VAriance) and a population phylogeny). The observed pattern is compatible with a history of separation into two glacial refugia that was better captured by the multilocus microsatellite data than by the mtDNA analysis.
Similar content being viewed by others


Introduction
Excluding Antarctica, South America is the only southern continent that extends significantly beyond 40°S. It thus represents a suitable area to study the biogeographic consequences of Quaternary glacial cycles in the Southern Hemisphere (Lessa et al., 2010; Sérsic et al., 2011). In this region, the long glacial climatic episodes allowed the formation of a single continuous ice sheet that almost completely covered the Patagonian Andean ranges and extended over the piedmont areas to the east and to sea level on the Pacific side. The continental shelf area was largely affected by events such as ice sheet calving into the ocean, retraction of the sea coast line and decrease in marine water temperature (Clapperton, 1993; Ponce et al., 2011; Rabassa et al., 2011).
A common expectation under this scenario is that the species affected by glaciations had retracted to lower latitudes during the glacial phases and then expanded poleward during interglacial periods (Knutsen et al., 2003; Hewitt, 2004; Maggs et al., 2008; Wilson and Eigenmann Veraguth, 2010; Provan, 2013). In the case of the coastal marine biota that inhabit the southern tip of South America, this general expectation would lead to a rather simple biogeographic scenario in comparison with the Northern Hemisphere ones, as the Atlantic and the Pacific coast of Southern South America converge at the southern tip in the archipelago of Tierra del Fuego. In this scenario, refugia during glaciations may have been located in the Pacific Ocean, the Atlantic Ocean or both, creating, in this last case, an opportunity for geographic isolation and genetic differentiation.
In biogeographic terms, the cold temperate waters around Southern South America were previously considered one entity, the Magellan Province, but currently some authors subdivide this Province into at least four biogeographic provinces: Southern Chile, Tierra del Fuego, Southern Argentina and Malvinas/Falklands (see Briggs and Bowen, 2012). Several marine coastal species are distributed across these four biogeographic provinces, but only a few of them had been subjected to population genetic studies. Most of these studies had been restricted to the Pacific or the Atlantic Ocean only and/or to a single molecular marker, usually a mitochondrial locus (Cárdenas et al., 2009; Fraser et al., 2010; González-Wevar et al., 2012; de Aranzamendi et al., 2014; Hüne et al., 2014; Nuñez et al., 2015). Contrastingly, many more phylogeographic studies have addressed the genetic consequences of Quaternary glacial cycles in the coastal marine fauna of the Northern Hemisphere (reviewed in Provan, 2013).
In a previous study, we analyzed the phylogeography of Eleginops maclovinus known as róbalo in Argentina and Chile, using a mitochondrial marker (Ceballos et al., 2012). This species, endemic to Southern South America (see Figure 1), is a coastal fish that belongs to the monotypic family Eleginopidae, considered the sister taxon of the Antarctic notothenioid clade (Matschiner et al., 2011; Near et al., 2012). On the Patagonian coast, E. maclovinus is a common fish species and an important component of many trophic webs, both as prey (Goodall and Galeazzi, 1985) and as predator, feeding mainly on benthic invertebrates such as crustaceans and polychaetes, and also on algae and fishes (Licandeo et al., 2006; Pequeño et al., 2010). Adults reach a maximum length of 80/90 cm (Brickle et al., 2005a; Licandeo et al., 2006) and are highly mobile, at least in aquaria (personal observations). E. maclovinus has been described as a protandrous hermaphrodite (Calvo et al., 1992; Brickle et al., 2005b; Licandeo et al., 2006) and is characterized by high fecundity and small pelagic eggs (Brickle et al., 2005b). Little is known about larval development of E. maclovinus, although it likely has extended pelagic phases, as found in many other notothenioid fishes. Thus, long-distance larval dispersal by marine currents would be likely, as has been suggested for a number of other notothenioid species in which population genetic studies commonly found nonsignificant differentiation across thousands of km (Matschiner et al., 2009; Damerau et al., 2012).

E. maclovinus were collected at five sites (circles) along the coast of Southern South America. The approximated range distribution of the species is represented in the map by the shadow coastal area. ‘PA’ indicates the number of private alleles from the nine microsatellite loci.
In Ceballos et al. (2012), we used mitochondrial DNA (mtDNA) sequences of 833 base pairs in length of cytochrome b from 261 individuals of E. maclovinus belonging to 9 localities distributed along the entire latitudinal range of the species. We found a weak geographic structure and a pattern of population expansion dated to be older than the Last Glacial Maximum (LGM). We suggested that this could be the consequence of a population expansion from a single refugium, probably located at relative low latitude on the Atlantic Ocean side. However, a study based on a single locus cannot accurately reflect the history of a species, and makes it nearly impossible to distinguish the relative impact of demographic and selective processes. We now reconsidered this scenario on the basis of studying nine microsatellite loci across the same geographic range of the species.
This is the first multilocus analysis on a coastal marine fish along both the Atlantic and the Pacific coasts of the southern tip of South America.
Materials and methods
Sample collection, DNA extraction and microsatellite amplification
E. maclovinus individuals were captured using trammel and gill nets at five sites along the Atlantic and Pacific Patagonian coasts: San Antonio Oeste (SAO) (40°50'S, 65°04'W), Rada Tilly (RT; 45°56'S, 67°32'W), Beagle Channel (BC; 54°49'S 68°10'W), Puerto Aysén (AY; 45°22'S, 72°51'W) and Concepción (CC; 36°44'S, 73°11'W) (Figure 1). Muscle samples were collected from each individual and preserved in 99% ethanol. Total DNA extractions were performed with a sodium dodecyl sulfate–proteinase K–NaCl–alcohol precipitation method modified from Miller et al. (1988).
Ten polymorphic microsatellite loci, isolated from a dinucleotide-enriched E. maclovinus genomic library, were surveyed in a total of 240 individuals (48 from each of the 5 sampled sites). Individuals from SAO are the same that were analyzed in Ceballos et al. (2011), where the protocol for the isolation of the microsatellite loci, PCR conditions and primers used are outlined. One locus (EmE9) and one individual from Puerto Aysén were not included in subsequent analyses because of amplification problems. Therefore, the final data set consisted of nine loci (EmA4, EmA5, EmC4, EmC11, EmE7, EmF6, EmG6, EmH9 and EmH10) and 239 individuals. The genotypes were determined using Genescan Service by Macrogen Korea (Seoul, Korea) and scored using the software Peak Scanner 1.0 (Applied Biosystems, Seoul, Korea). Allele binning was performed by overlapping all alleles from each locus and choosing one as reference. The fragment length of the reference was determined by rounding to the nearest number. The distance from the reference and the repeat pattern was then considered to define all other allele sizes.
Data analysis
The software Arlequin v3.11 (Excoffier et al., 2005) was used to perform the following calculations: number of alleles, observed and expected heterozygosity, tests for Hardy–Weinberg equilibrium, linkage disequilibrium, analysis of molecular variance (AMOVA), pairwise FST and Mantel test. The software MICRO-CHECKER 2.2.3 (Van Oosterhout et al., 2004) was used to detect evidence for scoring errors due to stuttering, large allele dropout or null alleles. The frequency of null alleles was estimated by maximum likelihood using the software ML-Null (Kalinowski and Taper, 2006). Confidence intervals for expected heterozygosity in each sampled locality were calculated using the package PopGenKit of R (R Core Team, 2013).
Population structure was explored using the Bayesian method of Pritchard et al. (2000) implemented in the software STRUCTURE 2.3.3 (Oxford, UK). Runs were performed with number of clusters (K) fixed between 1 and 5 using an admixture model with sample group information (Hubisz et al., 2009), correlated allele frequencies and presence of null alleles (Falush et al., 2007). Six independent runs were conducted for each K value. Preliminary runs showed that convergence was achieved after 50 000 iterations. Thus, this was used as burn-in and the estimations were based on 100 000 additional iterations. We inferred the K values that best captured the structure of the data by plotting the ‘Ln Probability of Data’ against K as suggested by the software developers. In addition, we implemented the method introduced by Evanno et al. (2005) to detect the optimal K using the software CLUMPAK (Kopelman et al., 2015).
Phylogenetic relationships between populations were assessed using the genetic distance DA (Nei et al., 1983) and the methods Neighbor Joining and UPGMA as implemented in the software POPTREE2 (Takezaki et al., 2010).
The program SAMOVA 1.0 (Spatial Analysis of MOlecular VAriance) was implemented to define groups of populations that are geographically 'homogeneous and maximally differentiated from each other (Dupanloup et al., 2002). Runs were performed with two groups of populations and 100 simulated annealing processes.
Migration direction (between SAMOVA-defined groups) was evaluated using MIGRATE 3.3.2 (Beerli, 2009). The Bayesian inference using Markov chain Monte Carlo was implemented with a search strategy consisted of 10 replicates with a heating scheme of four chains with different temperatures. Each chain consisted of a burn-in of 500 000 generations and 3 000 000 subsequent generations, from which a total of 30 000 steps were recorded by sampling every 100 generations. This searching scheme ensured an effective sample size of each parameter of at least 48 000.
Results
Microsatellite loci variability
Altogether, 239 individual fish were genotyped at 9 microsatellite loci. Genetic variability varied among loci, with number of alleles ranging from 8 (at locus EmF6) to 62 (EmH9) with an average of 33, and observed heterozygosity (averaged over samples) ranging from 0.57 (EmF6) to 0.93 (EmH9) (see Supplementary Table 1). No significant evidence of genetic linkage was detected after sequential Bonferroni correction for multiple comparisons. Significant deviation from Hardy–Weinberg equilibrium, evaluated at each locality and locus, was observed in 6 of the 45 tests performed (9 loci × 5 localities; Table 1). The software MICRO-CHECKER 2.2.3 (Van Oosterhout et al., 2004) found no evidence for scoring error due to stuttering or large allele dropout but detected the possible presence of null alleles at those loci that deviated significantly from Hardy–Weinberg equilibrium. The highest deviations were observed at locus EmF6 with an estimated frequency of null alleles ranging from 0.11 to 0.21 at 4 of the 5 sampled localities. Therefore, all subsequent analyses performed were duplicated, with and without locus EmF6. As no substantial differences were observed in runs with or without EmF6, we report only analyses including this locus.
Variation within localities
Observed heterozygosity within localities (averaged over loci) ranged from 0.85 to 0.88, whereas expected heterozygosity ranged from 0.87 to 0.91 (Table 2). Estimates of heterozygosity were very similar across localities. The highest overall allele number was observed at the northernmost locality on the Pacific side (Concepción) but no clear geographic pattern arose from the data (Table 2). On the other hand, the number of private alleles was negatively correlated with latitude (Table 2 and Figure 1; nonparametric Spearman’s rs test, P=0.017).
Geographic structure and gene flow
Among-population variance was low but significant (1.35%; P<0.0001, AMOVA). Pairwise FST values ranged from 0 to 0.019 (Table 3), indicating low genetic differentiation between populations (Hartl and Clark, 2007). However, the Mantel test revealed a significant correlation between geographic distances and FST (r=0.56, P=0.047; Figure 2).

Relationship between genetic differentiation (FST) and geographic distance (km). The Mantel test revealed a significant correlation (r=0.56, P=0.047). Black circles are statistically significant comparisons.
In the analysis with STRUCTURE, the value of K (number of genetic clusters) that best captured the structure of the data was K=3, as suggested by the plot of the ‘Ln Probability of Data’ (Supplementary Figure 1) and the method introduced by Evanno et al. (2005). However, also looking at K=2 allowed us to detect the uppermost hierarchical level of population structure (Figure 3). All runs at K=2 and K=3 produced identical clustering solutions with similar values of cluster membership (Q) for all individuals. Both values of K allowed the distinction of two main groups: one including the populations in/of the Pacific Ocean (CC and AY) plus the southernmost population (BC) and the other group including the two populations in/of the Atlantic Ocean (SAO and RT). When assuming K=2 individuals from CC, AY and BC showed a probability of membership corresponding almost exclusively to one cluster, whereas individuals from SAO and RT showed a mixed probability of membership from the two clusters. SAO and RT also differed from each other as most individuals from SAO showed a higher probability of membership to the cluster virtually absent in CC, AY and BC. Assuming K=3 allowed further differentiation between CC, AY and BC, revealing hierarchical structure (see Figure 3). SAMOVA analysis recovered an arrangement of populations similar to the STRUCTURE results, although a low percentage of variation between groups was found (0.85%, P=0.11; Table 4). In addition, the phylogenetic relationship between populations also separated CC, AY and BC in one clade and SAO and RT in another clade (Figure 4).

Analysis of population differentiation of E. maclovinus from five sampled localities using the software STRUCTURE. A total of 239 are represented by a vertical bar and colors indicate individual membership probabilities to belong to one of the inferred clusters (K). AY, Puerto Aysén; BC, Beagle Channel; CC, Concepción; RT, Rada Tilly; SAO, San Antonio Oeste. A full color version of this figure is available at the Heredity journal online.

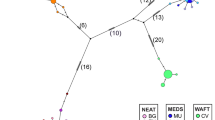
Phylogenetic relationship between populations inferred from the genetic distance DA (Nei et al., 1983). Phylogenetic methods: (a) Neighbor Joining; (b) UPGMA. Values on nodes are bootstrap support. AY, Puerto Aysén; BC, Beagle Channel; CC, Concepción; RT, Rada Tilly; SAO, San Antonio Oeste.
According to these results, we recognized two groups: (1) ‘Pacific’ that includes CC, AY and BC and (2) ‘Atlantic’ that includes SAO and RT. Migration rates between these groups, as assessed with MIGRATE, showed evidence of asymmetrical gene flow, with 98.1 estimated migrants per generation from Pacific to Atlantic and only 6.8 in the opposite direction.
Discussion
The results are in general agreement with the idea of lower latitude populations being less affected by Pleistocene glaciations than higher latitude populations. The main evidence for this is the latitudinal variation in the number of private alleles found at both the Atlantic and the Pacific coasts, with northern populations holding higher numbers of private alleles. This might suggest that northern populations would have been the source from which the southern populations originated (following a classical interpretation of the number of private alleles; see for example Bowcock et al., 1994). The geographic structure found could be interpreted under this scenario as well: if populations experienced a northward retraction during glaciations, then at least two separate refugia might have remained, one in the Atlantic Ocean and one in the Pacific Ocean, allowing differentiation in allopatric conditions that would explain the two groups recovered by SAMOVA and STRUCTURE (Table 4 and Figure 3), as well as the two clades shown by the population phylogenetic analysis (Figure 4). The asymmetrical pattern of migration rate estimated between these two groups could be attributed to higher subsequent gene flow from the Pacific to the Atlantic, in line with the west-to-east direction of the main currents at the southernmost tip of South America (Antezana, 1999; Miloslavich et al., 2011). This mostly unidirectional gene flow estimated with MIGRATE is also supported by STRUCTURE for K=2, as the Pacific group appears more homogeneous than the Atlantic group. Note also that only the pairwise FST comparisons within the Pacific group are nonsignificant. In concordance, two other studies carried out in the same region with mtDNA in the limpets Nacella magellanica (González-Wevar et al., 2012) and Siphonaria lessoni (Nuñez et al., 2015) also found a dominant pattern of gene flow from west to east.
In contrast, our previous work with a mtDNA marker (Ceballos et al., 2012) did not find any evidence for the structuring of genetic variation into an Atlantic and a Pacific group. Furthermore, the pattern compatible with a recent population expansion found in Ceballos et al. (2012) was likely generated by an expansion from a single refugium. This conclusion was mainly supported by the low geographic genetic structure, the unimodal global mismatch distribution as well as the negative and significant values of global neutrality tests (Ceballos et al., 2012). On the other hand, both mtDNA and microsatellites indicate that the fraction of genetic variance partitioned among populations is relatively low (1.62% and 1.35%, respectively). How can the data be reconciled to provide a cohesive scenario? We advance the following considerations. (1) Genetic structure: taking into account that all data point to limited divergence, it seems that a single mitochondrial locus did not allow us to recover significant subdivision into geographic regions that is achieved with the multilocus microsatellite data. (2) Timing of expansion: the start of the putative population expansion obtained by Ceballos et al. (2012) with the mitochondrial locus was estimated to have been about 125 kya, that is, long before the LGM (ca. 21 kya). This estimation was based on a mutation rate obtained from a notothenioid phylogeny. In addition to the general uncertainties tied to these calibrations, many-fold discrepancies have been reported between short-term population-level mutation rates and phylogenetic rates of substitution. The long-term substitution rate between species could be an order of magnitude lower than the molecular evolutionary rate operating in the time frame of ‘recent’ demographic processes within species (see Ho et al., 2011 for a review). In addition, populations with secondary contact that likely arose from two refugial sources coalescing into a single unit would also lead to overestimating the timing of expansion. Accounting for these two factors, it seems reasonable that the onset of the population expansion of E. maclovinus would have been after the LGM.
After these considerations we propose the following scenario in order to reconcile the results of our mitochondrial and nuclear studies: the results of the present study, which show the existence of one Pacific and one Atlantic group, rule out our previous hypothesis of one refugial population detected by a mtDNA marker (Ceballos et al., 2012) that is probably was not sensitive enough to detect the two groups. The apparent expansion pattern from single refugia observed with mtDNA could be the consequence of the expansion of one or a few formerly widespread ancestral haplotypes that persisted within two relatively large refugia. It seems therefore that a history of separation into two glacial refugia, probably during last glaciation, is best captured by the multilocus microsatellite data. However, as in any single locus analysis, we cannot rule out the possibility that the mitochondrial pattern reflects the action of natural selection on the mitochondrial genome.
Our results highlight the importance of multilocus approaches for resolving postglacial phylogeography, as has also been demonstrated for coastal species in the Northern Hemisphere (Wilson and Eigenmann Veraguth, 2010). We also contributed evidence of the role that past glacial cycles have had in the biodiversity of the region by allowing differentiation in allopatry within a species that now has a continuous distribution. In the case of E. maclovinus the achieved differentiation between putative refugia was low, probably because of relatively large population size and limited time in isolation. In other species with narrower environmental tolerance—which would have led to smaller population size—and/or lower dispersal ability, stronger genetic differentiation or even speciation could have been achieved. Similarly, other species could have persisted in the region during the LGM as has been proposed for some coastal species in the Northern Hemisphere (Marko et al., 2010). Unfortunately, most previous studies investigating postglacial phylogeography in coastal taxa in Patagonia, especially those involving both the Pacific and the Atlantic coast, have relied exclusively on the pattern of variation at a single mitochondrial locus. Most of them found a pattern of population expansion similar to the one detected with mtDNA in E. maclovinus. Common features are shallow geographic structure, star-like haplotype network, unimodal mismatch distribution and lack of a pattern of isolation by distance (Cárdenas et al., 2009; de Aranzamendi et al., 2011; González-Wevar et al., 2012; Ocampo et al., 2013; Hüne et al., 2014; Pérez-Barros et al., 2014). However, some species show signs of recent population expansion, also in conjunction with geographic breaks (Fraser et al., 2010; Sánchez et al., 2011; Nuñez et al., 2015), highlighting the great variance in species’ abilities to respond to the environmental change associated with glacial cycles.
To better understand the paleobiogeographical history of Southern South America, additional multilocus studies on other marine coastal species are required. Future efforts, in combination with the more abundant studies in marine biogeography that have already been carried out in the Northern Hemisphere (Marko et al., 2010), would provide a potentially useful global model for understanding how species will respond to climate change in the future.
In summary, our results suggest that two population refugia may have remained during last glaciation, one in the Pacific Ocean and the other in the Atlantic Ocean, and highlight the importance of multilocus approaches for resolving postglacial phylogeography.
Data archiving
Data available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.0pf75.
References
Antezana T . (1999). Hydrographic features of Magellan and Fuegian inland passages and adjacent Subantarctic waters. Sci Mar 63: 23–34.
Beerli P . (2009) How to use MIGRATE or why are Markov chain Monte Carlo programs difficult to use. In: Bertorelle G, Bruford MW, Hauffe HC, Rizzoli A, Vernesi C (eds). Population Genetics for Animal Conservation, volume 17 of Conservation Biology. Cambridge University Press: Cambridge UK. pp 42–79.
Bowcock AM, Ruiz-Linares A, Tomfohrde J, Minch E, Kidd JR, Cavalli-Sforza LL . (1994). High resolution of human evolutionary trees with polymorphic microsatellites. Nature 368: 455–457.
Brickle P, Arkhipkin AI, Shcherbich ZN . (2005a). Age and growth in a temperate euryhaline notothenioid, Eleginops maclovinus from the Falkland Islands. J Mar Biol Assoc UK 85: 1217.
Brickle P, Laptikhovsky V, Arkhipkin A . (2005b). Reproductive strategy of a primitive temperate notothenioid Eleginops maclovinus. J Fish Biol 66: 1044–1059.
Briggs JC, Bowen BW . (2012). A realignment of marine biogeographic provinces with particular reference to fish distributions. J Biogeogr 39: 12–30.
Calvo J, Morriconi E, Rae GA, San Roman NA . (1992). Evidence of protandry in a subantarctic notothenid, Eleginops maclovinus (Cuv. & Val., 1830) from the Beagle Channel, Argentina. J Fish Biol 40: 157–164.
Cárdenas L, Castilla JC, Viard F . (2009). A phylogeographical analysis across three biogeographical provinces of the south-eastern Pacific: the case of the marine gastropod Concholepas concholepas. J Biogeogr 36: 969–981.
Ceballos SG, Lessa EP, Victorio MF, Fernández DA . (2012). Phylogeography of the sub-Antarctic notothenioid fish Eleginops maclovinus: evidence of population expansion. Mar Biol 159: 499–505.
Ceballos SG, Victorio MF, Lyra ML, Fernández DA . (2011). Isolation and characterization of ten microsatellite loci from the Patagonian notothenioid fish Eleginops maclovinus. Conserv Genet Resour 3: 689–691.
Clapperton CM . (1993) Quaternary Geology and Geomorphology of South America. Elsevier: Amsterdam.
Damerau M, Matschiner M, Salzburger W, Hanel R . (2012). Comparative population genetics of seven notothenioid fish species reveals high levels of gene flow along ocean currents in the southern Scotia Arc, Antarctica. Polar Biol 35: 1073–1086.
de Aranzamendi MC, Bastida R, Gardenal CN . (2011). Different evolutionary histories in two sympatric limpets of the genus Nacella (Patellogastropoda) in the South-western Atlantic coast. Mar Biol 158: 2405–2418.
de Aranzamendi MC, Bastida R, Gardenal CN . (2014). Genetic population structure in Nacella magellanica: Evidence of rapid range expansion throughout the entire species distribution on the Atlantic coast. J Exp Mar Bio Ecol 460: 53–61.
Dupanloup I, Schneider S, Excoffier L . (2002). A simulated annealing approach to define the genetic structure of populations. Mol Ecol 11: 2571–2581.
Evanno G, Regnaut S, Goudet J . (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14: 2611–2620.
Excoffier L, Laval G, Schneider S . (2005). Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online 1: 47–50.
Falush D, Stephens M, Pritchard JK . (2007). Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes 7: 574–578.
Fraser CI, Thiel M, Spencer HG, Waters JM . (2010). Contemporary habitat discontinuity and historic glacial ice drive genetic divergence in Chilean kelp. BMC Evol Biol 10: 203.
González-Wevar Ca, Hüne M, Cañete JI, Mansilla A, Nakano T, Poulin E . (2012). Towards a model of postglacial biogeography in shallow marine species along the Patagonian Province: lessons from the limpet Nacella magellanica (Gmelin, 1791). BMC Evol Biol 12: 139.
Goodall R, Galeazzi A . (1985) A Review of the Food Habits of the Small Cetaceans of the Antarctic and Sub-Antarctic. In: Siegfried WR, Condy PR, Laws RM (eds). Antarctic Nutrient Cycles and Food Webs. Springer-Verlag: Berlin, Heidelberg, pp 566–572.
Hartl D, Clark A . (2007) Principles of Population Genetics, 4th edn. Sinauer: Sunderland, MA.
Hewitt GM . (2004). Genetic consequences of climatic oscillations in the Quaternary. Philos Trans R Soc Lond B Biol Sci 359: 183–195.
Ho SYW, Lanfear R, Bromham L, Phillips MJ, Soubrier J, Rodrigo AG et al. (2011). Time-dependent rates of molecular evolution. Mol Ecol 20: 3087–3101.
Hubisz MJ, Falush D, Stephens M, Pritchard JK . (2009). Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9: 1322–1332.
Hüne M, González-Wevar C, Poulin E, Mansilla A, Fernández DA, Barrera-Oro E . (2014). Low level of genetic divergence between Harpagifer fish species (Perciformes: Notothenioidei) suggests a Quaternary colonization of Patagonia from the Antarctic Peninsula. Polar Biol 38: 607–617.
Kalinowski ST, Taper ML . (2006). Maximum likelihood estimation of the frequency of null alleles at microsatellite loci. Conserv Genet 7: 991–995.
Knutsen H, Jorde PE, André C, Stenseth NC . (2003). Fine-scaled geographical population structuring in a highly mobile marine species: the Atlantic cod. Mol Ecol 12: 385–394.
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I . (2015). Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15: 1179–1191.
Lessa EP, D’Elía G, Pardiñas UFJ . (2010). Genetic footprints of late Quaternary climate change in the diversity of Patagonian-Fueguian rodents. Mol Ecol 19: 3031–3037.
Licandeo RR, Barrientos CA, González MT . (2006). Age, growth rates, sex change and feeding habits of notothenioid fish Eleginops maclovinus from the central-southern Chilean coast. Environ Biol Fishes 77: 51–61.
Maggs CA, Castilho R, Foltz D, Henzler C, Jolly MT, Kelly J et al. (2008). Evaluating signatures of glacial refugia for North Atlantic benthic marine taxa. Ecology 89: 108–122.
Marko PB, Hoffman JM, Emme Sa, McGovern TM, Keever CC, Nicole Cox L . (2010). The ‘expansion-Contraction’ model of Pleistocene biogeography: rocky shores suffer a sea change? Mol Ecol 19: 146–169.
Matschiner M, Hanel R, Salzburger W . (2009). Gene flow by larval dispersal in the Antarctic notothenioid fish Gobionotothen gibberifrons. Mol Ecol 18: 2574–2587.
Matschiner M, Hanel R, Salzburger W . (2011). On the origin and trigger of the notothenioid adaptive radiation. PLoS One 6: e18911.
Miller S, Dikes D, Polesky H . (1988). A simple salting procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16: 215.
Miloslavich P, Klein E, Díaz JM, Hernández CE, Bigatti G, Campos L et al. (2011). Marine biodiversity in the Atlantic and Pacific coasts of South America: knowledge and gaps. PLoS One 6: e14631.
Near TJ, Dornburg A, Kuhn KL, Eastman JT, Pennington JN, Patarnello T et al. (2012). Ancient climate change, antifreeze, and the evolutionary diversification of Antarctic fishes. Proc Natl Acad Sci USA 109: 3434–3439.
Nei M, Tajima F, Tateno Y . (1983). Accuracy of estimated phylogenetic trees from molecular data. J Mol Evol 19: 153–170.
Nuñez JD, Fernández Iriarte PJ, Ocampo EH, Iudica C, Cledón M . (2015). Deep phylogeographic divergence among populations of limpet Siphonaria lessoni on the east and west coasts of South America. Mar Biol 162: 595–605.
Ocampo EH, Robles R, Terossi M, Nuñez JD, Cledón M, Mantelatto FL . (2013). Phylogeny, phylogeography, and systematics of the American pea crab genus calyptraeotherescampos, 1990, inferred from molecular markers. Zool J Linn Soc 169: 27–42.
Pequeño G, Pavés H, Bertrán C, Vargas-Chacoff L . (2010). Seasonal limnetic feeding regime of the robalo Eleginops maclovinus (Valenciennes 1830), in the Valdivia river, Chile. Gayana 74: 47–56.
Pérez-Barros P, Lovrich GA, Calcagno JA, Confalonieri VA . (2014). Is Munida gregaria (Crustacea: Decapoda: Munididae) a truly transpacific species? Polar Biol 37: 1413–1420.
Ponce JF, Rabassa J, Coronato A, Borromei AM . (2011). Palaeogeographical evolution of the Atlantic coast of Pampa and Patagonia from the last glacial maximum to the Middle Holocene. Biol J Linn Soc 103: 363–379.
Pritchard JK, Stephens M, Donnelly P . (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Provan J . (2013). The effects of past, present and future climate change on range-wide genetic diversity in northern North Atlantic marine species. Front Biogeogr 5: 60–66.
R Core Team. (2013). R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria. Available from http://www.R-project.org/.
Rabassa J, Coronato A, Martínez O . (2011). Late Cenozoic glaciations in Patagonia and Tierra del Fuego: an updated review. Biol J Linn Soc 103: 316–335.
Sánchez R, Sepúlveda R, Brante A, Cárdenas L . (2011). Spatial pattern of genetic and morphological diversity in the direct developer Acanthina monodon (Gastropoda: Mollusca). Mar Ecol Prog Ser 434: 121–131.
Sérsic AN, Cosacov A, Cocucci AA, Johnson LA, Pozner R, Avila LJ et al. (2011). Emerging phylogeographical patterns of plants and terrestrial vertebrates from Patagonia. Biol J Linn Soc 103: 475–494.
Takezaki N, Nei M, Tamura K . (2010). POPTREE2: Software for constructing population trees from allele frequency data and computing other population statistics with Windows interface. Mol Biol Evol 27: 747–752.
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P . (2004). MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4: 535–538.
Wilson AB, Eigenmann Veraguth I . (2010). The impact of Pleistocene glaciation across the range of a widespread European coastal species. Mol Ecol 19: 4535–4553.
Acknowledgements
This work was funded by grants from the Agencia Nacional de Promoción Científicaca y Tecnológica (PICT906, PICT1596), CONICET (PIP 0321) and Proyectos Federales de Innovación Productiva 2007 (PFIP 1622/07). We specially thank Julian Ashford (ODU) for providing sampling support. We also thank María Natalia Paso Viola (MACN-CONICET), Alejandro D’Anatro and Carolina Abud from Universidad de la República, Montevideo, Uruguay, for their constant technical support and advice, and Sheryl Macnie, Sandy Becker and Neal Mathew for improving the English of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article

Cite this article
Ceballos, S., Lessa, E., Licandeo, R. et al. Genetic relationships between Atlantic and Pacific populations of the notothenioid fish Eleginops maclovinus: the footprints of Quaternary glaciations in Patagonia. Heredity 116, 372–377 (2016). https://doi.org/10.1038/hdy.2015.106
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2015.106
This article is cited by
-
Quaternary ice sheets and sea level regression drove divergence in a marine gastropod along Eastern and Western coasts of South America
Scientific Reports (2020)
-
Phylogenomics of an extra-Antarctic notothenioid radiation reveals a previously unrecognized lineage and diffuse species boundaries
BMC Evolutionary Biology (2019)
-
Effects of changes in salinity on oxygen and food consumption of the young sub-Antarctic notothenioid Eleginops maclovinus: possible implications of their use of an estuarine habitat
Polar Biology (2017)
-
Historical dimensions of population structure in a continuously distributed marine species: The case of the endemic Chilean dolphin
Scientific Reports (2016)
