
Abstract
The evolution of the mitochondrial genome and its potential adaptive impact still generates vital debates. Even if mitochondria have a crucial functional role, as they are the main cellular energy suppliers, mitochondrial DNA (mtDNA) introgression is common in nature, introducing variation in populations upon which selection may act. Here we evaluated whether the evolution of mtDNA in a rodent species affected by mtDNA introgression is explained by neutral expectations alone. Variation in one mitochondrial and six nuclear markers in Myodes glareolus voles was examined, including populations that show mtDNA introgression from its close relative, Myodes rutilus. In addition, we modelled protein structures of the mtDNA marker (cytochrome b) and estimated the environmental envelopes of mitotypes. We found that massive mtDNA introgression occurred without any trace of introgression in the analysed nuclear genes. The results show that the native glareolus mtDNA evolved under past positive selection, suggesting that mtDNA in this system has selective relevance. The environmental models indicate that the rutilus mitotype inhabits colder and drier habitats than the glareolus one that can result from local adaptation or from the geographic context of introgression. Finally, homology models of the cytochrome b protein revealed a substitution in rutilus mtDNA in the vicinity of the catalytic fraction, suggesting that differences between mitotypes may result in functional changes. These results suggest that the evolution of mtDNA in Myodes may have functional, ecological and adaptive significance. This work opens perspective onto future experimental tests of the role of natural selection in mtDNA introgression in this system.
Similar content being viewed by others

Introduction
Introgressive hybridization obscures species boundaries. Hence, the study of interspecific gene flow is fundamental to understand the molecular mechanisms of speciation and species delimitation, how species maintain their biological integrity despite exchanging genes and the role of adaptation in the process of genetic introgression (Mallet, 2005; Arnold, 2006; Pinho and Hey, 2010). Gene flow between closely related taxa may be a product of a neutral event that has no effect on an individual’s phenotype or fitness. For example, introgression is possible during species geographic expansion with replacement of the resident species (Excoffier et al., 2009). Simulations suggest that introgression can occur from the resident to the expanding species, and that the levels of introgression can be massive (Currat et al., 2008). However, (mal)adaptive consequences of hybridization and gene exchange have also been proposed (Rosas et al., 2010; Arnold et al., 2011; Pons et al., 2013). Indeed, gene flow can alter the performance of the recipient individual in a given environment by either increasing or decreasing fitness (Doi et al., 1999; Doiron et al., 2002; Arnold, 2006). Natural selection may thus promote, in some circumstances, the exchange of genes between hybridizing species (Arnold et al., 2011).
In general, if novel genetic variation, including that acquired by introgression, has positive adaptive effects, it leaves genetic signatures that can be detected with contemporary statistical tools (Arnold et al., 2011). First, introgression is expected to affect more dramatically locus affected by adaptive gene flow rather than any other neutral locus. Second, signs of past directional selection are predicted in markers involved in adaptive between-species gene flow, as opposed to neutral introgressive markers (Nielsen, 2005; Zhai et al., 2009; Behrman and Kirkpatrick, 2011), and after emerging, adaptations are maintained by ongoing stabilizing selection (see, for example Nespolo et al., 2011). Third, introgression might alter the ecological niche of the recipient if that genotypic variant and/or the linked mutations (for example, through direct or indirect epistatic effects) allow for expansion into additional habitat types (Schluter and Conte, 2009; Arnold et al., 2011). Fourth, the new genetic variant may contain structural, potentially functional, differences relative to the original alleles in the encoded protein (Doiron et al., 2002; Riise et al., 2007).
Mitochondrial DNA (mtDNA) is the most widely studied genetic material in animals and descriptions of interspecific gene flow involving mtDNA are common in the literature (Arnold, 2006; Toews and Brelsford, 2012). As mitochondria mediate important functions for the physiological performance of organisms (Ballard and Melvin, 2010), it may thus be shaped by natural selection (da Fonseca et al., 2008; Dowling et al., 2008; Cheviron and Brumfield, 2009; Hassanin et al., 2009). Several studies have shown that the transfer of mtDNA variants between closely related species or lineages influences the physiological properties of individuals (Doi et al., 1999; Doiron et al., 2002; Ballard and Melvin, 2010). Population genetic studies show that introgression of mtDNA often affects species over broad geographic ranges, sometimes leading to its fixation (Melo-Ferreira et al., 2012). Whether massive mtDNA introgression results from purely neutral processes or it can be significantly affected by natural selection remains a matter of debate (Cheviron and Brumfield, 2009; Arnold et al., 2011; Pons et al., 2013). For example, studies on the Eurasian bank vole Myodes (or Clethrionomys) glareolus have shown that its northeastern, and some eastern, populations harbour an mtDNA lineage that introgressed from its neighbour, Myodes rutilus, that has even become fixed in some populations (Tegelström, 1987; Deffontaine et al., 2005; Boratyński et al., 2011). This observation led to the hypothesis that colonization of northeastern Europe and western Asia by M. glareolus was facilitated by the introgression (before or during colonization of currently occupied regions), through hybridization, of physiological mechanisms that allowed it to inhabit more severe habitats (Boratyński et al., 2011; Šíchová et al., 2014).
The main aim of this study was to assess whether the mtDNA in M. rutilus and M. glareolus bares signs of evolution under natural selection (both past and post introgression) that may have affected mtDNA introgression between the species. To achieve this aim, we tested (1) whether introgression of mtDNA is more pronounced than of other nuclear markers, (2) whether structural differences could be found between the native and introgressed variants of the mitochondrial marker, cytochrome b gene, (3) whether such differences matched ecological niche changes and (4) whether signs of natural selection could be inferred from the phylogenetic (selection on long-term sequence evolution) and population genetic patterns (selection on population polymorphism) of DNA variation. Positive responses to these tests strengthen adaptive evolution hypotheses. Our results suggest that selection has played a role in the evolution of the mtDNA of these species that may have influenced mtDNA introgression.
Materials and methods
Laboratory procedures
Genetic variation in the complete mitochondrial cytochrome b gene (cyt b) and parts of six nuclear genes (LCAT (lecithin-cholesterol acyltransferase), G6pd (glucose-6-phosphate dehydrogenase) gene, BRCA1 (breast cancer 1) gene, GHR (growth hormone, receptor), vWF (von Willebrand factor) and ADRAB2 (α-2B adrenergic receptor)) were analysed in 131 voles from 6 localities across Finland (Figure 1). Total genomic DNA was extracted from ethanol-preserved tissues collected during field expeditions using a Qiagen (Valencia, CA, USA) extraction protocol. Automatic magnetic purification method in a KingFisher Magnetic Particle Processors (Thermo Scientific, Waltham, MA, USA) according to the manufacturer’s protocol was applied. The genes were amplified using primers specific to Myodes voles (Supplementary Table S1). PCR reactions for cyt b were performed in 30 μl mixes containing 3 μl of DNA (10–50 ng μl−1), 0.24 μl of Taq polymerase (0.025 U ml−1), 1.8 μl of F and R primers (5 μM), 3 μl of dNTP (0.2 mM), 3 μl of reaction buffer and 15.96 μl of H2O. Reactions for nuclear genes were performed with 1 × PCR buffer (BioTools, Madrid, Spain), 200 μM of dNTPs, 0.5 U of Taq polymerase and optimum MgCl2 concentrations for specific primers (Supplementary Table S1). PCR reactions were performed with 3 min of preliminary activation of Taq polymerase at 95 °C followed by 32 (34 for nuclear genes) three-step cycles: of denaturation at 94 °C (30 s), annealing at temperature specific for primer (30 s; Supplementary Table S1), extension at 72 °C (70 s) and a final extension at 72 °C (10 min). The products were purified in Exo-SAP apparatus (Amersham Biosciences, Orsay, France) and sequencing was performed with the BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystems, Foster City, CA, USA). Sequencing reactions were performed using the PCR primers in 16 μl mixes containing 1 μl of primer (3.3 μM) and 3 μl of PCR product according to ABI sequencing protocol. Two-direction sequencing was conducted with a BigDye Terminator kit (Applied Biosystems) on an Applied Biosystems 3130xl Genetic Analyzer. Forward and reverse strands were merged using SeqScape v 2.1.1 (Applied Biosystems, www.appliedbiosystems.com) and aligned using ClustalX v 2.0.10 (www.clustal.org). The allelic phase determinations for the nuclear genes were estimated with PHASE v2.1.1 (http://c4c.uwc4c.com/express_license_technologies/phase) separately for species. In the final analyses, only alleles with phase calls with posterior probabilities of >0.95 were used. This threshold led to the exclusion of three samples (and three haplotypes) of GHR, one (one) of LCAT, four (0) of ADRAB2 and one (0) of vWF genes. None of the genetic (phylogenetic reconstructions, neutrality tests, variability estimations) analyses were markedly affected by this exclusion. DnaSP v 5.10.01 (www.ub.edu/dnasp/) and ARLEQUIN 3.11 (cmpg.unibe.ch/software/arlequin3) were used to estimate descriptive statistics for each gene and clade.

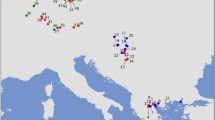
Locations (circles on large map) of the Myodes glareolus samples, with original (open) and introgressed (close) mtDNA used in the ecological modelling (all) and molecular analyses (six populations in Finland, indicated with arrows). The predicted areas for sympatric (blue) and allopatric distributions of GLA-type (red) and RUT-type (green) mtDNAs are presented. The inserted (small) map indicates the M. glareolus (red), M. rutilus (green) and sympatric (blue) species distributions (after International Union for Conservation of Nature (IUCN)).
Phylogenetic analyses
Analyses with Maximum Likelihood method on partial cyt b sequences were performed on a data set including sequences from GenBank (see Supplementary Information: Data_Sets.xls for detailed information about data sets) covering most of the Myodes glareolus distribution (see Figure 1). Analyses on the complete cyt b gene and on six nuclear genes were performed in 131 M. glareolus, 6 M. rutilus and 6 M. rufocanus voles. Additional reference sequences for all genes for Microtus and Myodes species were downloaded from GenBank (Accession numbers: AY167187, FJ881444, AY309414, AY309428, FM162067, AJ402709, FM162047, AJ891053, GQ267517, AB086024, AY295009 and AM910792). Phylogenetic relationships among complete cyt b haplotypes were reconstructed using three different methods. A hierarchical likelihood ratio test (LRT) and Akaike Information Criterion-based model selection were conducted in ModelTest v 3.0 (http://www.ctu.edu.vn/~dvxe/Bioinformatic/Software/BIT%20Software/modeltest.htm) and showed similar results. The chosen models were used for phylogenetic reconstructions, under the Maximum Likelihood method (Felsenstein, 1981), using PHYML v 2.4.4 (www.atgc-montpellier.fr/phyml), and neighbour-joining analyses using PHYLIP 3.68 (http://evolution.genetics.washington.edu/phylip.html), both assessed with 1000 bootstrap replicates. Bayesian inference was performed using MrBayes v 3.1.2 (mrbayes.sourceforge.net). Four Markov chain Monte Carlo methods were used with random starting trees that ran for multiple generations (10 and 1 million for nuclear genes and complete cyt b, respectively), sampling trees every 100 generations and the first trees, 30 000 for nuclear markers and 6000 complete cyt b, discarded from further analyses (burn-in). The remaining trees were used to construct the consensus trees and estimate posterior probabilities using the 50% majority rule. The relationships among haplotypes were analysed and visualized with the statistical parsimony method implemented in TCS 1.21 (darwin.uvigo.es/software/tcs.html).
Ecological models
Analyses were performed on a bar-coded data set of cyt b sequences of M. glareolus available from GenBank. The data set with 602 sequences of M. glareolus species with locations of origin was divided into two subsets with samples containing M. glareolus (GLA) and M. rutilus (RUT) types of mtDNA (Supplementary Information: Data_Sets.xls). The original resolution of locations was rescaled to 10 km and projected with Lambert Azimuthal Equal Area (ETRS-LAEA). Duplicated samples, of the same mtDNA type, from the same locality were removed, resulting in 194 sequences with locations for M. glareolus with the GLA-type mtDNA and 37 sequences with locations for M. glareolus with the RUT-type mtDNA (Figure 1 and Supplementary Information: Data_Sets.xls). The geographic coordinates of locations were determined following information obtained in the literature where the sequences were first published.
Environmental variables were downloaded from WorldClim database (www.worldclim.org/) and CGIAR-CSI website (www.cgiar-csi.org/) with a resolution of 5′, and further projected to ETRS-LAEA and rescaled to 10 km2 per square pixel. After a preliminary analysis, we retained eight bioclimatic variables with the lowest correlation within the study area (|r|<0.78; Supplementary Table S2). The least correlated variables were: actual evapotranspiration (the amount of water removed from a surface because of evaporation and transpiration), aridity index (AI=P/PET; the degree of dryness of the climate=the average annual precipitation/potential evapotranspiration, the ability of the atmosphere to remove water from the surface), annual precipitation (average of total yearly precipitation), precipitation seasonality (the coefficient of variation of monthly precipitation, the ratio between s.d. and average), precipitation of the warmest quarter of the year (sum of precipitation through warmest 3 months), mean diurnal range of temperature (yearly average of daily aptitude of temperature), maximum temperature of the warmest month and minimum temperature of the coldest month. To assess the differences in the spatial distribution and climatic requirements of M. glareolus with GLA and RUT mtDNA types, presence-only predictive modelling with a maximum entropy algorithm was applied. Models were constructed with Maxent ver. 3.3.3e (Phillips and Dudík, 2008) that performs well when compared with other techniques (Elith et al., 2006) even on limited data sets (Hernandez et al., 2006). This algorithm has been used to model various biological systems including analyses of the impact of climate change on the distribution of other small mammals (see, for example, Fløjgaard et al., 2009). The consensus models and uncertainty maps were built from 25 replicates for each mtDNA type separately. Each model was run with linear, quadratic, product and hinge features and with cross-validation subsampling. Response curves for each variable were evaluated with the jack-knife procedure. The minimum presence values threshold, which means that the location with the least predicted value defines the cutoff to classify the presence area, was applied to classify Maxent continuous logistic outputs into binary presence–absence data. The importance of each variable to the model was assessed by the average percentage of contribution of the 25 replicates. Multivariate response curves for each variable were extracted for each replicate and averaged. The performance of the models was assessed with receiver operating characteristics area under curve (ROC-AUC). To accomplish this, a new data set was created by randomly selecting, from the predicted models, the same number of locations as presences available. The new data set was used with the presence locations and predictions from the consensus model to obtain an ROC-AUC value. This method was repeated 100 times, creating a new data set in each repetition. The specificity for the chosen threshold using the same method as for ROC-AUC is reported. As the minimum presence value threshold was applied, the sensitivity is always 1. Principal component analysis and multivariate analysis of variance were applied to test the differences in reconstructed environmental parameters between the two mtDNA types. The principal component analysis was created using the scaled environmental geographic variables (the same 8 variables used for models) for the study area. A set of 50 predicted locations per mtDNA type were randomly chosen from each of the binary distributions resulting from Maxent. The values of the environmental geographic variables were extracted for these locations and used as the input to the multivariate analysis of variance analyses. This procedure was repeated 50 times. All analyses and graphs were made in R (R Development Core Team, http://www.r-project.org/).
Tests of neutral evolution
Strength and direction of past and/or ongoing selection from DNA sequence data were indirectly estimated with various types of neutrality tests (Nielsen, 2005; Zhai et al., 2009). Tests were conducted for seven markers on the same set of samples of M. glareolus collected in six populations in Finland (indicated by arrows on Figure 1). The descriptive statistics of molecular diversity and tests of neutrality based on differences among and frequencies of haplotypes (selection on genetic variants) were determined for all markers in ARLEQUINv.3.11. The assumption of selective neutrality and population equilibrium (as those statistics are sensitive to demographic deviation from mutation-drift equilibrium) were tested with Tajima’s D and Fu’s Fs determining its significance with 5000 coalescent simulations.
Multilocus tests of neutrality (also testing selection on genetic variants), comparing ratios of polymorphism within M. glareolus mtDNA types to divergence to its distinct outgroup, M. rufocanus, were conducted using the Hudson–Kreitman–Aguade (HKA) method with 10 000 simulated replicates (HKA software: genfaculty.rutgers.edu/hey/) (Nielsen, 2005; Zhai et al., 2009). This test looks for dissimilar patterns between markers but does not completely account for changes in population size. Given the presence of the two highly divergent mtDNA lineages within M. glareolus, with divergent coalescence histories that inflate the levels of observed cyt b polymorphism, the HKA test was applied to partitioned data sets of only GLA or RUT mtDNA types. Those analyses were conducted including sequences of all seven markers (cyt b, LCAT, G6pd, BRCA1, GHR, vWF and ADRAB2).
Past natural selection (on long-term sequence evolution) in the divergence of mitochondrial marker was estimated with a maximum likelihood approach that examines variation in the ratio of nonsynonymous to synonymous substitutions in PAML 4 (ω=dN/dS; Yang and Bielawski, 2000; Yang, 2007)). Primarily the branch model was applied to test for heterogeneity in ω among the inferred mtDNA types (including all M. glareolus haplotypes from phylogenetic analyses: Figure 2, Supplementary Appendix S1 and Supplementary Information). Branches were partitioned into original (GLA) and introgressed (RUT) mtDNA types (mitotypes) and analyses included outgroup sequences. LRTs were constructed based on log-likelihood scores for a model assuming a single ω ratio (model 0), a model allowing free ω ratios among branches (model 1) and a model assuming separate ω ratios for main mitotypes (model 2; for GLA and RUT mtDNA mitotypes separately). Thereafter, positive selection (as indicated by ω>1) was tested on the complete phylogenetic tree and on partitioned phylogenies including only one of the main mitotypes and an outgroup. For these analyses, the site models for heterogeneous selection pressure at amino acid sites were applied. LRTs were constructed based on log-likelihood scores for models assuming a single ω ratio among codons (M0), models allowing multiple ratios but ω ⩽1 (M7) and models allowing also ω ratios among codons larger than one (ω>1; M8).

Maximum likelihood (ML) trees for six nuclear markers and one mitochondrial marker. The numbers in the tree nodes indicate the bootstrap values for ML and neighbour-joining phylogenies and Bayesian posterior probabilities. The length of branches is proportional to the number of substitutions between haplotypes. The trees were rooted with Microtus (M) sequences (GenBank: M162047, GQ267517, AY295009, AM910792, AB086024, FM162067 and AY167187). The network nodes are outlined by the number of sampled individuals and the branches points (and line bends) by the hypothetical haplotypes. Myodes species are indicated by the following colours: white for M. glareolus, black for M. rutilus and grey for M. rufocanus. Two cytochrome b haplotypes of M. glareolus, one on introgressed and one on none-introgressed networks, are framed and in bold, referring to the haplotypes used in protein modelling.
McDonald–Kreitman test compares the proportion of nonsynonymous to synonymous mutations observed within lineages with the divergence between them to infer past or/and ongoing selection in the contemporary population (Egea et al., 2008). The variation within one mtDNA type, either GLA or RUT, was contrasted against the divergence from one haplotype to the other type. This test is robust to all standard demographic assumptions, although it might not depict correct sign of the selection and does not distinguish between past and ongoing selection.
Models of the cytochrome b protein
The structure of cyt b was homology modelled separately for M. glareolus samples belonging to either the original (GLA) or introgressed (RUT) mtDNA type. The cyt b basal nucleotide sequences of GLA and RUT (inferred from network) were translated to peptide sequences using Transeq in EMBOSS (Rice et al., 2000). The model of cyt b for GLA was built based on the alignment of the crystal structure of cytochrome bc1 complex from bovine (PDB: 1BGY, C chain, Iwata et al., 1998) and the GLA-type bank vole sequences using MALIGN in BODIL (Lehtonen et al., 2004) and MODELLER9v7 (Sali and Blundell, 1993). The level of similarity between bovine and Myodes sequences (≈82%) gives highly reliable protein models (see, for example, Hillisch et al., 2004). Ubiquinone was added to the quinine reduction site (Qi site) from another crystal structure of bovine cytochrome bc1complex (PDB: 1PP9, Huang et al., 2005) by superimposing the structures with VERTAA in BODIL. The model of cyt b for RUT-type sequences was built in a similar way. Sequences (n=100) resembling translated sequences of GLA (or RUT) were retrieved using a BLAST similarity search from UniProtKB/Swiss-Prot (au.expasy.org/tools/blast). Those sequences were aligned with cyt b of GLA (or RUT) in BODIL. Protein structure-based matrix was used with a gap penalty of 40 and default settings. The protein models were visualized using BODIL v. 0.81, and rendered with MOLSCRIPT v. 2.1.2 (www.avatar.se/molscript) and RASTER3D v. 2.7C (skuld.bmsc.washington.edu/raster3d).
Results
Phylogenetic analyses
In total, 131 vole samples collected in Finland (Figure 1) were analysed for variation in seven genes: one mitochondrial and six nuclear (Table 1 and Supplementary Information: Data_Sets.xls; GenBank Accession numbers: JX477265–JX477369 and JF930082–JF930131). The phylogenies of the nuclear genes were consistent across methods and coincided with the assignment of individuals to species (Figure 2). M. glareolus and M. rutilus formed monophyletic clades in most of the analyses, with the exception for the vWF gene where M. rutilus grouped within M. glareolus, possibly because of low variation in this marker (Table 1). The cyt b sequences were most likely of mitochondrial origin (not nuclear integrated copies), as no stop codons were found in the data set and the third position base composition was typical (A 40.4%, C 40.7%, G 2.5% and T 16.5%) of small mammals (A 39%, C 36%, G 3% and T 21%; Johns and Avise, 1998). Polymorphism was found at 137 sites of cyt b, defining 75 haplotypes (Table 1). Contrary to phylogenies based on the six nuclear genes, the cyt b haplotypes sampled in M. glareolus did not form a monophyletic clade. It was divided into two lineages, only partially overlapping with species assignment. Some individuals of M. glareolus (33) grouped together with M. rutilus-type mtDNA (mitotype RUT), suggesting mtDNA introgression. M. glareolus mitotype (GLA) grouped with the haplotypes specific for M. glareolus (Figure 2). Southern and Central Finland were inhabited by GLA-type individuals, whereas individuals in the North and East-Central regions carried the RUT-type cyt b. Samples from East-Central Finland included both mitotypes. The haplotypes of nuclear markers were shared between the populations but did not show geographic structure similar to mtDNA.
Ecological models
Ecological models were based on M. glareolus cyt b sequences retrieved from GenBank (Supplementary Information: Data_Sets.xls) and assigned to one of the two mtDNA types (mitotypes). The consensus models presented high average ROC-AUC. The average model for the GLA had a ROC-AUC (±s.d.) of 0.837±0.017 whereas that for the RUT was 0.942±0.022. The specificity for GLA was 0.404±0.036 with a threshold of 0.104. For RUT the reported specificity was 0.682±0.073 for the threshold value of 0.116. The area predicted to be occupied only by GLA extended over 33% of the study area (59 267 cells), covering most of the known range of the species. The predicted area for RUT covered only 5% (9117 cells) of the study area, particularly in the northeast. The predicted sympatric area between the two mtDNA types extends over 27% of the study area (47 921 cells) that corresponds to 44% of the total predicted area for GLA and 84% of the RUT predicted area (Figure 1).
The variables that mostly contributed to both models were: the minimum temperature of the coldest month (29% for GLA and 42% for RUT: ) and the actual evapotranspiration (15% for GLA and 18% for RUT; Supplementary Figure S1). Annual precipitation and precipitation of the warmest quarter greatly contributed to the GLA model, and the aridity index was important in defining the RUT distribution. The other variables (precipitation seasonality, mean diurnal range of temperature and maximum temperature of the warmest month) did not contribute markedly to any of the models. The relatively low s.d. values indicate that the contribution patterns were consistent among replicates (Supplementary Figure S1).
The two variables that contributed most to the models (minimum temperature of the coldest month and actual evapotranspiration) were also those that best described the differences between the environments inhabited by the two mtDNA types (Table 2). Response curves depicted detailed differences in habitat usage by GLA and RUT mtDNA types (Figure 3). Both types thrive at minimum temperatures higher than −20 °C, but GLA is more frequent in warmer places, whereas RUT has an optimum at −10 °C and is less supported as minimum temperature of the coldest month increases. A similar pattern was found for actual evapotranspiration, for which RUT has an optimum at lower values than GLA, although GLA spans over a larger range. The precipitation variables, both annual precipitation and precipitation of the warmest quarter of the year, indicate that GLA is precipitation dependent, occupying niches with relatively high rainfall, whereas RUT mitotype tolerates arid areas (Figure 3).

Multivariate response curves (±s.d.) for the environmental variables included for the GLA (solid) and RUT (dashed lines) mitotype models. AET, actual evapotranspiration; AI, aridity index; AP, annual precipitation; MDR, mean diurnal range; MTCM, minimum temperature of the coldest month; MTWM, maximum temperature of the warmest month; PS, precipitation seasonality; PWQ, precipitation of the warmest quarter.
Tests of neutral evolution
Selection on genetic variation
DNA sequence variation within six populations of M. glareolus from Finland was assessed for mitochondrial cyt b and six nuclear genes (Table 1). For most of the nuclear genes the frequency spectrum of mutations did not significantly differ from the neutral mutation-drift expectations, as determined by Tajima’s D and Fu’s Fs statistics (Table 1). However, for mitochondrial cyt b and the X-linked G6pd genes, the frequency distributions of mutations differed from neutral expectations, as Tajima’s D and Fu’s Fs values were significant and negative (Table 1 and Supplementary Table S3). Tajima’s D and Fu’s Fs statistics for cyt b gene were significant and negative when analysed separately for GLA- and RUT-type lineages (Table 1). For G6pd, the statistics for both the partitioned and the combined data set were negative and significant (Supplementary Table S3).
The neutral model was also rejected by the conservative HKA multi-gene test. The analyses showed an excess of within-species diversity in cyt b gene and an excess of between-species divergence (compared with the outgroup, M. rufocanus) in nuclear genes. The result based on the GLA data set was stronger (χ2=32.98, d.f.=6, P<0.000001, for number of copies of cyt b/nuclear: 98/196), but also had higher statistical power because of the larger sample size as compared with the RUT-type data set (χ2=16.67, d.f.=6, P<0.0027, for number of copies of cyt b/nuclear: 33/64). Pair-wise comparisons between the different combinations of markers showed that these statistics were only significant if the mitochondrial marker was included in the analysis (Table 3). After correcting for multiple HKA tests, none of the statistics between nuclear markers were significant (Table 3).
Long-term sequence evolution
The branch model analysis for detecting heterogeneity in past selection, based on dN/dS ratios for cyt b, showed significant heterogeneity among the mitotypes inferred from the phylogenetic analyses (Figure 2, Supplementary Appendix S1 and Supplementary Information). Both models, assuming free ratios and separate ratios for the detected haplotypes (0.0342, 0.0001 and 0.0102 for background and GLA- and RUT-type average ratios respectively), were significantly better than the one-ratio model (LRT: χ2>813, P<4 × 10−108). However, the free ratio model was not better than the simpler model assuming mitotype (GLA and RUT)-specific ω ratios (LRT: χ2=103, P=0.999). The site model analyses, which included data for both GLA and RUT mitotypes, showed that the model allowing multiple ratios (M7) was significantly better than the model assuming a single ratio across codons (M0; χ2=36.86, P<0.0001; Table 4). Moreover, the model assuming ω values of >1 (M8), inferring past positive selection, was better than the model allowing only multiple ratios (M7; χ2=9.18, P=0.01; Table 4). This ω ratio model (M8) was significantly better than the multiple ω ratio model (M7) for data set including all sequences as well as for data set including only the original M. glareolus mtDNA (GLA: χ2=10.44, P=0.005; Table 4). The analyses on the introgressed RUT mitotype showed no evidence of past positive selection (RUT: χ2<0.01, p≈1).
The McDonald–Kreitman test showed signs of selection on the cyt b gene, exhibiting higher ratio of within-species nonsynonymous to synonymous mutations versus such a ratio of between-lineage divergence (Supplementary Appendix S1 and Supplementary Information). The results were significant in the combined data set (χ2=7.694, NI=6.636, P=0.005) as well as in the partitioned data sets (GLA: χ2=7.738, NI=7.009, P=0.005; RUT: χ2=9.854, NI=9.818, P=0.001).
Models of the cytochrome b protein
Protein models for GLA and RUT cyt b mitotypes were built using the crystal structure of the bovine cytochrome bc1 complex as a template. Sequence identities between the template structure and the basal sequences of GLA and RUT cyt b types were 82.1% and 82.3%, respectively. As the sequence identities are very high and there are no insertion/deletions in the vicinity of the substrate binding site, the models should describe the structural features of these two enzymes with high accuracy. The two cyt b types differed only in two amino acids (Ala17 and Val327 in RUT and Ser17 and Ile327 in GLA). Alignment of different vole cyt b gene sequences (n=100) resembling those of the GLA and RUT mitotypes showed serine to be the predominant, conserved amino acid at position 17. According to a BLAST similarity search, no other vole species besides M. rutilus (and M. glareolus with the introgressed RUT mtDNA type) had alanine at position 17 as in the RUT mitotype.
Discussion
In this study we used two closely related but ecologically distinct species of Myodes voles between which introgression of the mitochondrial genome has been described (Tegelström, 1987; Boratyński et al., 2011) in order to examine signs of evolution of mtDNA under natural selection.
If mtDNA introgression has contributed to local adaptation, different mitotypes should populate regions with different ecological envelopes (Figure 1). We found that M. glareolus with introgressed M. rutilus (RUT)-type mitochondria inhabits more severe environments, with lower precipitation and lower temperature, than M. glareolus species with its own (GLA) mtDNA (Table 2, Figure 3 and Supplementary Figure S1). Therefore, similar to other small mammals (Fløjgaard et al., 2009) the distribution of M. glareolus appears to be related to productivity in the environment (Stephenson, 1998). This result suggest that introgressed M. rutilus mitotype in M. glareolus background could have facilitated the invasion of this species to more severe environments, assuming that hybridization happened before the invasion (as suggested by some of the previous studies: Tegelström, 1987; Deffontaine et al., 2005). These evidences also suggest that different mitotypes (and likely also key OXPHOS nuclear units) populate different climatic regions that may have allowed local adaptation. However, given the distribution of M. glareolus and M. rutilus, and the contact of the species in the northeastern range of the distribution of M. glareolus (Figure 2), we cannot discard that this ecological pattern of introgression may simply result from a geographic coincidence, namely if introgression is recent enough to not have yet allowed neutral diffusion of the introgressed haplotypes to southern regions.
It is striking, however, to note that mtDNA introgression is massive, almost fixed in the northern range of M. glareolus, and no signs of these reticulation events were found in the six nuclear markers analysed here (Figure 2). Of course, these six markers represent a small subsample of the nuclear genome, and a more thorough sampling of this genomic compartment is needed to properly address this question, but nuclear introgression seems at this point absent or rare at most. Such pattern of massive mtDNA introgression with little signs of nuclear DNA flow is becoming frequent in studies of hybridization (Melo-Ferreira et al., 2009; Pons et al., 2013). This pattern can result from the relative demography of the species involved in the reticulation events. Phenomena such as competitive replacement of a resident species by an invading one, with hybridization in the invasion front, are expected to promote massive mtDNA introgression (Currat et al., 2008), and that may have been the case here. Gender asymmetries in the allospecific crosses, caused for instance by male competition or frequency-dependent assortative mating of females (Toews and Brelsford, 2012), may also account for this direction of mtDNA introgression. However, direct selection either on mtDNA or because of cytonuclear coevolution may have also contributed to the inferred pattern.
Therefore, we assessed whether the patterns of sequence evolution conformed to neutral expectations, both in the long term, by analysing ratios of synonymous and nonsynonymous substitutions, and in the short term, by analysing site frequency spectra. Our analyses of dN/dS ratios (Yang and Bielawski, 2000) suggested several instances of evolution under natural selection. Unlike the population genetic methods, which might be sensitive to demographic processes (Nielsen, 2005), dN/dS ratios are insensitive to demographic assumptions, even though selection has to be recurrent enough to be detected. These tests suggested past positive selection on sequence polymorphism within the GLA mtDNA type (dN/dS ratio test), and negative selection on sequences within both mitotypes at the population level (dN/dS and McDonald–Kreitman tests; Table 4). Even if these results most likely reflect pre-introgression evolution, they suggest that mtDNA evolution in this system has been governed by natural selection and may thus have adaptive value. Note that even if only one mtDNA marker was analysed here, it can be used as a proxy of the evolution of the whole mtDNA, where other important causal mutations of adaptation may have occurred. The detected signs of past positive selection and stabilizing selection on sequence polymorphism suggest that mtDNA may have played an important role in the adaptive evolution of M. glareolus.
The adaptive significance of cyt b evolution is also suggested by protein models. They revealed that the amino acid substitution at position 17 hypothetically changes the function of the protein, as it is located in close vicinity to the catalytic region of the enzyme (Figure 4). It is plausible that the hydroxyl group of serine in the GLA-type mtDNA anchors the position of a conserved histidine (His201, Figure 4a). This histidine has a crucial role in the binding and catalysis at Qi site of the cytochrome bc1 enzymatic complex (Kolling et al., 2003). The methyl group in the same position of the derived RUT-type cyt b cannot form a hydrogen bond with His201 (Figure 4b). As the side chain of this histidine in RUT cyt b is not tightly fixed, it may be relatively free to move away from the substrate binding site (Figure 4b). Thus, the entrance channel into the catalytic site could be wider, enabling easier enter for ubiquinone and exit for products. Accordingly, lesser movement of enzyme, that is, induced fit, is required upon substrate binding process into the catalytic site. Such importance of the flexibility and size of the entrance channels have been previously described (Toyota et al., 2002; Riise et al., 2007). It is possible that additional functional differences, similar to those in cyt b, exist in other parts of the mtDNA genome, and therefore this result must be interpreted as a proxy of processes that may affect the complete mtDNA. If so, these different mitotypes may result in structurally distinct proteins, questioning the possible influence in function.

(a) Protein models of introgressed and original cytochrome b. Serine in position 17 of amino acid sequence of Myodes glareolus cyt b hydrogen bound to the catalytically crucial amino acid His201 located near ubiquinone. (b) Alanine, in the same position, is shown unbound to His201. Hydrogen bonds are shown as green dotted lines.
Population-based neutrality tests (that test selection on gene variants) also showed that variation in cyt b differs from neutral (and/or mutation-drift) expectations, suggesting that selection has influenced its haplotypes frequencies, either directly or indirectly via linkage with other adaptive genes or gene complexes. The frequency distribution-based neutrality tests (Tajima’s D and Fu’s Fs) suggested non-neutral evolution of mtDNA, because common demographic effects among unlinked genetic markers were not found (Table 1 and Supplementary Table S3; but see Fay and Wu, 1999 for special bottleneck+expansion situation). The negative values of Tajima’s D and Fu’s Fs may thus indicate positive selection of the mtDNA variants (or past bottleneck; Tajima, 1989), an expected signal if variants in the northeast have been favoured by natural selection. The multi-gene HKA analyses, testing also selection on genetic variants, confirmed significant deviation from expectations of neutral (and/or constant population) molecular evolution. It has however to be kept in mind that the power for detecting deviations from neutral expectation assumptions vary among markers, as for example, the amount of nuclear DNA variation is usually reduced compared with that of the mtDNA. Similarly, the different strength of selection detected between mtDNA types, implying stronger effects within the GLA as opposed to the RUT mitotype (Table 3), might have resulted from variable demographic histories between two mitotypes (Pons et al., 2013) or from variable power of the analyses between them.
Conclusions
We used several indirect approaches to test whether evolution of sequence polymorphisms and population variability of mtDNA conformed to the neutral expectations. Results from multiple approaches suggest that neutral expectations are often rejected. Even if the results do not suggest that sequence polymorphism within the rutilus mitotype evolved under positive selection, we show that the glareolus type likely did, and that some structural differences between mtDNA variants may have functional consequences. The differences between mtDNA types and selection signals from population genetic neutrality tests are especially important in the context of introgression, both suggesting that introgressed variant might be beneficial for individuals in local conditions, as showed by their ecological envelopes. The fact that there is extensive mtDNA introgression but not a single shared allele at any of the six nuclear loci may also reflect a selective nature of introgression (Pons et al., 2013). Whether cyt b itself or other linked genes or gene complexes may have been the target of selection is not clear, as the signals of natural selection are likely the result of the cumulative effects of linked mtDNA mutations, mutational combinations and/or gene complexes. Even if the case for adaptive introgression is not yet established, our results are promising in that respect. Future mitogenomic, genomic, functional and physiological analyses (Boratyński et al., 2010, 2013; Ketola et al., 2014) are needed to provide a complete understanding of the bases of potentially adaptive mtDNA evolution in Myodes rodents.
Haplotype, mtDNA type, species and geographic affiliations of included sequences are presented in Supplementary Information: Data_Sets.xls.
Data archiving
Sequence data have been submitted to GenBank: accession numbers JX477265- JX477369, JF930082-JF930131.
References
Arnold ML . (2006) Evolution Through Genetic Exchange. Oxford University Press: Oxford, New York.
Arnold ML, Ballerini ES, Brothers AN . (2011). Hybrid fitness, adaptation and evolutionary diversification: lessons learned from Louisiana Irises. Heredity 108: 159–166.
Ballard JWO, Melvin RG . (2010). Linking the mitochondrial genotype to the organismal phenotype. Mol Ecol 19: 1523–1539.
Behrman KD, Kirkpatrick M . (2011). Species range expansion by beneficial mutations. J Evol Biol 24: 665–675.
Boratyński Z, Alves PC, Berto S, Koskela E, Mappes T, Melo-Ferreira J . (2011). Introgression of mitochondrial DNA among Myodes voles: consequences for energetics? BMC Evol Biol 11: 355.
Boratyński Z, Koskela E, Mappes M, Oksanen TA . (2010). Sex-specific selection on energy metabolism—selection coefficients for winter survival. J Evol Biol 23: 1969–1978.
Boratyński Z, Koskela E, Mappes T, Schroderus E . (2013). Quantitative genetics and fitness effects of basal metabolism. Evol Ecol 27: 301–314.
Cheviron ZA, Brumfield RT . (2009). Migration-selection balance and local adaptation of mitochondrial haplotypes in rufous-collared sparrows (Zonotrichia capensis) along an elevational gradient. Evolution 63: 1593–1605.
Currat M, Ruedi M, Petit RJ, Excoffier L . (2008). The hidden side of invasions: massive introgression by local genes. Evolution 62: 1908–1920.
da Fonseca RR, Johnson WE, O’Brien SJ, Ramos MJ, Agostinho A . (2008). The adaptive evolution of the mammalian mitochondrial genome. BMC Genomics 9: 119.
Deffontaine V, Libois R, Kotlík P, Sommer R, Nieberding C, Paradis E et al. (2005). Beyond the Mediterranean peninsulas: evidence of central European glacial refugia for a temperate forest mammal species, the bank vole (Clethrionomys glareolus). Mol Ecol 14: 1727–1739.
Doi A, Suzuki H, Matsuura ET . (1999). Genetic analysis of temperature-dependent transmission of mitochondrial DNA in Drosophila. Heredity 82: 555–560.
Doiron S, Bernatchez L, Blier PU . (2002). A comparative mitogenomic analysis of the potential adaptive value of arctic charr mtDNA introgression in brook charr populations (Salvelinus fontinalis Mitchill). Mol Biol Evol 19: 1902–1909.
Dowling DK, Friberg U, Lindell J . (2008). Evolutionary implications of non-neutral mitochondrial genetic variation. Trends Ecol Evol 23: 546–554.
Egea R, Casillas S, Barbadilla A . (2008). Standard and generalized McDonald–Kreitman test: a website to detect selection by comparing different classes of DNA sites. Nucleic Acids Res 36: W157–W162.
Elith J, Graham CH, Anderson RP, Dudıḱ M, Ferrier S, Guisan A et al. (2006). Novel methods improve prediction of species’ distributions from occurrence data. Ecography 29: 129–151.
Excoffier L, Foll M, Petit JR . (2009). Genetic consequences of range expansions. Annu Rev Ecol Evol S 40: 481–501.
Fay JC, Wu CI . (1999). A human population bottleneck can account for the discordance between patterns of mitochondrial versus nuclear DNA variation. Mol Biol Evol 16: 1003–1005.
Felsenstein J . (1981). Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 17: 368–376.
Fløjgaard C, Normand S, Skov F, Svenning J-Ch . (2009). Ice age distributions of European small mammals: insights from species distribution modelling. J Biogeogr 36: 1152–1163.
Hassanin A, Ropiquet A, Couloux A, Cruaud C . (2009). Evolution of the mitochondrial genome in mammals living at high altitude: new insights from a study of the tribe Caprini (Bovidae, Antilopinae). J Mol Evol 68: 293–310.
Hernandez PA, Graham CH, Master LL, Albert DL . (2006). The effect of sample size and species characteristics on performance of different species distribution modeling methods. Ecography 29: 773–785.
Hillisch A, Pineda LF, Hilgenfeld R . (2004). Utility of homology models in the drug discovery process. Drug Discov Today 9: 659–669.
Huang L, Cobessi D, Tung EY, Berry EA . (2005). Binding of the respiratory chain inhibitor antimycin to the mitochondrial bc1 complex: a new crystal structure reveals an altered intramolecular hydrogen-bonding pattern. J Mol Biol 351: 573–597.
Iwata S, Lee JW, Okada K, Lee JK, Iwata M, Rasmussen B et al. (1998). Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex. Science 281: 64–71.
Johns GC, Avise JC . (1998). A comparative summary of genetic distances in the vertebrates from the mitochondrial cytochome b gene. Mol Biol Evol 15: 1481–1490.
Ketola T, Boratyński Z, Kotiaho J . (2014). Manipulating genetic architecture to reveal fitness relationships. Proc PoS 1: e1.
Kolling DRJ, Samoilova RI, Holland JT, Berry EA, Dikanov SA, Crofts AR . (2003). Exploration of ligands to the Qi site semiquinone in the bc1 somplex using high-resolution EPR. J Biol Chem 278: 39747–39754.
Lehtonen JV, Still DJ, Rantanen VV, Ekholm J, Björklund D, Iftikhar Z et al. (2004). BODIL: a molecular modeling environment for structure-function analysis and drug design. J Comput Aided Mol Des 18: 401–419.
Mallet J . (2005). Hybridization as an invasion of the genome. Trends Ecol Evol 20: 229–237.
Melo-Ferreira J, Boursot P, Carneiro M, Esteves PJ, Farelo L, Alves PC . (2012). Recurrent introgression of mitochondrial DNA among hares (Lepus spp.) revealed by species-tree inference and coalescent simulation. Syst Biol 61: 367–381.
Melo-Ferreira J, Alves PC, Freitas H, Ferrand N, Boursot P . (2009). The genomic legacy from the extinct Lepus timidus to the three hare species of Iberia: contrast between mtDNA, sex chromosomes and autosomes. Mol Ecol 18: 2643–2658.
Nespolo RF, Bacigalupe LD, Figueroa CC, Koteja P, Opazo JC . (2011). Using new tools to solve an old problem: the evolution of endothermy in vertebrates. Trends Ecol Evol 26: 414–423.
Nielsen R . (2005). Molecular signatures of natural selection. Annu Rev Genet 39: 197–218.
Phillips SJ, Dudík M . (2008). Modeling of species distributions with Maxent: new extensions and a comprehensive evaluation. Ecography 31: 161–175.
Pinho C, Hey J . (2010). Divergence with gene flow: models and data. Annu Rev Ecol Evol S 41: 215–230.
Pons J-M, Sonsthagen S, Dove C, Crochet P . (2013). Extensive mitochondrial introgression in North American Great Black-backed Gulls (Larus marinus) from the American Herring Gull (Larus smithsonianus) with little nuclear DNA impact. Heredity 112: 226–239.
Rice P, Longden I, Bleasby A . (2000). EMBOSS: The European molecular biology open software suite. Trends Genet 16: 276–277.
Riise EK, Lorentzen MS, Helland R, Smalås AO, Leiros H-S, Willassen NP . (2007). The first structure of a cold-active catalase from Vibrio salmonicida at 1.96 Å reveals structural aspects of cold adaptation. Acta Crystallogra D 63: 135–148.
Rosas U, Barton NH, Copsey L, de Reuille PB, Coen E . (2010). Cryptic variation between species and the basis of hybrid performance. PLoS Biol 8: e1000429.
Sali A, Blundell TL . (1993). Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234: 779–815.
Schluter D, Conte GL . (2009). Genetics and ecological speciation. Proc Natl Acad Sci USA 106: 9955–9962. 9.
Šíchová K, Koskela E, Mappes T, Lantová P, Boratyński Z . (2014). On personality, energy metabolism, and mtDNA introgression in bank voles, Myodes glareolus. Anim Behav in press.
Stephenson N . (1998). Actual evapotranspiration and deficit: biologically meaningful correlates of vegetation distribution across spatial scales. J Biogeogr 25: 855–870.
Tajima F . (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595.
Tegelström H . (1987). Transfer of mitochondrial DNA from the northern red-backed vole (Clethrionomys rutilus) to the bank vole (C. glareolus). J Mol Evol 24: 218–227.
Toews DPL, Brelsford A . (2012). The biogeography of mitochondrial and nuclear discordance in animals. Mol Ecol 21: 3907–3930.
Toyota E, Ng KK, Kuninaga S, Sekizaki H, Itoh K, Tanizawa K et al. (2002). Crystal structure and nucleotide sequence of an anionic trypsin from Chum Salmon (Oncorhynchus keta) in comparison with Atlantic Salmon (Salmo salar) and bovine trypsin. J Mol Biol 324: 391–397.
Yang Z . (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24: 1586–1591.
Yang Z, Bielawski JP . (2000). Statistical methods for detecting molecular adaptation. Trends Ecol Evol 15: 496–503.
Zhai W, Nielsen R, Slatkin M . (2009). An investigation of the statistical power of neutrality tests based on comparative and population genetic data. Mol Biol Evol 26: 273–283.
Acknowledgements
We acknowledge Barbara Tschirren for providing samples from her study populations, Sami Kyröläinen for laboratory efforts, Matti Koivula and Tom Hoogesteger for help in field work, Jackson H Jennings (also for revising the language), Maciej Pabijan and Sara Rocha for suggestions to manuscript, and Konnevesi Research Station and the Experimental Animal Unit of the University of Jyväskylä for providing the facilities. The study was financially supported by the Academy of Finland (Grant no. 257340 to EK; 132190 to TM), the University of Jyväskylä (grants to ZB and MY), the National Doctoral Programme in Informational and Structural Biology (to MY), the Centre of Excellence in Evolutionary Research of the Academy of Finland and Portuguese Foundation for Science and Technology, FCT (Project Grant PTDC/BIA-EVF/111931/2009, partially funded by Fundo Europeu de Desenvolvimento Regional through the COMPETE program FCOMP-01-0124-FEDER-014266). ZB, JM-F and PT are postdoctoral grantees of the FCT, Portugal (ZB: SFRH/BPD/84822/2012, JM-F: SFRH/BPD/43264/2008 and PT: SFRH/BD/42480/2007, both POPH-QREN funds from the European Social Fund and Portuguese Ministry of Education and Science). MY is grantee of the National Doctoral Programme in Informational and Structural Biology, Finland. ZB was a Marie Curie Host Fellow (fellow/contract number: 00-00133-18/HPMT-CT-2000-00133), Finland.
Author contributions
The work was conducted as a part of ZB postdoctoral research. ZB designed and carried out fieldwork, genetic analyses and prepared the manuscript. EK and TM participated in designing study, fieldwork, laboratory work and in manuscript preparation. SB conducted PCR and sequencing analyses and participated in manuscript preparation. PCA and JM-F participated in the interpretation of results and manuscript preparation. PT conducted ecological modelling and participated in manuscript preparation. OTP and MY modelled and analysed the protein structures and participated in manuscript preparation. All authors read and approved the final version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Rights and permissions
About this article
Cite this article
Boratyński, Z., Melo-Ferreira, J., Alves, P. et al. Molecular and ecological signs of mitochondrial adaptation: consequences for introgression?. Heredity 113, 277–286 (2014). https://doi.org/10.1038/hdy.2014.28
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2014.28
